
Abstract
N-methyl-D-aspartate receptors (NMDARs) are ionotropic glutamate receptors that play an essential role in mediating excitatory neurotransmission in the mammalian central nervous system (CNS). Functional NMDARs are tetramers composed of GluN1, GluN2A-D, and/or GluN3A-B subunits, giving rise to a wide variety of NMDAR subtypes with unique functional properties. Here, we examined the surface delivery and functional properties of NMDARs containing mutations in the glycine-binding sites in GluN1 and GluN3A subunits expressed in mammalian cell lines and primary rat hippocampal neurons. We found that the structural features of the glycine-binding sites in both GluN1 and GluN3A subunits are correlated with receptor forward trafficking to the cell surface. In addition, we found that a potentially clinically relevant mutation in the glycine-binding site of the human GluN3A subunit significantly reduces surface delivery of NMDARs. Taken together, these findings provide novel insight into how NMDARs are regulated by their glycine-binding sites and may provide important information regarding the role of NMDARs in both physiological and pathophysiological processes in the mammalian CNS.
Similar content being viewed by others
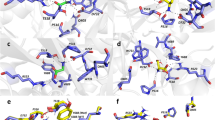


Introduction
N-methyl-D-aspartate receptors (NMDARs) are a subclass of glutamate receptors that play an essential role in synapse development, excitatory neurotransmission as well as synaptic plasticity in the mammalian central nervous system (CNS)1,2. It has been well established that dysregulation of NMDARs plays a critical role in the aetiology of many neuropsychiatric and neurological disorders and conditions, including Huntington’s disease3,4, schizophrenia5, cocaine addiction6, and nicotine dependence7. In addition, a growing number of studies suggest that many neuropsychiatric disorders are associated with mutations in genes that encode various NMDAR subunits, including the GluN18,9, and GluN3A10,11 subunits. Thus, understanding the molecular mechanisms that regulate NMDARs is an essential step towards designing effective therapies for these patients.
Structurally, NMDARs are heterotetramers composed of GluN1 (with eight splice variants), GluN2 (GluN2A through GluN2D), and/or GluN3 (GluN3A and GluN3B) subunits2,12. All GluN subunits have a common membrane topology, including an extracellular amino-terminal domain (ATD), extracellular ligand-binding domains (LBDs) formed by S1 and S2 segments, four membrane domains (M1 through M4), and an intracellular C-terminal domain (CTD)2,12. The conventional NMDAR subtype ‒ GluN1/GluN2 ‒ is activated by the binding of agonists to the glutamate-binding site in the LBD of GluN2 together with the simultaneous binding of a co-agonist to the glycine-binding site in the LBD of GluN12,13,14,15. Interestingly, unconventional NMDAR subtypes ‒ namely, GluN1/GluN3A and GluN1/GluN3B receptors ‒ are activated by the binding of agonist to the glycine-binding site in the LBD of the GluN3 subunit, whereas binding of a co-agonist to the glycine-binding site in the LBD of GluN1 drives the desensitisation of glycine-induced currents in GluN1/GluN3 receptors16,17,18,19. Thus, the glycine-binding sites in the LBD of various GluN subunits play distinct functional roles in NMDARs.
Both the number and type of NMDARs present at the neuronal surface are regulated at multiple levels20,21,22, including their synthesis23,24, subunit assembly25,26,27,28, processing within the endoplasmic reticulum (ER)29,30,31,32,33,34,35,36, trafficking to the cell membrane37,38,39, lateral diffusion40,41, internalisation/recycling42,43,44,45, and degradation45,46. The ER quality control retains unassembled GluN1 (except the GluN1-2, GluN1-3, and GluN1-4 splice variants)31,32,33, GluN234,35, and GluN3 subunits30,36. This is supported by experiments in mice that lack the GluN1 subunit in the hippocampus, in which GluN2 subunits accumulate in the ER47. In addition, a wide variety of regions in GluN subunits ‒ including the LBD ‒ are critical for regulating the surface delivery of NMDARs48,49. Specifically, Kenny et al. previously reported that disrupting the glycine-binding site in the LBD of GluN1 by introducing the D732A mutation reduces the surface delivery of GluN1/GluN2A receptors49. Similarly, She et al. reported that the glutamate-binding site in the LBD of GluN2B regulates the surface delivery of GluN1/GluN2B receptors48; this finding was supported by a recent study using human GluN1/GluN2B receptors with known pathogenic mutations50. However, whether structural changes in the glycine-binding sites in the GluN1 and/or GluN3A subunits regulate the surface delivery of functional GluN3A-containing NMDARs is currently unknown.
Here, we used a combination of microscopy, quantitative assays and electrophysiology in mammalian cell lines and primary rat hippocampal neurones in order to investigate whether disrupting the structure of the glycine-binding sites in the LBDs of GluN1 and/or GluN3A subunits affects the surface delivery of GluN3A-containing NMDARs. We found that mutant GluN1 and GluN3A subunits have reduced glycine sensitivity, and this reduction is correlated with reduced forward trafficking of NMDARs to the cell surface. These results were supported by additional experiments using the human GluN3A subunit. Taken together, our results provide novel insight into the mechanisms that regulate the number, type, and function of NMDARs at the neuronal surface.
Results
The S1 segment of the LBD in GluN3A subunit is essential for the surface delivery of GluN1/GluN3A receptors
Previous studies revealed that different domains in GluN1 and GluN2 regulate the surface expression of NMDARs2,20. We therefore examined whether the N-terminal and/or C-terminal regions of GluN3A subunits regulate the surface expression of NMDARs by expressing wild-type and mutant GluN1 and GluN3A subunits in COS-7 cells (a cell line derived from African green monkey kidney fibroblast; acronym “COS” is derived from the cells being CV-1 (simian) in Origin, and carrying the SV40 genetic material) and Human Embryonic Kidney 293 (HEK293) cells (which do not express endogenous GluN subunits), and in cultured hippocampal neurones. Our group51 and others52,53 previously reported that GluN1-4a/GluN3A receptors produce larger glycine-induced currents compared to GluN1-1a/GluN3A receptors; therefore, we used the GluN1-4a subunit in our experiments with COS-7 and HEK293 cells.
We first generated rat GluN3A subunits that lack the entire CTD (GluN3A-ΔCTD) or the entire ATD and S1 segment (GluN3A-ΔATD + ΔS1; Fig. 1a) and expressed each GluN3A subunit in COS-7 cells either alone or together with GluN1-4a subunit (Fig. 1b). We found that GluN1-4a/GluN3A-ΔCTD receptors are delivered to the surface at wild-type levels; in contrast, GluN1-4a/GluN3A-ΔATD + ΔS1 receptors failed to traffic to the cell surface (Fig. 1b,c). Next, we examined receptors containing GluN3A subunits that lack either the ATD (GluN3A-ΔATD) or the S1 segment (GluN3A-ΔS1; Fig. 1a) ‒ and found that GluN1-4a/GluN3A-ΔATD receptors are expressed at the cell surface (albeit at reduced levels compared to wild-type GluN1-4a/GluN3A receptors), whereas GluN1-4a/GluN3A-ΔS1 receptors failed to traffic to the cell surface (Fig. 1b,c; see also the Discussion). Consistent with our above-mentioned findings, when expressed alone (i.e. without GluN1) the truncated GluN3A subunits failed to reach the cell surface (Fig. 1c). The total expression levels were not significantly different among the studied GluN subunit combinations (Supplementary Fig. S1). These findings indicate that the S1 segment of the GluN3A subunit is required for the surface delivery of GluN1/GluN3A receptors.

The S1 segment in the GluN3A subunit is essential for the surface expression of GluN1/GluN3A receptors. (a) Schematic diagram of the GluN3A subunit, with the amino-terminal domain (ATD), ligand-binding domain (LBD) composed of S1 and S2 segments, and C-terminal domain (CTD) indicated; the black rectangles indicate membrane domains. (b) Representative images of total and surface GluN1-4a/GFP-GluN3A receptors lacking the indicated domains in the GluN3A subunit expressed in COS-7 cells and labelled 24 h after transfection. (c) Summary of the relative surface expression of the indicated GluN subunits measured using fluorescence microscopy (n ≥ 20 cells per group); *p < 0.05 vs. GluN1-4a/GluN3A (ANOVA).
The glycine-binding sites in both GluN1 and GluN3A subunits regulate the number of NMDARs at the cell surface and their function in both cell lines and hippocampal neurones
Previous studies showed that the surface delivery of GluN1/GluN2 receptors is regulated by the glycine-binding site in the GluN1 subunit49 and the glutamate-binding site in the GluN2 subunit48. Here, we examined whether the glycine-binding sites in the GluN1 or GluN3A subunits also regulate the surface delivery of GluN3A-containing NMDARs.
We first used mutations in the GluN1 subunit that were shown previously to alter the EC50 for glycine binding to GluN1/GluN2 receptors17,54 (Fig. 2a). Specifically, we introduced the A714L mutation in GluN1-4a subunit, which stabilises the open conformation of the LBD and slightly reduces the receptor’s sensitivity to glycine; other mutations in GluN1-4a subunit included F484A, T518L17, and D732A49,54, all of which reduce glycine affinity by two orders of magnitude. In addition, we used the double mutant GluN1-4a-F484A + T518L subunit, which was previously reported to be glycine-insensitive at concentrations up to 30 mM when co-expressed with the GluN2A subunit17.

Mutations in the glycine-binding site in the GluN1 subunit alter the surface expression and desensitisation kinetics of GluN1/GluN3A receptors. (a) Schematic representation of the glycine-binding site in the GluN1 subunit (PDB code: 1PB7); the amino acid residues studied here, and a glycine molecule, are shown. (b) Representative images of COS-7 cells transfected with the indicated wild-type or mutant GluN1-4a subunits together with GFP-GluN3A (GluN3A) subunit and labelled 24 h after transfection. (c) Summary of the relative surface expression of the indicated GluN subunits measured using fluorescence microscopy (n ≥ 24 cells per group); *p < 0.05 vs. GluN1-4a/GluN3A (ANOVA). (d) Quantification of relative surface expression of the indicated GluN1-4a/GFP-GluN3A (GluN1-4a/GluN3A) receptors using quantitative colorimetric assay is shown (n = 6); *p < 0.05 relative to GluN1-4a/GluN3A (ANOVA). (e) Representative whole-cell patch-clamp recordings from HEK293 cells transfected with the indicated wild-type or mutant GluN1-4a/GluN3A receptors. Currents were elicited by applying glycine at the indicated concentrations. (f) Summary of the τw of desensitisation for the indicated GluN1-4a/GluN3A receptors (n ≥ 5 cells per group). (g) Peak concentration-response curves for the indicated GluN1-4a/GluN3A receptors. Each data point represents the mean relative current from ≥5 independent cells. The EC50 values and Hill coefficients are listed in Table 1.
First, we co-expressed each mutant GluN1-4a subunit together with GluN3A subunit in COS-7 cells and measured surface expression using fluorescence microscopy (Fig. 2b,c) and in HEK293 cells using quantitative assay of surface expression (Fig. 2d). The total expression levels were not significantly different among the studied GluN subunit combinations (Supplementary Fig. S1). We found that the surface expression was highest for GluN1-4a/GluN3A receptors, followed by (in decreasing order of surface expression) GluN1-4a-A714L/GluN3A, GluN1-4a-F484A/GluN3A, GluN1-4a-T518L/GluN3A, GluN1-4a-D732A/GluN3A, and GluN1-4a-F484A + T518L/GluN3A receptors; interestingly, this rank order corresponds to the previously determined order of glycine EC50 values for GluN1/GluN2 receptors containing these mutant GluN1 subunits (Supplementary Fig. S2).
Because the previously assessed EC50 values for glycine were determined using GluN1/GluN2 receptors expressed in Xenopus oocytes, they might not necessarily reflect the change in glycine affinity of GluN1/GluN3A receptors expressed in mammalian cells. We therefore measured the whole-cell currents induced by increasing concentrations of glycine (ranging from 10 µM to 10 mM) in HEK293 cells co-expressing GluN1/GluN3A subunits (Fig. 2e); importantly, using a rapid solution exchange system enabled us to detect the peak glycine-induced current prior to receptor desensitisation. In these experiments, we transfected HEK293 cells using different amounts of GluN cDNAs, as some GluN1-4a/GluN3A combinations otherwise yielded small glycine-induced currents (the following ratios of cDNAs encoding the GluN1-4a and GluN3A subunits were used: GluN1-4a/GluN3A (1:1), GluN1-4a-A714L/GluN3A (1:1), and GluN1-4a-F484A/GluN3A (1:1); GluN1-4a-T518L/GluN3A (2:1), GluN1-4a-D732A/GluN3A (2:1), and GluN1-4a-F484A + T518L/GluN3A (2:1)); therefore, we were unable to directly compare the absolute peak current amplitudes among the various GluN1-4a/GluN3A receptors. Nevertheless, we found that the time constant of desensitisation (τw) differed among the various mutant receptors tested and corresponded with the same rank order that we observed for surface delivery (Fig. 2f and Supplementary Fig. S2). Maximum steady-state currents carried by wild-type GluN1-4a/GluN3A receptors activated by 1 mM glycine were in range of 5 to 25 pA (n = 10) which precluded reliable concentration-response analysis of steady-state currents. Our analysis of the peak concentration-response relationship revealed that only the GluN1-4a-F484A mutation caused a rightward shift of the curve (Fig. 2g), resulting in an ~5-fold decrease in glycine potency compared to wild-type GluN1-4a/GluN3A receptors (Table 1); this finding is consistent with previous reports that activation of GluN1/GluN3A receptors is regulated primarily by the glycine-binding site in GluN3A subunit16,17,18. In contrast, the Hill coefficient (h) was increased for all receptors containing mutant GluN1-4a subunits compared to wild-type receptors (Table 1), which indicates that the mutations change the degree of cooperativity between the glycine-binding sites in the GluN1 subunits55. In contrast to our results described above, the determined EC50 values for the mutant GluN1-4a/GluN3A receptors tested did not correlate with their surface expression levels (Supplementary Fig. S2). Cells expressing GluN1-4a-D732A/GluN3A receptors had no detectable currents, even at the highest glycine concentration tested; moreover, at all glycine concentrations tested, currents measured through GluN1-4a-F484A + T518L/GluN3A receptors did not desensitise (Fig. 2e). Thus, we were unable to perform detailed functional analyses of these two mutant receptors (see also the Discussion). Taken together, these data support the hypothesis that structural changes in the glycine-binding site in the GluN1 subunit are associated with changes in the surface delivery and the desensitisation properties of GluN1/GluN3A receptors.
Given that the glycine-binding sites in the LBD of GluN1 and GluN3A subunits are highly conserved, we next asked whether similar structural changes in the glycine-binding site in the GluN3A subunit can also affect the surface delivery of GluN1/GluN3A receptors. Accordingly, we introduced point mutations in GluN3A subunit at the positions analogous to GluN1 subunit, yielding four mutant GluN3 subunits (GluN3A-T825L, GluN3A-Y605A, GluN3A-S633L, and GluN3A-D845A) and the double mutant GluN3A-Y605A + S633L subunit (Fig. 3a); each subunit was then co-expressed together with GluN1-4a subunit in COS-7 cells and surface expression was examined using microscopy (Fig. 3b,c) and in HEK293 cells using quantitative assay of surface expression (Fig. 3d). The total expression levels were not significantly different among the studied GluN subunit combinations (Supplementary Fig. S1). Interestingly, the rank order of surface expression for the receptors containing mutant GluN3A subunits was similar to the order that we observed with the analogous mutant GluN1 subunits (Supplementary Fig. S2). Specifically, receptors containing the GluN3A-T825L subunit had the highest surface expression, followed by (in decreasing order) GluN3A-Y605A, GluN3A-S633L, GluN3A-D845A, and GluN3A-Y605A + S633L subunits.

Mutations in the glycine-binding site in the GluN3A subunit regulate the surface expression and glycine affinity of GluN1/GluN3A receptors. (a) Schematic representation of the glycine-binding site in the GluN3A subunit (PDB code: 2RC7; in orange), with a glycine molecule shown; for comparison, the binding site in the GluN1 subunit is shown in blue. (b) Representative images of COS-7 cells transfected with the indicated wild-type or mutant GFP-GluN3A subunit together with GluN1-4a subunit and labelled 24 h after transfection. (c) Summary of the relative surface expression of the indicated GluN subunits measured using fluorescence microscopy (n ≥ 20 cells per group); *p < 0.05 vs. GluN1-4a/GluN3A (ANOVA). (d) Heterologous HEK293 cells co-expressing the indicated GluN1-4a and GFP-GluN3A (GluN3A) subunits were labeled with primary anti-GFP and secondary antibodies in non-permeabilizing and permeabilizing conditions. The bar graphs show quantification of relative surface expression of the indicated GluN subunit combinations obtained using quantitative colorimetric assay (n = 6); *p < 0.05 relative to GluN1-4a/GluN3A (ANOVA). (e,h) Representative whole-cell patch-clamp recordings from HEK293 cells transfected with the indicated wild-type or mutant GluN1-4a/GluN3A receptors. Currents were elicited by applying the indicated concentrations of glycine. (f) Summary of the τw of desensitisation for the indicated GluN1-4a/GluN3A (n ≥ 5 cells per group). (g) Peak concentration-response curves for the indicated wild-type and mutant GluN1-4a/GluN3A receptors. Each data point represents the mean relative current from ≥5 cells. The EC50 values and Hill coefficients are listed in Table 1. (i) The pharmacological analysis with CGP-78608 at the GluN1/GluN3A receptors. Representative whole-cell patch-clamp recordings from HEK293 cells transfected with the indicated wild-type or mutant GluN1-4a/GluN3A receptors at a membrane potential of −60 mV. Currents were elicited by applying 1,000 µM glycine; 0.5 µM CGP78608 was applied as indicated. (j) Summary of current densities (pA/pF) obtained from the HEK293 cells expressing the indicated GluN1-4a/GluN3A receptors (n ≥ 6 cells per group).
Next, we measured the functional properties of GluN1-4a/GluN3A receptors containing wild-type or mutant GluN3A subunits expressed in HEK293 cells in order to examine the effect of these mutations on glycine affinity. We found that both GluN1-4a/GluN3A-T825L and GluN1-4a/GluN3A-Y605A receptors produced detectable currents (1:2 ratios of cDNAs encoding the GluN1-4a and GluN3A subunits were used), whereas the other three mutant receptors failed to produce measurable currents, even with 10 mM glycine (Fig. 3e). Moreover, these two mutant receptors had a τw of desensitisation similar to wild-type GluN1-4a/GluN3A receptors (Fig. 3f), which is consistent with the notion that the desensitisation of GluN1/GluN3A receptors is regulated by the glycine-binding site in the GluN1 subunit. An analysis of the peak concentration-response relationship revealed that glycine is 9-fold and 29-fold less potent at activating GluN1-4a/GluN3A-T825L and GluN1-4a/GluN3A-Y605A receptors, respectively, compared to wild-type GluN1-4a/GluN3A receptors (Fig. 3g and Table 1), which clearly correlates with their surface expression levels (Supplementary Fig. S2). In addition, the decreased glycine potency in GluN1-4a/GluN3A-T825L receptors was accompanied by an increase in the Hill coefficient (Table 1), suggesting a change in the degree of cooperativity between the glycine-binding sites in the GluN3A subunits. These data support the hypothesis that activation of GluN1/GluN3A receptors is mediated by glycine binding to the GluN3A subunit.
As noted above, we measured no detectable currents in cells expressing GluN1-4a/GluN3A-S633L, GluN1-4a/GluN3A-D845A, or GluN1-4a/GluN3A-Y605A + S633L receptors (Fig. 3e). This finding may be explained by the greatly reduced number of receptors at the cell surface, a loss of glycine binding, and/or a change in the desensitisation properties of these mutant receptors. To address these possibilities, we co-expressed GluN3A-S633L, GluN3A-D845A, and GluN3A-Y605A-S633L subunits together with the mutant GluN1-4a-F484A subunit, which slows the receptor’s desensitisation kinetics (Fig. 2f) and does not prevent surface delivery of the GluN1/GluN3A receptors (Fig. 2). We found that cells expressing GluN1-4a-F484A/GluN3A-S633L receptors had detectable currents when glycine was applied at 1, 3, or 10 mM, whereas no currents were detected in cells expressing GluN1-4a-F484A/GluN3A-D845A or GluN1-4a-F484A/GluN3A-Y605A + S633L receptors, even with 10 mM glycine (Fig. 3h). In addition, we performed electrophysiological recordings with compound CGP-78608 using HEK293 cells expressing GluN3A, GluN3A-S633L, GluN3A-D845A, and GluN3A-Y605A-S633L subunits together with the GluN1-4a subunit. In the agreement with recent study56, we observed profound potentiating effect of CGP-78608 at the wild-type GluN1-4a/GluN3A receptors exposed to 1 mM (Fig. 3i,j) or 10 mM (Supplementary Fig. S3) glycine. However, by using the CGP-78608 with 1 mM glycine (Fig. 3i,j) or even with 10 mM glycine (Supplementary Fig. S3) we were not able to observe responses from the HEK293 cells expressing the GluN1-4a/GluN3A-S633L, GluN1-4a/GluN3A-D845A and GluN1-4a/GluN3A-Y605A + S633L receptors. Together, our data suggest that the glycine-binding sites in both the GluN1 and GluN3A subunits regulate the surface delivery of GluN1/GluN3A receptors in the mammalian cell lines.
In order to separate the relative contributions of forward trafficking from endocytosis, we employed a mutated version of K44A HA-dynamin 2 (dynamin-K44A) which inhibits clathrin-dependent endocytosis, using similar approach as reported previously for the mutated GluN1/GluN2 receptors48. We performed the quantitative assays on the HEK293 cells expressing the GluN1/GluN3A receptor combinations which exhibited significantly reduced surface expression (Figs 2d and 3d), in the presence of dynamin-K44A. In these experiments, we did not observe altered pattern of surface delivery or total expression of the respective mutated GluN1/GluN3A receptors (Fig. 4a,b and Supplementary Fig. S1; compare with Figs 2d and 3d and Supplementary Fig. S1), indicating that glycine binding regulates forward trafficking rather than internalization of GluN1/GluN3A receptors. To further corroborate this conclusion, we next labelled the COS-7 cells expressing the mutated GluN1/GluN3A receptors which exhibited reduced surface delivery in the experiments described above, with a Golgi apparatus (GA) marker Golgi matrix protein 130 (GM130; Fig. 4c,e). Our microscopical analysis revealed significantly decreased colocalisation of the GluN1-4a-T518L/GluN3A, GluN1-4a-D732A/GluN3A and GluN1-4a-F484A + T518L/GluN3A and GluN3A subunit combinations with the GM130 when compared with the wild-type GluN1-4a/GluN3A receptors (Fig. 4d). Concerning the GluN1-4a-F484A/GluN3A receptors, we did not observe significant tendency for their reduced colocalisation with the GM130, which resembles previously reported finding with the GluN1/GluN2B-S664G receptors48. Our microscopical analysis further revealed decreased colocalisation of the GluN1-4a/GluN3A-S633L, GluN1-4a/GluN3A-D845A and GluN1-4a/GluN3A-Y605A + S633L receptors with the GM130 when compared with the wild-type GluN1-4a/GluN3A receptors (Fig. 4f). Together, these data indicate that impaired glycine binding reduces forward trafficking of GluN1/GluN3A receptors to the cell surface.

The glycine-binding sites in the GluN1 and GluN3A subunits regulate forward trafficking of GluN1/GluN3A receptors (a,b) HEK293 cells co-expressing the indicated GluN1-4a and GFP-GluN3A (GluN3A) subunits together with the dynamin-K44A were labelled with primary anti-GFP and secondary antibodies in non-permeabilizing and permeabilizing conditions. The bar graphs show quantification of relative surface expression of the indicated GluN subunit combinations obtained using quantitative colorimetric assay (n = 6); *p < 0.05 relative to GluN1-4a/GluN3A (ANOVA). (c,e) Representative confocal microscopy images of the COS-7 cells transfected with the indicated GluN1-4a and GFP-GluN3A (GluN3A) subunits; the anti-GM130 antibody was used to label the GA. (d,f) Summary of the average intensity of GFP-GluN3A (GluN3A) subunit signal colocalised with GM130 over the average intensity of GFP-GluN3A (GluN3A) subunit signal outside GM130 signal, calculated for the indicated GluN subunit combinations (n ≥ 10 cells); *p < 0.05 vs. GluN1-4a/GluN3A; ANOVA).
Next, we asked whether the introducing of mutations in the glycine-binding sites of GluN subunits also affects the trafficking of NMDARs in neurones. We therefore transfected cultured rat hippocampal neurones at DIV10 with YFP-tagged wild-type or mutant GluN1-1a subunits (we chose this splice variant because it is not delivered to the cell surface unless associated with the GluN2 and/or GluN3 subunit) or GFP-tagged wild-type or mutant GluN3A subunits. Four days after transfection (i.e. at DIV14), we then measured the surface and total pools of recombinant GluN subunits using labelling with an anti-GFP antibody and confocal microscopy. Our analysis revealed the following rank order for the surface delivery of mutant GluN1-1a subunits (in decreasing order): GluN1-1a-A714L, GluN1-1a-F484A, GluN1-1a-T518L, GluN1-1a-D732A, and GluN1-1a-F484A + T518L (Fig. 5a,b). Similarly, all five mutant GluN3 subunits exhibited the following rank order of surface delivery (in decreasing order): GluN3A-T825L, GluN3A-Y605A, GluN3A-S633L, GluN3A-D845A, and GluN3A-Y605A + S633L (Fig. 5c,d). The total expression levels were not significantly different among the studied GluN subunits (Supplementary Fig. S1). These results are consistent with our cell surface expression data in cell lines and support the notion that the glycine-binding sites in both the GluN1 and GluN3A subunits are critical for surface delivery of NMDARs.

The glycine-binding sites in the GluN1 and GluN3A subunits regulate the surface expression of NMDARs in hippocampal neurones. (a,c) Representative images of cultured rat hippocampal neurones transfected at DIV10 with the indicated YFP-GluN1-1a (a) or GFP-GluN3A (c) subunits; at DIV14, surface and total subunits were stained using an anti-GFP antibody. (b,d) Summary of the relative surface expression of the indicated subunits measured in 10-µm segments of secondary or tertiary dendrites (n ≥ 30 segments in ≥ 6 different cells per group); *p < 0.05 vs. GluN1-1a (b) or GluN3A (d) (ANOVA).
Clinically relevant mutation in the glycine-binding site of GluN3A subunit alters the surface delivery of NMDARs
A growing body of data indicates that many neuropsychiatric disorders are associated with mutations in genes that encode NMDAR subunits, including the GluN3A subunit10,11; however, in most cases how these mutations affect the trafficking and/or function of NMDARs is unknown. To address this question, we performed a database search and identified two clinically relevant mutations in the human GluN3A (hGluN3A) subunit; both of these residues are conserved between rat and human GluN3A subunits (Fig. 6a). The N549S mutation is located within the LBD but is likely not involved in forming of the glycine-binding site, whereas the D845N mutation is present within the glycine-binding site (Fig. 6b; see the Discussion for further information).

Clinically relevant mutation in the LBD of the GluN3A subunit alters the surface delivery of NMDARs. (a) Sequence alignment of the rat and human GluN3A subunits, with two clinically relevant mutations in the LBD shown in black rectangles. The numbers refer to the amino acid residues. (b) Schematic representation of the part of the LBD of the GluN3A subunit (PDB code: 2RC7); the amino acid residues studied here, and a glycine molecule, are shown. (c) Representative images of COS-7 cells transfected with the indicated human GFP-GluN3A (hGluN3A) and GluN1-4a (hGluN1-4a) subunits. (d) Summary of the relative surface expression of the indicated hGluN1-4a/hGluN3A subunits measured using fluorescence microscopy (n ≥ 18 cells per group); *p < 0.05 vs. hGluN1-4a/hGluN3A (ANOVA). (e,g) Examples of whole-cell patch-clamp recordings from HEK293 cells transfected with the indicated hGluN1-4a/hGluN3A receptors. Currents were elicited by applying the indicated concentration of glycine. (f) Summary of the τw of desensitisation for the indicated hGluN1-4a/hGluN3A receptors (n ≥ 5 cells per group). (h) Peak concentration-response curves for the indicated wild-type and mutant hGluN1-4a/hGluN3A receptors. Each data point represents the mean relative currents recorded from ≥4 cells. The EC50 values and Hill coefficients are listed in Table 2. (i) Representative images of hippocampal neurones transfected at DIV10 with wild-type or mutant hGluN3A subunits and labelled at DIV14. (j) Summary of the relative surface expression of the indicated subunits measured in 10-µm segments of secondary or tertiary dendrites (n ≥ 50 segments from ≥10 different cells per group); *p < 0.05 vs. hGluN3A (ANOVA).
We first generated the human GluN1-4a subunit (hGluN1-4a) as well as GFP-tagged wild-type hGluN3A, and mutated hGluN3A-N549S, and hGluN3A-D845N subunits. We then co-expressed these hGluN1-4a and hGluN3A subunits in COS-7 cells and measured surface expression of the receptors containing either wild-type or mutant hGluN3A subunits using an anti-GFP antibody and fluorescent microscopy. We found that although hGluN1-4a/hGluN3A-N549S receptors were expressed at the cell surface at the same level as wild-type hGluN1-4a/hGluN3A receptors; the surface expression of hGluN1-4a/hGluN3A-D845N receptors was significantly reduced (Fig. 6c,d). The total expression levels were not significantly different among the studied hGluN subunit combinations (Supplementary Fig. S1).
Next, we measured the functional properties of mutant hGluN3A subunits co-expressed in HEK293 cells together with hGluN1-4a subunit (1:2 ratios of cDNAs encoding the hGluN1-4a and hGluN3A subunits were used). Consistent with robust expression at the cell surface, hGluN1-4a/hGluN3A-N549S receptors produced glycine-induced currents at all concentrations tested (Fig. 6e), with a τw of desensitisation similar to wild-type hGluN1-4a/hGluN3A receptors (Fig. 6f). In contrast ‒ and consistent with our surface delivery findings obtained with COS-7 cells ‒ no detectable glycine-induced currents were measured in HEK293 cells expressing hGluN1-4a/hGluN3A-D845N receptors, even at the highest glycine concentration tested (Fig. 6e). We therefore introduced the F484A mutation into the hGluN1-4a subunit and co-expressed this mutant hGluN1-F484A subunit together with wild-type or mutant hGluN3A subunits in HEK293 cells. As expected, both hGluN1-4a-F484A/hGluN3A and hGluN1-4a-F484A/hGluN3A-N549S receptors produced glycine-induced currents with reduced desensitisation kinetics (Fig. 6g). However, no detectable glycine-induced currents were measured in cells expressing hGluN1-4a-F484A/hGluN3A-D845N receptors (Fig. 6g), supporting our previous finding that the D845N mutation drastically reduces the surface delivery of hGluN1/hGluN3A receptors. Our analysis of the peak concentration-response relationship for wild-type and mutant hGluN1-4a/hGluN3A receptors revealed that glycine is ~2-fold less potent at activating both hGluN1-4a/hGluN3A-N549S and hGluN1-4a-F484A/hGluN3A-N549S receptors compared to their respective wild-type receptors (Fig. 6h and Table 2). The reduced glycine potency in both the hGluN1-4a/hGluN3A-N549S and hGluN1-4a-F484A/hGluN3A-N549S receptors was accompanied by a decrease in the Hill coefficient (Table 2), consistent with the altered cooperativity between the glycine-binding sites in the hGluN3A subunits.
Next, we measured the surface expression of wild-type and mutant hGluN3A subunits expressed in cultured hippocampal neurones, using the same strategy we used above with rat GluN subunits (see Fig. 5). Consistent with our data from COS-7 and HEK293 cells, we found that the hGluN3A-N549S subunit was delivered to the neuronal cell surface at the same level as the wild-type hGluN3A subunit, whereas the surface expression of the hGluN3A-D845N subunit was significantly reduced (Fig. 6i,j). The total expression levels were not significantly different among the studied hGluN3A subunits (Supplementary Fig. S1). Taken together, these data provide compelling evidence that the glycine-binding site in the GluN3A subunit plays a critical role in the surface delivery of both rat and human NMDARs.
Discussion
The relatively long N- and C-termini of GluN subunits is believed to allow the receptor to interact with a variety of proteins during its journey to the cell surface, at the cell surface, and during internalisation (for more details see Introduction section). We found that GluN1/GluN3A receptors with a truncated CTD in the GluN3A subunit are delivered to the cell surface at normal levels. Similarly, GluN1/GluN2B receptors in which the GluN2B subunit lacks most of its CTD are expressed at the cell surface34, whereas GluN1/GluN2C receptors lacking the CTD are expressed at the surface at reduced numbers57. Nevertheless, we cannot exclude the possibility that the CTD in the GluN3A subunit regulates other steps in the trafficking of GluN3A-containing NMDARs, including surface mobility and/or stability; here, however, we focused on the surface delivery of GluN1/GluN3A receptors containing mutations in the subunits’ glycine-binding sites.
In GluN1/GluN3A receptors, the glycine-binding site in GluN3A subunit activates the receptor, whereas the glycine-binding site in GluN1 subunit mediates the rapid desensitisation kinetics of glycine-induced currents16,17,18,19. Here, we analysed glycine-induced currents in order to measure the functional properties of various GluN1/GluN3A receptors containing mutations in the glycine-binding sites of both GluN1 and GluN3A subunits. We found that the changes in τw values for desensitisation of mutant GluN1/GluN3A receptors is consistent with previously measured changes of EC50 values for glycine binding to mutant GluN1/GluN2 receptors17,54. This finding is also consistent with previous studies that used mutant GluN1 subunits (e.g. GluN1-F484A subunit) to reduce the desensitisation kinetics of GluN1/GluN3A receptors17,52,53,56,58,59. The observed order of changes in EC50 values for the mutant GluN1-4a-F484A/GluN3A receptors based on peak current amplitude is similar to previous reports of steady-state currents16,17. Moreover, an analysis of peak currents enabled us to estimate the EC50 value for wild-type GluN1-4a/GluN3A receptors as well, which was previously not possible due to the virtually negligible steady-state currents when these receptors were expressed in Xenopus oocytes16,17,60. However, we observed an apparent rightward shift in the concentration-response curve for GluN1-4a/GluN3A receptors compared to previously reported estimates of glycine EC50 values for steady-state currents based on bell-shaped curves17 as well as to previously reported Kd values from binding studies using soluble LBDs61; this shift could be due to a variety of factors, including: i) decreased glycine affinity of the peak current compared to the steady-state current, ii) the fast desensitisation kinetics of GluN1/GluN3A receptors, and/or iii) the use of different expression systems. Furthermore, we found that both the GluN1/GluN3A-T825L and GluN1/GluN3A-Y605A receptors have similar desensitisation kinetics but markedly decreased glycine affinity compared to wild-type receptors. These findings support the conclusion that the glycine-binding site in GluN3A subunit is required for activating the GluN1/GluN3A receptors but does not contribute to the desensitisation of GluN1/GluN3A receptors. This conclusion is in agreement with recent elegant study using CGP-78608, that in addition revealed the presence of functional GluN1/GluN3A receptors in juvenile rat hippocampus56.
Our microscopy and quantitative assay data showed that analogous mutations in the glycine-binding sites of GluN1 and GluN3A subunits reduce the surface delivery of their respective NMDARs when expressed in both mammalian cell lines and hippocampal neurones. Given that endogenous GluN1, GluN2A-B, and GluN3 subunits are likely expressed in hippocampal neurones12,21,22,56,62, our data support the view that the structural changes in the glycine-binding sites in exogenous GluN1 and GluN3A subunits dominate the trafficking of NMDARs that include endogenous GluN subunits (for example, in the case of tri-heteromeric GluN1/GluN2/GluN3A receptors). Although the underlying mechanism by which mutations in the glycine-binding sites alters surface delivery is currently unknown, it is likely that the quality control machinery in the ER plays a role, as was shown previously for GluN1/GluN2 receptors as well as α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors (AMPARs)48,49,63. Glutamate may be present in the ER in the millimolar range64,65 and may be important for detecting properly assembled functional GluN1/GluN2 receptors and AMPARs via an ER-specific quality control mechanism. The precise concentration of glycine in the ER is unknown; however, given the role that glycine plays in various metabolic pathways66, it is likely present in sufficient amounts to interact with newly assembled receptors. In theory, a neurone could regulate the number of NMDARs at the cell surface by controlling the production of agonists such as glycine and D-serine or competitive antagonists such as kynurenic acid, which are found naturally in the mammalian CNS2,67. In addition, certain pharmacological compounds that act on the glycine-binding sites of GluN1 and/or GluN3A subunits may alter the surface expression of NMDARs, thereby providing a possible therapeutic strategy for certain pathological conditions. In this respect, it is important to note that treating cells with up to 1 mM glycine for 48 hours did not affect the surface expression of either wild-type or mutant GluN1/GluN3A receptors (Supplementary Fig. S4). This suggests that: i) intracellular glycine is already present at saturating concentrations in cultured cells, ii) high levels of extracellular glycine do not sufficiently alter the concentration of glycine within the ER, and/or iii) high levels of glycine do not regulate the intracellular processing of GluN1/GluN3A receptors.
Importantly, we also examined two putative pathogenic mutations in the LBD of the human GluN3A subunit. The N549S mutation is associated with the risk of nicotine dependence (rs75981117)68, and the D845N mutation has been classified by the UCSC browser as “clinically associated” (rs146060776). We found that the N549S mutation had no effect on the surface delivery or desensitisation kinetics of NMDARs and only slightly reduced the receptor’s affinity for glycine, consistent with the idea that the N549 residue in GluN3A subunit likely does not contribute directly to the subunit’s glycine-binding site. These conclusions are also in the agreement with our recent study which showed that the GluN3A-N549Q mutation (which should disrupt the predicted glycosylation of this residue) does not alter the surface delivery or desensitisation kinetics of the GluN1/GluN3A receptors51. In contrast, the D845N mutation drastically reduced the surface delivery of NMDARs, consistent with the high degree of homology between the rat and human GluN3A subunits (~89%) and the fact that we observed a similar reduction in the surface expression of the NMDARs containing the rat GluN3A-D845A subunit. Indeed, a growing number of reports have identified pathogenic mutations within GluN subunits, and new pathogenic mutations within the LBD ‒ particularly the glycine-binding site ‒ of both GluN1 and GluN3A subunits will likely be identified in future studies.
Taken together, our findings regarding the role of glycine-binding sites in GluN1 and GluN3A subunits in the surface delivery of NMDARs provide new previously overlooked insight into the physiological and pathophysiological regulation of these receptors in the mammalian CNS.
Methods
Molecular biology
Generation of the untagged rat GluN1-4a (GenBank: U08267.1; pcDNA1 vector), YFP-tagged rat GluN1-1a (GenBank: U08261; pcDNA3.1 vector)69,70, and GFP-tagged rat GluN3A (Gene ID: 191573; pCINeo vector)30 subunits have been described previously. The cDNA encoding the human GluN3A subunit was purchased from Origene, and the GFP-tagged human GluN3A (hGluN3A) construct was generated by cloning the human version of the GRIN3A gene into the rat GFP-GluN3A expression vector. The human version of the GluN1-4a subunit (hGluN1-4a) was generated by changing the four amino acid residues (N159S, R212K, I267L, M415L) that differ between the rat and human GluN1-4a subunits (differences in the signal peptides were ignored). All point mutations were introduced using the Quick-Change site-directed mutagenesis kit (Agilent Technologies) and were verified by DNA sequencing. K44A HA-dynamin 2 pcDNA3.1 (dynamin-K44A) was a gift from Sandra Schmid (Addgene plasmid # 34685; http://n2t.net/addgene:34685; RRID:Addgene_34685).
Mammalian cell culture
COS-7 and HEK293 cells were obtained from ATCC and cultured in Opti-MEM I (Thermo Fisher Scientific) containing 5% fetal bovine serum (Thermo Fisher Scientific)69,70. We employed the COS-7 cells for microscopy as these cells were also employed previously in two relevant papers concerning this study48,49. In addition, COS-7 cells are larger than HEK293 cells which is advantagenous for performing localisation studies. For electrophysiology, we used HEK293 cells because they are (i.) smaller than the COS-7 cells that enables us to better compensate patch-clamp recordings, (ii.) routinely used for electrophysiology of NMDARs in most laboratories. Finally, we employed the HEK293 cells for quantitative assays to compare the surface delivery of GluN1/GluN3A receptors in both cell lines. HEK293 cells and COS-7 cells were transfected with 2 µl Lipofectamine 2000 (Thermo Fisher Scientific) plus 900 ng of total cDNAs encoding the GluN1 and GluN3A subunits. For electrophysiology, transfected cells were trypsinised and grown at low density; cells intended for microscopy and quantitative assays were grown without the trypsinisation step. The experiments were performed 24–72 h after transfection.
Primary hippocampal neurones
All animal experiments were approved by the Animal Care and Use Committee of the Institute of Physiology of the Czech Academy of Sciences (CAS) and were conducted in accordance with the guidelines of the European Union directive 2010/63/EU. The Institute of Physiology CAS possesses the National Institutes of Health Statement of Compliance with Standards for Humane Care and Use of Laboratory Animals. Primary cultures of hippocampal neurones were prepared from embryonic day 18 Wistar rats and cultured in Neurobasal media supplemented with B-27 (Thermo Fisher Scientific) and L-glutamine (Thermo Fisher Scientific) using established protocols69. At DIV10, the neurones were transfected using Lipofectamine 2000 as described previously69.
Microscopy
COS-7 cells grown in 12-well plates were transfected with a mixture of Lipofectamine 2000 and 2:1 ratios of cDNAs encoding the GluN1-4a and GFP-GluN3A subunits were used (to minimize occurrence of GFP-positive cells lacking the expression of GluN1-4a subunits). The YFP-GluN1 or GFP-GluN3A subunits present at the cell surface of live cells were labelled using rabbit anti-GFP as the primary antibody (AB3080P, 1:1000; Merck) and an anti-rabbit antibody conjugated to Alexa Fluor 555 (A21429, 1:1000; Thermo Fisher Scientific) or Alexa Fluor 647 (A21244, 1:1000; Thermo Fisher Scientific) for COS-7 cells and hippocampal neurones, respectively; the antibodies were diluted in blocking solution containing 3% normal goat serum (Thermo Fisher Scientific)69,70. The stained cells were then fixed in 4% paraformaldehyde (PFA; Sigma-Aldrich) in phosphate-buffered saline (PBS) for 20 min, and mounted using ProLong Gold Antifade reagent (Thermo Fisher Scientific). For labelling the total pool of GluN subunits, fixed cells were permeabilised with 0.25% Triton X-100 (Serva) in PBS for 5 min, and then incubated with anti-GFP followed by an anti-rabbit antibody conjugated to Alexa Fluor 488 (A11034, 1:1000; Thermo Fisher Scientific)69,70. Colocalisation experiments were performed on fixed COS-7 cells labelled with rabbit anti-GM130 antibody (G7295, 1:1000; Sigma-Aldrich) followed by an anti-rabbit antibody conjugated to Alexa Fluor 647 (1:1000). COS-7 cells and hippocampal neurones were imaged at room temperature using an Olympus Cell R microscope with a 60x/1.35 oil immersion objective (surface expression experiments on COS-7 cells) or a Leica SP8 confocal scanning microscope with a 63x/1.40 oil immersion apochromatic objective (colocalisation experiments on COS-7 cells; hippocampal neurons), respectively. The z-stack for all images was 0.3 µm, and the resolution was 1344 × 1024 pixels and 1024 × 1024 pixels for COS-7 cells and hippocampal neurones, respectively. The images were analysed using ImageJ 1.52N software (NIH) as described previously69,71. In brief, the surface and total fluorescence intensities of COS-7 cells were analysed on whole-cell areas. For hippocampal neurones, the fluorescence intensity of the total and surface signals was analysed on 5 separate 10-µm segments of secondary or tertiary dendrites per neurone. Prior to the intensity analysis, a z-stack projection was made with maximal intensity from the bottom of the cell to the top of the cell. Colocalisation of GluN subunit combinations with GM130 marker was analysed from single z-stack using an automated ImageJ macro to batch process the data. First, the background was subtracted using ImageJ function Subtract background with rolling factor set to 64. Mask of the entire cell area was generated from native GFP signal by thresholding. For GA area localisation a mask was generated from GM130 signal by thresholding. The thresholds were determined from fluorescent intensity histograms using ImageJ function Threshold with Otsu algorithm and the obtained thresholds were used for batch processing across the all data. A GM130-negative cell area was generated by subtraction of the GM130 mask from the GFP signal mask. The GA colocalisation was then calculated as a ratio of the average intensity of GFP signal through the generated GM130 mask over the average intensity of GFP signal through the GM130-negative cell area.
Electrophysiology
Whole-cell patch-clamp recordings were performed on transfected HEK293 cells expressing GluN1/GluN3A receptors using an Axopatch 200B amplifier (Molecular Devices) as described previously51,58. The extracellular solution contained (in mM): 160 NaCl, 2.5 KCl, 10 HEPES, 10 glucose, 0.2 EDTA, and 0.7 CaCl2 (pH adjusted to 7.3 with NaOH). The intracellular solution contained (in mM): 125 gluconic acid, 15 CsCl, 5 BAPTA, 10 HEPES, 3 MgCl2, 0.5 CaCl2, and 2 ATP-Mg salt (pH adjusted to 7.2 with CsOH). CGP-78608 (Tocris) was prepared according to manufacturer’s instructions. Glass patch pipettes (3–6 MΩ tip resistance) were prepared using a model P-97 micropipette puller (Sutter Instrument Co.). A microprocessor-controlled multi-barrel rapid perfusion system (with a time constant for solution exchange around the cell of ~10 ms) was used to apply the extracellular solutions57. All electrophysiology experiments were performed at room temperature. pCLAMP 9 software (Molecular Devices) was used to record and analyse the glycine-induced currents at a membrane potential of −60 mV.
Calculation of glycine potency
Data were analysed using SigmaPlot 10.0 (Systat Software, Inc.). The normalised peak concentration-response data for each HEK293 cell were best-fitted using the following equation:
where Imax is the maximum peak current in response to glycine, EC50 is the glycine concentration that elicited the half-maximal response, [Glycine] is glycine concentration, and h is the apparent Hill coefficient. The peak current (I) through GluN1/GluN3A receptors was elicited by applying glycine at concentration of 10–10,000 μM and was normalised to the maximum peak current recorded in that cell.
Quantitative assay of surface expression
HEK293 cells grown in 12-well plates were transfected with a mixture of Lipofectamine 2000 and equal amounts of cDNA vectors containing dynamin-K44A and/or GluN subunits, as described52. After 38–40 hr, cells were fixed for 20 min in 4% PFA in PBS and incubated for 60 min in PBS containing 0.2% bovine serum albumin (BSA) without (surface expression) or with (total expression) 0.1% Triton X-100. Next, cells were incubated in primary rabbit anti-GFP antibody (AB3080P; Merck; 1:500 for surface expression and 1:1000 for total expression) and then with secondary horseradish peroxidase-conjugated donkey anti-rabbit IgG (NA934V; GE Healthcare; 1:1000), both diluted in PBS with 0.2% BSA for 1 hr. The color reaction was done with ortho-phenylenediamine (OPD; 0.4 mg/ml) dissolved in phosphate-citrate buffer containing sodium phosphate (both from Merck) for 30 min (surface expression) or 15 min (total expression) and was terminated with 3 M HCl. The optical density was determined at 492 nm using a Personal Densitometer SI (GE Healthcare). Data were obtained from three independent experiments (each included two different wells for surface or total expression for each GluN subunit combination). The background signal was subtracted and data were normalized to average data obtained from the cells expressing control GluN1-4a/GFP-GluN3A receptors.
Statistical analysis
The quantitative microscopy/assays and electrophysiology data were compared to the respective wild-type group, and summary data are presented as the mean ± the standard error of the mean (SEM). Group differences were analysed using a Student’s t-test or a one-way ANOVA followed by the Dunnett’s Method using SigmaStat 3.5 (Systat Software, Inc.), and differences with a p-value < 0.05 were considered significant.
Data Availability
All materials, data, and associated protocols will be promptly made available to readers upon request without undue qualifications for material transfer agreements.
References
Perez-Otano, I., Larsen, R. S. & Wesseling, J. F. Emerging roles of GluN3-containing NMDA receptors in the CNS. Nat Rev Neurosci 17, 623–635, doi:nrn.2016.92 (2016).
Traynelis, S. F. et al. Glutamate receptor ion channels: structure, regulation, and function. Pharmacol Rev 62, 405–496, doi:62/3/405 (2010).
Mahfooz, K. et al. GluN3A promotes NMDA spiking by enhancing synaptic transmission in Huntington’s disease models. Neurobiol Dis 93, 47–56, doi:S0969-9961(16)30074-2 (2016).
Marco, S. et al. Suppressing aberrant GluN3A expression rescues synaptic and behavioral impairments in Huntington’s disease models. Nat Med 19, 1030–1038, doi:nm.3246 (2013).
Mueller, H. T. & Meador-Woodruff, J. H. NR3A NMDA receptor subunit mRNA expression in schizophrenia, depression and bipolar disorder. Schizophr Res 71, 361–370, doi:S0920996404000908 (2004).
Yuan, T. et al. Expression of cocaine-evoked synaptic plasticity by GluN3A-containing NMDA receptors. Neuron 80, 1025–1038, doi:S0896-6273(13)00709-5 (2013).
Chen, J. et al. Demonstration of critical role of GRIN3A in nicotine dependence through both genetic association and molecular functional studies. Addict Biol, https://doi.org/10.1111/adb.12718 (2019).
Lemke, J. R. et al. Delineating the GRIN1 phenotypic spectrum: A distinct genetic NMDA receptor encephalopathy. Neurology 86, 2171–2178, doi:WNL.0000000000002740 (2016).
Chen, W. et al. GRIN1 mutation associated with intellectual disability alters NMDA receptor trafficking and function. J Hum Genet, doi:jhg201719 (2017).
Takata, A. et al. A population-specific uncommon variant in GRIN3A associated with schizophrenia. Biol Psychiatry 73, 532–539, https://doi.org/10.1016/j.biopsych.2012.10.024 (2013).
Shen, Y. C. et al. Exomic sequencing of the ionotropic glutamate receptor N-methyl-D-aspartate 3A gene (GRIN3A) reveals no association with schizophrenia. Schizophr Res 114, 25–32, doi:S0920-9964(09)00323-5 (2009).
Paoletti, P., Bellone, C. & Zhou, Q. NMDA receptor subunit diversity: impact on receptor properties, synaptic plasticity and disease. Nat Rev Neurosci 14, 383–400, https://doi.org/10.1038/nrn3504 (2013).
Kleckner, N. W. & Dingledine, R. Requirement for glycine in activation of NMDA-receptors expressed in Xenopus oocytes. Science 241, 835–837 (1988).
Clements, J. D. & Westbrook, G. L. Activation kinetics reveal the number of glutamate and glycine binding sites on the N-methyl-D-aspartate receptor. Neuron 7, 605–613, doi:0896-6273(91)90373-8 (1991).
Patneau, D. K. & Mayer, M. L. Structure-activity relationships for amino acid transmitter candidates acting at N-methyl-D-aspartate and quisqualate receptors. J Neurosci 10, 2385–2399 (1990).
Awobuluyi, M. et al. Subunit-specific roles of glycine-binding domains in activation of NR1/NR3 N-methyl-D-aspartate receptors. Mol Pharmacol 71, 112–122, doi:mol.106.030700 (2007).
Kvist, T., Greenwood, J. R., Hansen, K. B., Traynelis, S. F. & Brauner-Osborne, H. Structure-based discovery of antagonists for GluN3-containing N-methyl-D-aspartate receptors. Neuropharmacology 75, 324–336, doi:S0028-3908(13)00360-2 (2013).
Madry, C. et al. Principal role of NR3 subunits in NR1/NR3 excitatory glycine receptor function. Biochem Biophys Res Commun 354, 102–108, doi:S0006-291X(06)02825-7 (2007).
Kehoe, L. A., Bernardinelli, Y. & Muller, D. GluN3A: an NMDA receptor subunit with exquisite properties and functions. Neural Plast 2013, 145387, https://doi.org/10.1155/2013/145387 (2013).
Horak, M., Petralia, R. S., Kaniakova, M. & Sans, N. ER to synapse trafficking of NMDA receptors. Front Cell Neurosci 8, 394, https://doi.org/10.3389/fncel.2014.00394 (2014).
Sanz-Clemente, A., Nicoll, R. A. & Roche, K. W. Diversity in NMDA receptor composition: many regulators, many consequences. Neuroscientist 19, 62–75, doi:1073858411435129 (2013).
Hansen, K. B., Yi, F., Perszyk, R. E., Menniti, F. S. & Traynelis, S. F. NMDA Receptors in the Central Nervous System. Methods Mol Biol 1677, 1–80, https://doi.org/10.1007/978-1-4939-7321-7_1 (2017).
Chazot, P. L. & Stephenson, F. A. Biochemical evidence for the existence of a pool of unassembled C2 exon-containing NR1 subunits of the mammalian forebrain NMDA receptor. J Neurochem 68, 507–516 (1997).
Huh, K. H. & Wenthold, R. J. Turnover analysis of glutamate receptors identifies a rapidly degraded pool of the N-methyl-D-aspartate receptor subunit, NR1, in cultured cerebellar granule cells. J Biol Chem 274, 151–157 (1999).
Meddows, E. et al. Identification of molecular determinants that are important in the assembly of N-methyl-D-aspartate receptors. J Biol Chem 276, 18795–18803, https://doi.org/10.1074/jbc.M101382200 (2001).
Schuler, T., Mesic, I., Madry, C., Bartholomaus, I. & Laube, B. Formation of NR1/NR2 and NR1/NR3 heterodimers constitutes the initial step in N-methyl-D-aspartate receptor assembly. J Biol Chem 283, 37–46, doi:M703539200 (2008).
Atlason, P. T., Garside, M. L., Meddows, E., Whiting, P. & McIlhinney, R. A. N-Methyl-D-aspartate (NMDA) receptor subunit NR1 forms the substrate for oligomeric assembly of the NMDA receptor. J Biol Chem 282, 25299–25307, doi:M702778200 (2007).
Farina, A. N. et al. Separation of domain contacts is required for heterotetrameric assembly of functional NMDA receptors. J Neurosci 31, 3565–3579, doi:31/10/3565 (2011).
McIlhinney, R. A. et al. Assembly intracellular targeting and cell surface expression of the human N-methyl-D-aspartate receptor subunits NR1a and NR2A in transfected cells. Neuropharmacology 37, 1355–1367 (1998).
Perez-Otano, I. et al. Assembly with the NR1 subunit is required for surface expression of NR3A-containing NMDA receptors. J Neurosci 21, 1228–1237, doi:21/4/1228 (2001).
Standley, S., Roche, K. W., McCallum, J., Sans, N. & Wenthold, R. J. PDZ domain suppression of an ER retention signal in NMDA receptor NR1 splice variants. Neuron 28, 887–898, doi:S0896-6273(00)00161-6 (2000).
Okabe, S., Miwa, A. & Okado, H. Alternative splicing of the C-terminal domain regulates cell surface expression of the NMDA receptor NR1 subunit. J Neurosci 19, 7781–7792 (1999).
Horak, M. & Wenthold, R. J. Different roles of C-terminal cassettes in the trafficking of full-length NR1 subunits to the cell surface. J Biol Chem 284, 9683–9691, doi:M807050200 (2009).
Hawkins, L. M. et al. Export from the endoplasmic reticulum of assembled N-methyl-d-aspartic acid receptors is controlled by a motif in the c terminus of the NR2 subunit. J Biol Chem 279, 28903–28910, https://doi.org/10.1074/jbc.M402599200 (2004).
Qiu, S. et al. An endoplasmic reticulum retention signal located in the extracellular amino-terminal domain of the NR2A subunit of N-Methyl-D-aspartate receptors. J Biol Chem 284, 20285–20298, doi:M109.004960 (2009).
Matsuda, K., Fletcher, M., Kamiya, Y. & Yuzaki, M. Specific assembly with the NMDA receptor 3B subunit controls surface expression and calcium permeability of NMDA receptors. J Neurosci 23, 10064–10073, doi:23/31/10064 (2003).
Washbourne, P., Bennett, J. E. & McAllister, A. K. Rapid recruitment of NMDA receptor transport packets to nascent synapses. Nat Neurosci 5, 751–759, https://doi.org/10.1038/nn883 (2002).
Washbourne, P., Liu, X. B., Jones, E. G. & McAllister, A. K. Cycling of NMDA receptors during trafficking in neurons before synapse formation. J Neurosci 24, 8253–8264, https://doi.org/10.1523/JNEUROSCI.2555-04.2004 (2004).
Jeyifous, O. et al. SAP97 and CASK mediate sorting of NMDA receptors through a previously unknown secretory pathway. Nat Neurosci 12, 1011–1019, https://doi.org/10.1038/nn.2362 (2009).
Groc, L. et al. Differential activity-dependent regulation of the lateral mobilities of AMPA and NMDA receptors. Nat Neurosci 7, 695–696, https://doi.org/10.1038/nn1270 (2004).
Dupuis, J. P. & Groc, L. Surface trafficking of neurotransmitter receptors: From cultured neurons to intact brain preparations. Neuropharmacology, doi:S0028-3908(19)30168-6 (2019).
Roche, K. W. et al. Molecular determinants of NMDA receptor internalization. Nat Neurosci 4, 794–802, https://doi.org/10.1038/90498 (2001).
Perez-Otano, I. et al. Endocytosis and synaptic removal of NR3A-containing NMDA receptors by PACSIN1/syndapin1. Nat Neurosci 9, 611–621, https://doi.org/10.1038/nn1680 (2006).
Lavezzari, G., McCallum, J., Dewey, C. M. & Roche, K. W. Subunit-specific regulation of NMDA receptor endocytosis. J Neurosci 24, 6383–6391, https://doi.org/10.1523/JNEUROSCI.1890-04.2004 (2004).
Scott, D. B., Michailidis, I., Mu, Y., Logothetis, D. & Ehlers, M. D. Endocytosis and degradative sorting of NMDA receptors by conserved membrane-proximal signals. J Neurosci 24, 7096–7109, https://doi.org/10.1523/JNEUROSCI.0780-04.2004 (2004).
Kato, A., Rouach, N., Nicoll, R. A. & Bredt, D. S. Activity-dependent NMDA receptor degradation mediated by retrotranslocation and ubiquitination. Proc Natl Acad Sci USA 102, 5600–5605, doi:0501769102 (2005).
Fukaya, M., Kato, A., Lovett, C., Tonegawa, S. & Watanabe, M. Retention of NMDA receptor NR2 subunits in the lumen of endoplasmic reticulum in targeted NR1 knockout mice. Proc Natl Acad Sci USA 100, 4855–4860, https://doi.org/10.1073/pnas.0830996100 (2003).
She, K., Ferreira, J. S., Carvalho, A. L. & Craig, A. M. Glutamate binding to the GluN2B subunit controls surface trafficking of N-methyl-D-aspartate (NMDA) receptors. J Biol Chem 287, 27432–27445, doi:M112.345108 (2012).
Kenny, A. V., Cousins, S. L., Pinho, L. & Stephenson, F. A. The integrity of the glycine co-agonist binding site of N-methyl-D-aspartate receptors is a functional quality control checkpoint for cell surface delivery. J Biol Chem 284, 324–333, doi:M804023200 (2009).
Swanger, S. A. et al. Mechanistic Insight into NMDA Receptor Dysregulation by Rare Variants in the GluN2A and GluN2B Agonist Binding Domains. Am J Hum Genet 99, 1261–1280, doi:S0002-9297(16)30438-4 (2016).
Skrenkova, K. et al. N-Glycosylation Regulates the Trafficking and Surface Mobility of GluN3A-Containing NMDA Receptors. Front Mol Neurosci 11, 188, https://doi.org/10.3389/fnmol.2018.00188 (2018).
Smothers, C. T. & Woodward, J. J. Expression of glycine-activated diheteromeric NR1/NR3 receptors in human embryonic kidney 293 cells Is NR1 splice variant-dependent. J Pharmacol Exp Ther 331, 975–984, doi:jpet.109.158493 (2009).
Cummings, K. A. & Popescu, G. K. Protons Potentiate GluN1/GluN3A Currents by Attenuating Their Desensitisation. Sci Rep 6, 23344, doi:srep23344 (2016).
Williams, K., Chao, J., Kashiwagi, K., Masuko, T. & Igarashi, K. Activation of N-methyl-D-aspartate receptors by glycine: role of an aspartate residue in the M3-M4 loop of the NR1 subunit. Mol Pharmacol 50, 701–708 (1996).
Weiss, J. N. The Hill equation revisited: uses and misuses. FASEB J 11, 835–841 (1997).
Grand, T., Abi Gerges, S., David, M., Diana, M. A. & Paoletti, P. Unmasking GluN1/GluN3A excitatory glycine NMDA receptors. Nat Commun 9, 4769, https://doi.org/10.1038/s41467-018-07236-4 (2018).
Lichnerova, K., Kaniakova, M., Skrenkova, K., Vyklicky, L. & Horak, M. Distinct regions within the GluN2C subunit regulate the surface delivery of NMDA receptors. Front Cell Neurosci 8, 375, https://doi.org/10.3389/fncel.2014.00375 (2014).
Kaniakova, M. et al. 7-Methoxyderivative of tacrine is a ‘foot-in-the-door’ open-channel blocker of GluN1/GluN2 and GluN1/GluN3 NMDA receptors with neuroprotective activity in vivo. Neuropharmacology 140, 217–232, doi:S0028-3908(18)30487-8 (2018).
Madry, C., Betz, H., Geiger, J. R. & Laube, B. Supralinear potentiation of NR1/NR3A excitatory glycine receptors by Zn2+ and NR1 antagonist. Proc Natl Acad Sci USA 105, 12563–12568, doi:0805624105 (2008).
Chatterton, J. E. et al. Excitatory glycine receptors containing the NR3 family of NMDA receptor subunits. Nature 415, 793–798, https://doi.org/10.1038/nature715 (2002).
Yao, Y. & Mayer, M. L. Characterization of a soluble ligand binding domain of the NMDA receptor regulatory subunit NR3A. J Neurosci 26, 4559–4566, doi:26/17/4559 (2006).
Rozeboom, A. M. et al. Evidence for glycinergic GluN1/GluN3 NMDA receptors in hippocampal metaplasticity. Neurobiol Learn Mem 125, 265–273, doi:S1074-7427(15)00185-9 (2015).
Penn, A. C., Williams, S. R. & Greger, I. H. Gating motions underlie AMPA receptor secretion from the endoplasmic reticulum. EMBO J 27, 3056–3068, doi:emboj2008222 (2008).
Berger, S. J., Carter, J. C. & Lowry, O. H. The distribution of glycine, GABA, glutamate and aspartate in rabbit spinal cord, cerebellum and hippocampus. J Neurochem 28, 149–158 (1977).
Meeker, R. B., Swanson, D. J. & Hayward, J. N. Light and electron microscopic localization of glutamate immunoreactivity in the supraoptic nucleus of the rat hypothalamus. Neuroscience 33, 157–167 (1989).
Wang, W. et al. Glycine metabolism in animals and humans: implications for nutrition and health. Amino Acids 45, 463–477, https://doi.org/10.1007/s00726-013-1493-1 (2013).
Moroni, F., Russi, P., Lombardi, G., Beni, M. & Carla, V. Presence of kynurenic acid in the mammalian brain. J Neurochem 51, 177–180 (1988).
Yang, J. et al. The contribution of rare and common variants in 30 genes to risk nicotine dependence. Mol Psychiatry 20, 1467–1478, doi:mp2014156 (2015).
Lichnerova, K. et al. Two N-glycosylation Sites in the GluN1 Subunit Are Essential for Releasing N-methyl-d-aspartate (NMDA) Receptors from the Endoplasmic Reticulum. J Biol Chem 290, 18379–18390, doi:M115.656546 (2015).
Kaniakova, M. et al. Key amino acid residues within the third membrane domains of NR1 and NR2 subunits contribute to the regulation of the surface delivery of N-methyl-D-aspartate receptors. J Biol Chem 287, 26423–26434, doi:M112.339085 (2012).
Kaniakova, M., Lichnerova, K., Vyklicky, L. & Horak, M. Single amino acid residue in the M4 domain of GluN1 subunit regulates the surface delivery of NMDA receptors. J Neurochem 123, 385–395, https://doi.org/10.1111/jnc.12002 (2012).
Acknowledgements
This work was supported by the Czech Science Foundation (18-04329S), and the Grant Agency of Charles University (GAUK: 468217; M.Ko.). We also acknowledge the Light Microscopy Core Facility, IMG CAS, Prague, Czech Republic, which was supported by MEYS (LM2015062), OPPK (CZ.2.16/3.1.00/21547) and NPU I (LO1419).
Author information
Authors and Affiliations
Contributions
M.H. and K.S. designed the study; K.S., K.H., M.Ko., and M.Ka. performed the experiments; K.S., K.H., M.Ko., S.K. and B.K. analysed the data; and K.S., B.K. and M.H. wrote the paper.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Skrenkova, K., Hemelikova, K., Kolcheva, M. et al. Structural features in the glycine-binding sites of the GluN1 and GluN3A subunits regulate the surface delivery of NMDA receptors. Sci Rep 9, 12303 (2019). https://doi.org/10.1038/s41598-019-48845-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-48845-3
This article is cited by
-
GluN2B subunit selective N-methyl-D-aspartate receptor ligands: Democratizing recent progress to assist the development of novel neurotherapeutics
Molecular Diversity (2024)
-
Activation of non-classical NMDA receptors by glycine impairs barrier function of brain endothelial cells
Cellular and Molecular Life Sciences (2022)
-
Shenzhi Jiannao formula ameliorates vascular dementia in vivo and in vitro by inhibition glutamate neurotoxicity via promoting clathrin-mediated endocytosis
Chinese Medicine (2021)
-
The pathogenic S688Y mutation in the ligand-binding domain of the GluN1 subunit regulates the properties of NMDA receptors
Scientific Reports (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
