
Abstract
Radioactive particles were released into the environment during the Fukushima Dai-ichi Nuclear Power Plant (FDNPP) accident. Many studies have been conducted to elucidate the chemical composition of released radioactive particles in order to understand their formation process. However, whether radioactive particles contain nuclear fuel radionuclides remains to be investigated. Here, we report the first determination of Pu isotopes in radioactive particles. To determine the Pu isotopes (239Pu, 240Pu and 241Pu) in radioactive particles derived from the FDNPP accident which were free from the influence of global fallout, radiochemical analysis and inductively coupled plasma-mass spectrometry measurements were conducted. Radioactive particles derived from unit 1 and unit 2 or 3 were analyzed. For the radioactive particles derived from unit 1, activities of 239+240Pu and 241Pu were (1.70–7.06) × 10−5 Bq and (4.10–8.10) × 10−3 Bq, respectively and atom ratios of 240Pu/239Pu and 241Pu/239Pu were 0.330–0.415 and 0.162–0.178, respectively. These ratios were consistent with the simulation results from ORIGEN code and measurements from various environmental samples. In contrast, Pu was not detected in the radioactive particles derived from unit 2 or 3. The difference in Pu contents is clear evidence towards different formation processes of radioactive particles, and detailed formation processes can be investigated from Pu analysis.
Similar content being viewed by others


Introduction
On 11 March 2011, a major earthquake of magnitude 9 occurred in eastern Japan and resulted in a severe nuclear accident at the Fukushima Daiichi Nuclear Power Plant (FDNPP). This accident was classified at the maximum level of 7 on the International Nuclear and Radiological Event Scale (INES)1. Following this accident, large amounts of radionuclides generated in the reactor core were released into the environment through hydrogen explosions in unit 1 and 3, and vent operations and leakages in unit 1, 2, and 32. After 8 years, among the released high volatile radionuclides such as 129mTe, 132Te, 131I, 133Xe, 134Cs, and 137Cs3,4,5,6, the long-lived radionuclide 137Cs (half-life T1/2 = 30.2 y) still remains and derives a great social influence. Most of the radioactive Cs was released in soluble form such as CsI or CsOH, transported with aerosols, and deposited on soil by wet and dry deposition processes7. Among the other long-lived radionuclides, the amounts of 90Sr (T1/2 = 28.7 y) and actinides such as 239Pu (T1/2 = 24100 y), 240Pu (T1/2 = 6560 y) and 241Pu (T1/2 = 14.4 y) have been of concern despite their low volatile property. The amount of 90Sr has been quantified and found to be generally lower than that of 137Cs in the soil8,9.
In a nuclear accident, Pu is one of the most notable radionuclides due to its high radiotoxicity and chemical toxicity10,11. The most significant contribution to Pu contamination in the environment is derived from the global fallout due to the nuclear weapon tests during 1945–198012. The other notable contribution is the past nuclear accident in Chernobyl. Contributions from these events are mixed in an environmental sample. To identify the Pu source, the Pu isotopic ratio provides important information because the atom ratio of 240Pu/239Pu differs according to the Pu source13. The 240Pu/239Pu atom ratio of global fallout was reported as 0.180 ± 0.00714,15,16. On the other hand, in the Chernobyl accident, Muramatsu et al.17 reported that the 240Pu/239Pu atom ratio was approximately 0.408 ± 0.003, and higher values of 0.45–0.52 were reported for Chernobyl hot particles18; such a high value resulted from the long burn-up time of the nuclear fuel. Before the FDNPP accident, the ratios in the soils ranged from 0.14 to 0.24 in Japan19,20 and the Pu contribution mainly originated from global fallout16. After the FDNPP accident, the 240Pu/239Pu atom ratio in various environmental samples around the FDNPP increased compared to the values reported previously. The reported values ranged from 0.286 to 0.381 for the soils, litter, black substances (road dust), vegetation, river water, and air dust samples21,22,23,24,25,26,27. Nishihara et al.28 estimated the 240Pu/239Pu ratio of the fuel inventory in the reactor cores of unit 1–3 as 0.320–0.356 using the ORIGEN2 code and the fuel burn-up data from Tokyo Electric Power Company Holdings, Inc. (TEPCO). The detected 240Pu/239Pu ratio in the environmental samples were close to that of the fuel inventory calculated by Nishihara et al. In addition, the 241Pu/239Pu atom ratio is also important for identifying Pu source. 241Pu/239Pu atom ratio of global fallout was reported as 0.00194 ± 0.00014 (241Pu decay corrected to 1 January, 2000)14. Before the FDNPP accident, the 241Pu/239Pu atom ratio in fallout materials was 0.00287 ± 0.00028 (241Pu decay corrected to 1 January, 2000) in Japan29. After the FDNPP accident, a higher value of 241Pu/239Pu atom ratio (0.103–0.135(241Pu decay corrected to 15 March, 2011)) was reported from soil and litter samples around the FDNPP as well as 240Pu/239Pu atom ratio21. However, these environmental samples were influenced by the contamination from the global fallout existing before the FDNPP accident, which hampered the accurate assessment of the environmental impact of the released Pu isotopes.
After the FDNPP accident, small particles containing highly concentrated radioactive Cs (radioactive particles) were detected in various environmental samples such as air dust30, soil31, vegetation32, river water33, and dust in residences34. The radioactive particles were several micrometres to several hundred micrometres in diameter, mainly comprised of amorphous SiO2 and were insoluble in water30,32. Previous studies revealed that the radioactive particles also contain Fe, Cr, Mn, Zr, and Zn which were the same as the elemental composition in the structure of the reactor, such as the fuel cladding, vessel, and primary cooling water, and estimated that the radioactive particles were formed in the reactor during the meltdown and directly released from the reactor35,36. Physical and chemical properties of radioactive particles play an important role in evaluating the reaction and accident circumstances in a reactor during meltdown37. Satou et al.38 classified the radioactive particles into two types, ‘Type A’ and ‘Type B’, by particle size, Cs concentration, and radioactivity ratio of 134Cs/137Cs, and estimated that the difference in these types reflected each accident event in the reactor38. For ‘Type A’ particles, the diameter is 1–10 µm, Cs is clearly detected by an energy dispersive X-ray spectrometer (EDS), and the 134Cs/137Cs activity ratio is approximately 1.0438. On the other hand, for ‘Type B’ particles, the diameter is 70–400 µm, the Cs concentration is too low to be detected by an EDS, and the 134Cs/137Cs activity ratio is approximately 0.9338. Kogure et al.39 investigated the distribution of substantial elements, such as Si, O, Cs, Fe, and Zn in ‘Type A’ radioactive particles and revealed that Si, O, Fe, and Zn were homogeneously distributed, while Cs was highly concentrated in the vicinity of the surface. Regarding trace elements in the radioactive particles, the fuel element U was detected in ‘Type A’ by synchrotron radiation (SR) X-ray analysis35,40 and secondary ion mass spectrometry (SIMS)36, and 90Sr was detected in ‘Type B’ by radiochemical analysis41. Following the detection of U in ‘Type A’, some studies have suggested that the radioactive particles were formed from the mist which included SiO2, Cs, U, and other constituent elements when the molten core dropped on the concrete under the reactor vessel36,42,43. In addition, the elemental composition in the radioactive particles are not changed after emission from the reactor due to their insolubility in water. The chemical property of the radioactive particles was stable in the environment and retained the original property at the time of emission from the reactor. In addition, the radioactive particles were free from the influence of the global fallout. Therefore, the amount of Pu in radioactive particles is important to estimate the detailed formation process of radioactive particles in the reactor, for example, the reaction temperature condition.
In this study, we perform radiochemical analysis to determine the amount of Pu and the atom ratios of 240Pu/239Pu and 241Pu/239Pu for the radioactive particles from the FDNPP accident. In addition, the formation process of radioactive particles in the reactor is discussed based on the amount of Pu.
Methods
Sampling locations
Two environmental samples were collected near the FDNPP. One is a bulk dust sample collected on 23 January 2015 from an open garage building in Futaba town, Fukushima Prefecture, 3 km north-west of the FDNPP (37°44′N and 141°02′E). The other is a bulk soil sample collected on 1 July 2016 in Okuma town, Fukushima Prefecture, 3 km south-south-west of the FDNPP (37°40′N and 141°02′E). Details of the sample collection were described in our previous study38,41.
Extraction and isolation of a radioactive particle
We obtained the radioactivity distributions in the dust and soil samples by autoradiography using an imaging plate (IP) (BAS-SR 2025, FUJIFILM Corporation., Japan) sensitive to β-rays and gamma-rays to identify the radioactive particles from these samples. The IP was set on the sample with an exposure time of 10 min and high radioactivity spots were identified. By separating the spots in the bulk samples and repeating the IP measurements, we reduced the volume of the samples. Then, the radioactive particles were isolated from the environmental samples by the multiple wet separation method developed by Miura et al.33. Finally, 1 mL water containing only a radioactive particle was obtained and the solution was dried on a carbon tape of size 2 mm × 2 mm. The shape and major elements of the radioactive particle were identified and measured by scanning electron microscopy (SEM) (VE-9800, Keyence Inc., Japan) equipped with an EDS (EDAX, AMETEK Co., Ltd., USA). In the analysis, acceleration voltage of the scanning electron beam was set to 20 keV.
Gamma-ray measurement
The gamma-rays from the radioactive particles attached to the carbon tape were measured by a germanium semiconductor detector (GEM40-76, ORTEC, USA). The activities of 134Cs and 137Cs were measured by the gamma-ray intensities of 604 keV and 662 keV, respectively. The detection efficiencies of the gamma-rays from the radioactive particles were determined by a standard sample quantified at University of Tsukuba38,41.
Radiochemical analysis for quantifying Sr and Pu in the radioactive particles
In this study, we quantified 90Sr in the radioactive particles before quantifying Pu. Strontium-90 in the radioactive particles was purified and concentrated by solid-phase extraction and was measured by liquid scintillation counter41. In the first step, the radioactive particle was dissolved by alkali fusion. The radioactive particle with the carbon tape was put into a zirconium crucible, and 2 mL of high purity 3 M NaOH solution (Kanto Chemical Co., Inc., Japan) was added. The crucible was heated to 80 °C in an electric furnace to evaporate the solution, and then it was further heated to 350 °C for 30 min. After cooling, the residue in the crucible was dissolved by 90 mL of 3 M HNO3 solution. The solution was passed through the Sr Rad Disk (3 M, Inc., USA) to trap the Sr. The passed solution was later used for Pu identification. The trapped Sr was eluted from the Disk using a 0.02 M EDTA solution. The solution was then passed through the ion-exchange resin followed by 2% (w/v) EDTA solution to remove natural radionuclide of 210Pb which would interfere with β-ray measurement from the resin. The 90Sr fraction eluted by 3 M HCl solution from the resin was set into the liquid scintillation counter (1220 Quantulus, Perkin Elmer Inc.). Cherenkov light from 90Y, a daughter nuclide of 90Sr fraction was continuously measured over 2 weeks. Detailed procedures are described in Zhang et al.41,44.
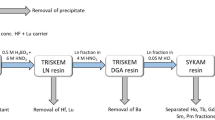
To quantify Pu in the radioactive particle, we performed radiochemical analysis to purify and concentrate Pu and measured Pu isotopes by inductively coupled plasma-mass spectroscopy (ICP-MS). As a chemical yield carrier, 0.57 pg of 242Pu was added to the solution passing through the Sr Rad Disk. To adjust Pu to Pu (III) state which undergoes better co-precipitation during the following CaF2/LaF3 co-precipitation procedure, 2 mL of 20% TiCl3 solution was added to the sample solution. Following the addition of 100 mg Ca and 100 mg La, 7 mL HF was pipetted into the sample solution and the centrifuge tube was shaken to facilitate the co-precipitation. After allowing 10 min for the reaction, the precipitate was separated from the supernatant by centrifugation at 3000 rpm for 15 min. The supernatant was discarded, and the precipitate was dissolved in 20 mL of 3 M HNO3 with the addition of 0.5 g H3BO3. Finally, 0.3 g NaNO2 was added to the sample solution for valence adjustment of Pu to the tetravalent state by heating at 40 °C for 30 min. This solution was ready for the following extraction chromatographic separation. An extraction chromatography was performed with a combination of TEVA, UTEVA, and DGA resin cartridges (2 mL each, Eichrom Technologies, Inc., USA) to separate Pu from the interfering elements, such as U, Pb, Tl, Bi and Th45. After the chromatographic separation, the purified Pu solution was evaporated to dryness at 200 °C, and the residue was dissolved in 4 mL aqua regia and heated to dryness again. Finally, the sample residue was dissolved in 0.7 mL of 4% HNO3 solution for sector field (SF)-ICP-MS measurement. The whole procedure of Pu separation is shown in Fig. 1, and further step-by-step details were described by Wang et al.45.

Procedure of radiochemical analysis for quantifying Pu in radioactive particles.
Estimating total chemical yield of quantifying Sr and Pu in the radioactive particle
In the Pu quantification process, chemical yield of Pu was identified by 242Pu carrier added for passed solution of the Sr-Rad Disk. The Pu loss by the Sr-Rad Disk was investigated to estimate the total chemical yield. For this purpose, we prepared a blank solution (3 M HNO3 solution) with 0.23 pg of 239Pu and performed the same radiochemical separation as that for radioactive particle Pu analysis.
SF-ICP-MS measurement
SF-ICP-MS (Element XR, Thermo Fisher Scientific, Germany) was used to detect Pu. The APEX-Q sample introduction system (Elemental Scientific, USA) was combined with SF-ICP-MS to improve the sensitivity and suppress the formation of UH+ and UH2+, which interfere with the accurate Pu measurement. The amount of Pu isotopes was determined from the mass region of 239, 240 and 241. The mass region of 242 was also measured to obtain the chemical yield from the spiked 242Pu. From these measurements, the radioactivities of 239+240Pu and 241Pu, and atom ratios of 240Pu/239Pu and 241Pu/239Pu were determined. The detailed settings of the measurement were reported by Zheng et al.46.
Results
Shape and radioactivity of the radioactive particle
Three radioactive particles named DPs were obtained from the dust samples, and one radioactive particle named SP was obtained from soil samples. Figure 2 shows the SEM images of representative DPs (DP2) and SP. The SEM images of other particles are shown in Fig. S1. The diameter of DPs were several hundred micrometers. For the SP, the diameter was determined to be 120 µm from the SEM image. Figure 3 shows the EDS spectra of representative DPs (DP2) and SP. The EDS spectra of other particles were shown in Fig. S2. From these spectra, for the whole of the particles, it is observed that DPs and SP were mainly composed of SiO2. Moreover, the Cs peak was not clearly observed. The 137Cs radioactivity of these radioactive particles identified from gamma-ray spectrometry was 234 ± 1 Bq, 887 ± 4 Bq, 736 ± 4 Bq and 224 ± 1 Bq for DP1, DP2, DP3 and SP, respectively on 11 March 2011. The 134Cs/137Cs activity ratios of DPs ranging from 0.895 ± 0.005 to 0.922 ± 0.006 were clearly different from that of SP, 1.03 ± 0.01 as shown in Table 1.

SEM images of the radioactive particles (a) DP2 and (b) SP.

EDS spectra of the radioactive particles of DP2 and SP.
Quantifying the amount of Pu in the radioactive particle
The yield of radiochemical analysis of Pu for each sample obtained from the 242Pu amount added after Sr separation was 91.0 ± 5.0%, 92.9 ± 5.3%, 95.5 ± 5.7% and 89.5 ± 3.9% for DP1, DP2, DP3 and SP, respectively. The total chemical yield including Sr separation method was determined from the analysis of operational blank sample with 239Pu spike as 92.3 ± 2.8%. Therefore, the loss of Pu in Sr separation was negligible.
Figure S3 shows ICP-MS spectra for radioactive particles and operational blank sample. The counting rates in the mass region of 239 (239Pu) and 240 (240Pu) were 166 and 66 cps, 289 and 121 cps and 938 and 357 cps, for DP1, DP2 and DP3, respectively. These rates were much higher than those for the operational blank sample (less than 15 cps), thereby indicating that we successfully identified the Pu contained in radioactive particles for the first time. The results of quantifying Pu in the radioactive particles are summarized Table 1. The radioactivity of 239+240Pu was (1.70 ± 0.20) × 10−5 Bq, (3.48 ± 0.45) × 10−5 Bq and (7.06 ± 0.57) × 10−5 Bq for DP1, DP2 and DP3, respectively. The 240Pu/239Pu atom ratio was 0.330 ± 0.077, 0.415 ± 0.069 and 0.373 ± 0.045 for DP1, DP2 and DP3, respectively. On the other hand, the counting rates for the SP sample were 39 and 18 cps for 239Pu and 240Pu, respectively. These rates were just slightly higher than those for the operational blank sample, thereby limiting an accurate quantification of Pu isotopes. Nevertheless, an upper limit of 239+240Pu activity contained in SP was estimated as 4.44 × 10−6 Bq. In addition, 241Pu was also quantified from counting rates in the mass region of 241 for DP2 and DP3. The radioactivity of 241Pu was (4.10 ± 0.68) × 10−3 Bq and (8.10 ± 1.27) × 10−3 Bq, for DP2 and DP3 respectively and the 241Pu/239Pu atom ratio was 0.178 ± 0.016 and 0.162 ± 0.028 for DP2 and DP3, respectively on 11 March 2011. On the other hand, the counting rates of 241Pu for DP1 and SP were very close to the operational blank sample limiting its accurate quantification. We could estimate an upper limit of 241Pu as 1.97 × 10−3 Bq and 2.32 × 10−3 Bq, for DP1 and SP, respectively.
Discussion
Different activity ratios of radioactive Cs were detected for DPs and SP (Table 1). Using the ORIGEN code, Nishihara et al.28 calculated the 134Cs/137Cs activity ratio as 0.95, 1.08, 1.05 for the units 1, 2, and 3, respectively. The 134Cs/137Cs activity ratios of DPs and SP were compared with the calculated values, and we estimated that DPs were derived from unit 1 and SP was derived from unit 2 or 3. DPs were identified as ‘Type B’ due to the similarity in particle size, Cs concentration, and 134Cs/137Cs activity ratio, according to the classification by Satou et al.38. SP was similar to ‘Type B’ with respect to the particle size and Cs concentration. However, with respect to the 134Cs/137Cs activity ratio, SP was similar to ‘Type A’ rather than ‘Type B’. We conclude that SP had differences with the features of both ‘Type A’ and ‘Type B’ and was classified as a new type of radioactive particle41.
The 240Pu/239Pu and 241Pu/239Pu atom ratios in the radioactive particles in this study were compared with the value calculated using ORIGEN code. The 240Pu/239Pu ratios in units 1, 2, and 3 reactor cores were 0.344, 0.320, and 0.350 calculated from fuel inventories by Nishihara et al.28. These ratios of DPs (DP1: 0.330 ± 0.077, DP2: 0.415 ± 0.069, DP3: 0.373 ± 0.045) generally agreed well with the calculated values of each reactor. The 241Pu/239Pu ratios in units 1, 2, and 3 reactor cores were 0.192, 0.192, and 0.183 also calculated by Nishihara et al.28. These ratios of DPs (DP2: 0.178 ± 0.016, DP3: 0.162 ± 0.028) are also consistent with these calculated values of each reactor. However, due to the relatively large error in the measured values, we cannot distinguish the source unit of the radioactive particles only from these atom ratios. The obtained 240Pu/239Pu and 241Pu/239Pu atom ratios in the radioactive particle were also compared with those of various environmental samples, such as soil21,22, litter21,23, black substance23, vegetation24,25, river water26, and air dust27, as shown in Figs 4 and 5. The 240Pu/239Pu atom ratio detected in environmental samples ranged from 0.14 to 0.381. In particular, the ratios (the numbers in brackets) in litters obtained by Yamamoto et al. (0.335 ± 0.007)23 and Zheng et al. (0.323 ± 0.017–0.330 ± 0.032)21, in black substances reported by Yamamoto et al. (0.335 ± 0.004–0.365 ± 0.015)23, and in vegetable reported by Dunne et al. (0.324 ± 0.015 – 0.359 ± 0.07)25 showed relatively high values among them. It was inferred that these samples were less contaminated by the global fallout source of Pu due to the extremely low transfer factor of global fallout Pu from soil to plant47. The ratios in DPs were consistent with the ratios in these environmental samples. In contrast, the values of 240Pu/239Pu atom ratio in soils reported by Zheng et al.21 and Yang et al.22 were lower than that of DPs. The 241Pu/239Pu atom ratios in DPs agreed better with litters obtained by Zheng et al. (0.128 ± 0.034–0.135 ± 0.012)21 than soil obtained by Zheng et al. (0.103 ± 0.013)21 as well as the 240Pu/239Pu atom ratio. As pointed out by previous studies21,22, these environmental samples were strongly influenced by global fallout, the value of which was 0.180 ± 0.007 for 240Pu/239Pu atom ratio14,15,16 and 0.00194 ± 00014 for 241Pu/239Pu atom ratio14. The Pu contribution from the FDNPP accident can be estimated using the two end-member model48, and the extent of Pu contribution in soil samples was estimated as follows: 87% by Zheng et al.21 and 31–59% by Shibahara et al.49. In the radioactive particle, the Pu injection occurred only in the reactor unlike other environmental samples. Thus, radioactive particle samples are completely free from the influence of the global fallout and the 240Pu/239Pu and 241Pu/239Pu atom ratio reflect the ratio in the core of the FDNPP directly. Therefore, both atom ratio of radioactive particles could provide us more accurate information on the ratio in the core of the FDNPP than other environmental samples and has great potential for source identification of the released radioactive particles in the environment, which should be further explored in a future study.

Comparison of atom ratios of 240Pu/239Pu in various environmental samples, radioactive particles, and source terms: global fallout15, soils during 1966–1977 in Japan16, soil after the FDNPP accident21,22, litter21,23, black substance23, vegetation24,25, river water26, average of air dusts27, and calculated result of reactor core28.

Figure 6 shows the relationship between the activity ratio of 239+240Pu/137Cs and the distance to the sampling site from the FDNPP. The 239+240Pu/137Cs activity ratio of DPs was (7.26 ± 0.86) × 10−8, (3.92 ± 0.51) × 10−8 and (9.58 ± 0.78) × 10−8 for DP1, DP2 and DP3, respectively. These ratios were two orders of magnitude smaller than those in soils reported by Yamamoto et al.23. In contrast, these values agreed well with those in black substances and litters reported by Yamamoto et al.23 and Zheng et al.21, respectively. This result is consistent with the discussion on 240Pu/239Pu and 241Pu/239Pu atom ratio. The typical value of 239+240Pu/137Cs activity ratio of the global fallout was of the order of 10−3 50. Therefore, we conclude that the soil samples were strongly influenced by the global fallout compared with the black substances and litters as mentioned in the discussion on 240Pu/239Pu and 241Pu/239Pu atom ratio. The relationship between the 239+240Pu/137Cs activity ratio and the distance from the FDNPP is important for understanding the dispersion and deposition behavior of the volatile element such as Cs and non-volatile elements such as Pu in the environment. The 239+240Pu/137Cs activity ratios in black substances and litters agreed with those in the radioactive particles, although the distance from the FDNPP were completely different. The ratios of radioactive particles directly reflected the released ratio of Pu to Cs from the reactor. The agreement indicates that the behavior of dispersion and deposition of Pu in the environment after being released from the reactor was similar to that of Cs within a distance of 25 km, as pointed out by Yamamoto et al.23.

The determination of Pu isotopes contained in the radioactive particles can reveal not only the accurate 240Pu/239Pu and 241Pu/239Pu atom ratio derived from the FDNPP accident but also provide insight into the formation process of the radioactive particles. In case of the Chernobyl accident, ‘hot particles’ containing U and other actinides were released. Lancsaris et al.51 reported that the 239+240Pu activities of the hot particles were in the range (4.2–92) × 10−3 Bq. On the other hand, the 239+240Pu activities of the radioactive particles released from the FDNPP accident were very small ((1.70–7.06) × 10−5 Bq) compared to that of the hot particles released from the accident in Chernobyl. Such a difference in Pu activity among the particles could be attributed to the formation process of these particles in the reactor between the radioactive and hot particle. The hot particles were considered to be originating from the fragments of the fuel core, and the particles were formed during the explosive break-up of the reactors52. In the case of radioactive particles released from the FDNPP accident, however, a previous study related to the formation process of the ‘Type B’ (which is of the same type as DPs) indicated that fission product gases were incorporated into the silicate-rich materials such as glass fiber insulator materials surrounding the reactor pressure vessel53. Our first identification of Pu in the radioactive particles indicated that fuel elements such as Pu were introduced to the silicate materials at the time of the formation of ‘Type B’ radioactive particles. Aberecht et al.54 estimated that refractory species having the least oxygen, such as Pu2O3, were evaporated by the reductive reaction in the reductive atmosphere of the reactor due to the large amounts of hydrogen gas during the meltdown. During the formation process of ‘Type B’ radioactive particle, we estimated that the mist, including the evaporated Pu2O3 and other fission products such as Cs, were incorporated into the insulator materials in the reductive atmosphere. The contents of the mist should change with rising temperature of the mist according to the volatile pressure of each element. Therefore, investigating the amounts of various elements contained in the radioactive particles enabled us to understand the detailed formation process of the particles. The 239+240Pu/137Cs activity ratio was different for DPs ((3.92–9.58) × 10−8) and SP (<1.98 × 10−8), which implies that the formation processes of both were different. We estimated that the temperature in unit 1 was higher than that in unit 2 or 3 during the formation of the radioactive particles.
Similar to Pu, Sr is one of the trace elements contained in the radioactive particles. Zhang et al.41 reported that the activity ratio of 90Sr/137Cs in radioactive particles was of the order of 10−4. The activities of 90Sr were 0.171 ± 0.010 Bq, 0.589 ± 0.034 Bq and 0.0455 ± 0.0040 Bq for DP1, DP2 and SP, respectively. The relationship between DPs and SP for the 90Sr/137Cs activity ratio and 90Sr activity was similar, as summarised in Table 1. Pontillon et al.55 and Lewis et al.56 observed that the release fraction from the core inventory of Sr was much lower than that of Cs and that of Pu was even lower. Therefore, we considered that the amount of Pu in the radioactive particles, which is less volatile than Sr, would be clearly different between DPs and SP due to the rising fuel temperature in the core. Pontillon et al.55 also categorised Ag as a volatile element along with Cs and showed that the emission of Ag increased with the rise in temperature, while Cs was released from the fuel pellet until 2300 K. Satou57 considered that the release rate of 110mAg reflected the fuel temperature in the core and investigated the activity ratio of 110mAg/137Cs in soils. From the result of his investigation, he estimated that the temperature of unit 1 was higher than that of unit 2 or 3 when the radionuclides were released into the environment, which is characterized by the high 110mAg/137Cs activity ratio. Therefore, we estimated that DPs derived from unit 1 were formed under a high temperature atmosphere, where non-volatile radionuclides such as Pu were more volatile compared to SP derived from unit 2 or 3.
In this study, we first investigated the amounts of Pu in DP (Type B) radioactive particles. On the other hand, Pu was not detected from radioactive particles that were not categorized as ‘Type A’ or ‘Type B’. The amount of Pu in ‘Type A’ particles still remains to be investigated. The formation process of radioactive particles can be investigated by measuring Pu amount and comparing between different particle types. In this study, the error of obtained Pu isotopic ratio was relatively large because we quantified trace amounts of Pu in the radioactive particles. To obtain more accurate ratio, it is necessary to quantify Pu from the Pu-bearing particles that contain more concentrates Pu than the radioactive particles. For this purpose, we consider investigating the Pu-bearing particles and quantifying Pu in these particles by identification methods such as the alpha particle imaging detector developed by Morishita et al.58 in the future.
Data Availability
The data that support the findings of this study are available upon request from the corresponding author [JI].
References
Steinhauser, G. et al. Comparison of the Chernobyl and Fukushima nuclear accidents: A review of the environmental impact. Sci. Total Environ. 470, 800–817 (2014).
Yamamoto, M. Overview of the Fukushima Dai-ichi Nuclear Power Plant (FDNPP) accident, with amounts and isotopic compositions of the released radionuclides. J. Radioanal. Nucl. Chem. 303, 1277–1231 (2015).
Kinoshita, N. et al. Assessment of individual radionuclide distributions from the Fukushima nuclear accident covering central-east. Proc. Natl. Acad. Sci. USA 108, 19526–19529 (2011).
Tagami, K. et al. Estimation of Te-132 distribution in Fukushima Prefecture at the early stage of the Fukushima Daiichi Nuclear Power Plant reactor failures. Environ. Sci. Technol. 47, 5007–5012 (2013).
Chino, M. et al. Preliminary Estimation of Release Amounts of 131I and 137Cs accidentally discharged from the Fukushima Daiichi Nuclear Power Plant into the atmosphere. J. Nucl. Sci. Technol. 48, 1129–1134 (2011).
Stohl, A. et al. Xenon-133 and Caesium-137 releases into the atmosphere from the Fukushima Dai-ichi nuclear power plant: determination of the source term, atmospheric dispersion, and deposition. Atmos. Chem. Phys. 12, 2313–2343 (2012).
Kaneyasu, N. et al. Sulfate aerosol as a potential transport medium of radiocesium from the Fukushima nuclear accident. Environ. Sci. Technol. 46, 5720–5726 (2012).
Igarashi, Y. et al. Atmospheric radioactivity over Tsukuba, Japan: a summary of three years of observations after the FDNPP accident. Prog. Earth Planet. 2, 1–19 (2015).
Zhang, Z. et al. Atmospheric activity concentration of 90Sr and 137Cs after the Fukushima Daiichi nuclear accident. Environ. Sci. Technol. 52, 9917–9925 (2018).
Taylor, D. M. Environmental plutonium in humans. Appl. Radiat. Isot. 46, 1245–1252 (1995).
Perelygin, V. P. et al. Man-made plutonium in environment-possible serious hazard. Radiat. Meas. 28, 385–392 (1997).
UNSCEAR, United Nations Scientific Committee on the Effect of Atomic Radiation. Ionizing Radiation: Sources and Biological Effects. Report to the General Assembly, United Nations Scientific Committee on the Effects of Atomic Radiation, P. 773 (1982).
Zheng, J. et al. The key role of atomic spectrometry in radiation protection. J. Anal. At. Spectrom. 28, 1676–1699 (2013).
Krey, P. W. et al. Mass isotopic composition of global fall-out plutonium in soil. IAEA-SM-199-39. 671–678 (IAEA, 1976).
Kelly, J. et al. Global distribution of Pu isotopes and 237Np. Sci. Total. Environ. 238/238, 483–500 (1999).
Yang, G. et al. Plutonium concentration and isotopic ratio in soil samples from central-eastern Japan collected around the 1970s. Sci. Rep. 5, 9636 (2015).
Muramatsu, Y. et al. Concentrations of 239Pu and 240Pu and Their Isotopic Ratios Determined by ICP-MS in Soils Collected from the Chernobyl 30-km Zone. Environ. Sci. Technol. 34, 2913–2917 (2000).
Oughton, D. et al. Accelerator mass spectrometry measurement of 240Pu/239Pu isotope ratios in Novaya Zemlya and Kara Sea sediments. Appl. Radiat. Isot. 61(2–3), 249–253 (2004).
Ohtsuka, Y. et al. Evaluation of 239+240Pu, 137Cs and natural 210Pb fallout in agricultural upland fields in Rokkasho, Japan. J. Radioanal. and Nucl. Chem. 261(3), 625–630 (2004).
Muramatsu, Y. et al. Determination of Pu concentration and its isotope ratio in Japanese soils by HR-ICP-MS. J. Radioanal. and Nucl. Chem. 255(3), 477–480 (2003).
Zheng, J. et al. Isotopic evidence of plutonium release into the environment from the Fukushima DNPP accident. Sci. Rep. 2, 0304 (2012).
Yang, G. et al. Isotopic compositions of 236U, 239Pu, and 240Pu in soil contaminated by the Fukushima Daiichi Nuclear Power plant accident. Sci. Rep. 7, 13619 (2017).
Yamamoto, M. et al. Isotopic Pu, Am and Cm signatures in environmental samples contaminated by the Fukushima Dai-ichi Nuclear Power Plant accident. J. Environ. Radioact. 132, 31–46 (2014).
Schneider, S. et al. Plutonium release from Fukushima Daiichi fosters the need for more detailed investigations. Sci. Rep. 3, 2988 (2013).
Dunne, J. A. et al. Spatial pattern of plutonium and radiocaesium contamination released during the Fukushima Daiichi nuclear power plant disaster. Sci. Rep. 8, 16799 (2018).
Sakaguchi, A. et al. Isotopic determination of U, Pu, and Cs in environmental waters following the Fukushima Daiichi Nuclear Power Plant accident. Geochem. J. 46, 355–360 (2012).
Shinonaga, T. et al. Air bone plutonium and non-natural uranium from the Fukushima DNPP found at 120 km distance a few days after reactor hydrogen explosions. Environ. Sci. Thechnol. 48, 3808–3814 (2014).
Nishihara, K. et al. Estimation of fuel composition in Fukushima-Daiichi Nuclear Power Plant. JAEA-Data/Code 2012-018, 65–117 (2012).
Zhang, Y. et al. Characterization of Pu concentration and its isotopic composition in a reference fallout material. Sci. Total. Environ. 408, 1139–1144 (2010).
Adachi, K. et al. Emission of spherical cesium-bearing particles from an early stage of the Fukushima nuclear accident. Sci. Rep. 3, 2554 (2013).
Satou, Y. et al. First successful isolation of radioactive particles from soil near the Fukushima Daiichi Nuclear Power plant. Anthropocene. 14, 71–76 (2016).
Yamaguchi, N. et al. Internal structure of cesium-bearing radioactive microparticles released from Fukushima nuclear power plant. Sci. Rep. 6, 20548 (2016).
Miura, H. et al. Discovery of radiocesium-bearing microparticles in river water and their influence on the solid-water distribution coefficient (Kd) of radiocesium in the Kuchibuto River in Fukushima. Geochem. J. 52, 145–154 (2018).
Higaki, S. et al. Discovery of non-spherical heterogeneous radiocesium-bearing particles not derived from Unit 1 of the Fukushima Dai-ichi Nuclear Power Plant, in residences five years after the accident. J. Environ. Radioact. 177, 65–70 (2018).
Abe, Y. et al. Detection of Uranium and chemical state analysis of individual radioactive microparticles emitted from the Fukushima nuclear accident using multiple synchrotron radiation X-ray analyses. Anal. Chem. 86, 8521–8525 (2014).
Imoto, J. et al. Isotopic signature and nano-texture of cesium-rich micro particles: release of uranium and fission products from the Fukushima Daiichi Nuclear Power Plant. Sci. Rep. 7, 5409 (2017).
Chen, F. et al. Application of synchrotron radiation and other techniques in analysis of radioactive microparticles emitted from the Fukushima Daiichi Nuclear Power Plant accident-A review. J. Environ. Radioact. 196, 29–39 (2019).
Satou, Y. et al. Analysis of two forms of radioactive particles emitted during the early stages of the Fukushima Dai-ichi Nuclear Power Station accident. Geochem. J. 52, 0514 (2018).
Kogure, T. et al. Constituent elements and their distribution in the radioactive Cs-bearing silicate glass microparticles released from Fukushima nuclear plant. Microscopy. 65, 451–459 (2016).
Ono, T. et al. Investigation of the chemical characteristics of individual radioactive microparticles emitted from reactor 1 by the Fukushima Daiichi Nuclear Power Plant accident by using multiple synchrotron radiation X-ray analyse. Bunseki Kagaku. 66, 251–261 (2017).
Zhang, Z. et al. Activity of 90Sr in Cs-rich microparticles collected in the difficult-to-return zone around the Fukushima Daiichi Nuclear Power Plant. Environ. Sci. Technol. 53(10), 5868–5876 (2019).
Furuki, G. et al. Cesium-rich micro-particles: A window into the meltdown events at the Fukushima Daiichi Nuclear Power Plant. Sci. Rep. 7, 42731 (2017).
Ochiai, A. et al. Uranium dioxides and debris fragments released to the environment with Cesium-rich microparticles from the Fukushima Daiichi Nuclear Power Plant. Environ. Sci. Technol. 52, 2586–2594 (2018).
Zhang, Z. et al. Rapid isolation method for radioactive strontium using EmporeTM strontium rad disk. J. Nucl. Radiochem. Sci. 16, 15–21 (2016).
Wang, Z. et al. High-performance method for determination of Pu isotopes in soil and sediment samples by sector field-inductively coupled plasma mass spectrometry. Anal. Chem. 89, 2221–2226 (2017).
Zheng, J. et al. Evaluation of a new sector-field ICP-MS with Jet Interface for ultra-trace determination of Pu isotopes: from femtogram to attogram levels. J. Nucl. Radiochem. Sci. 1, 7–13 (2015).
Ni, Y. et al. The transfer of fallout plutonium from paddy soil to rice: A field study in Japan. J. Environ. Radioact. 196, 22–28 (2019).
Krey, P. et al. Remote Pu contamination and total inventories from Rocky Flats. Health Phys. 30, 209–214 (1976).
Shibahara, Y. et al. Determination of isotopic ratios of plutonium and uranium in soil samples by thermal ionization mass spectrometry. J. Radioanal. Nucl. Chem. 307, 2281–2287 (2016).
Warneke, T. et al. A new ground-level fallout record of uranium and plutonium isotopes for northern temperate latitudes. Earth Planet. Sci. Letters. 203, 1047–1057 (2002).
Lancsarics, G. et al. Transuranium elements in the hot particles emitted during the Chernobyl accident. Radiat. Protect. Dosim. 23, 111–113 (1988).
Salbu, B. et al. Hot particles in accidental releases from Chernobyl and Windscale nuclear installations. Analyst 123, 843–849 (1998).
Martin, P. et al. Analysis of external surface irregularities on Fukushima-derived fallout particles. Frontiers Energy Res. 5, 25 (2017).
Aberecht, D. et al. Linear free energy correlations for fission product release from the Fukushima-Daiichi nuclear accident. Environ. Sci. Technol. 49, 3158–3166 (2015).
Pontillon, Y. & Ducors, G. Behavior of fission products under severe PWR accident conditions - the VERCORS experimental programme - part 2: release and tarnsport of fission gases and volatile fission products. Nucl. Eng. and Design. 240, 1853–1866 (2010).
Lewis, B. J. et al. Fission product chemistry in oxide fuels. In Comprehensive Nuclear Materials, Konings, R. J. M. Ed. 2, 515–546 (Elsevier, Amsterdam, 2012).
Satou, Y. Study of relationship between deposition of radioactive materials and radioactive particles in the difficult-to-return zone caused by the Fukushima Dai-ichi Nuclear Power Plant accident. PhD Thesis, University of Tsukuba (2017).
Morishita, Y. et al. Detection of alpha particle emitters originating from nuclear fuel inside reactor building of Fukushima Daiichi Nuclear Power Plant. Sci. Rep. 9, 581 (2019).
Acknowledgements
This work was supported by the Grant of Fukushima Prefecture related to Research and Development in Radiological Sciences, the Research fund for Advanced Decommissioning Science (Eichi Program), and the Japan Society for the Promotion of Science (JSPS) KAKENHI (Grant numbers JP16H01777 and JP17K00537). We acknowledge Dr. Y. Yamaguchi (Osaka University) for his kind support in the management of analytical instruments.
Author information
Authors and Affiliations
Contributions
J.I., Z.Z., K.N., A.S., J.Z. and T.A. designed the study. Z.Z., K.N., and Y.S. carried out sample collection in Fukushima Prefecture. J.I. and Z.Z. extracted radioactive particles from bulk samples and performed non-destructive analysis. J.I., M.F., Y. Ni. and J.Z. analysed Pu isotopes in radioactive particle samples. J.I. wrote the main body of this paper under the guidance of Z.Z., K.N., A.S., J.Z. and T.A. All authors reviewed the analytical results and commented on the manuscript.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
41598_2019_48210_MOESM1_ESM.pdf
First determination of Pu isotopes (<sup>239</sup>Pu, <sup>240</sup>Pu and <sup>241</sup>Pu) in radioactive particles derived from Fukushima Daiichi Nuclear Power Plant accident
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Igarashi, J., Zheng, J., Zhang, Z. et al. First determination of Pu isotopes (239Pu, 240Pu and 241Pu) in radioactive particles derived from Fukushima Daiichi Nuclear Power Plant accident. Sci Rep 9, 11807 (2019). https://doi.org/10.1038/s41598-019-48210-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-48210-4
This article is cited by
-
Analysis of particles containing alpha emitters in stagnant water in Fukushima Daiichi Nuclear Power Station’s Unit 3 reactor building
Scientific Reports (2024)
-
Origin of Cs-bearing silicate glass microparticles observed during Fukushima accident and recommendations on nuclear safety
Journal of Radioanalytical and Nuclear Chemistry (2023)
-
Analysis of particles containing alpha-emitters in stagnant water at torus room of Fukushima Dai-ichi Nuclear Power Station’s Unit 2 reactor
Scientific Reports (2022)
-
Local and global trace plutonium contributions in fast breeder legacy soils
Nature Communications (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
