
Abstract
The tumor suppressor p53 is involved in the DNA damage response and induces cell cycle arrest or apoptosis upon DNA damage. Drosophila p53 encodes two isoforms, p53A and p53B, that induce apoptosis in somatic cells. To investigate the roles of Drosophila p53 isoforms in female germline cells, the DNA damage response was analyzed in the adult ovary. Early oogenesis was sensitive to irradiation and lok-, p53-, and hid-dependent cell death occurred rapidly after both low- and high-dose irradiation. Both p53 isoforms were responsible for this cell death. On the other hand, delayed cell death in mid-oogenesis was induced at a low level only after high-dose irradiation in a p53-independent manner. The daily egg production, which did not change after low-dose irradiation, was severely reduced after high-dose irradiation in p53 mutant females due to the loss of germline stem cells. When the p53A or p53B isoform was expressed in the germline cells in the p53 mutant females at levels that do not affect normal oogenesis, p53A, but not p53B, restored the fertility of the irradiated female. In summary, moderate expression of p53A is critical to maintain the function of germline stem cells during normal oogenesis as well as after high-dose irradiation.
Similar content being viewed by others

Introduction
Ionizing radiation (IR) activates the DNA damage response resulting in cell cycle arrest, DNA repair, apoptosis, and senescence1. Chk2 and p53 are required for irradiation-induced cell death and function as tumor suppressor genes by preventing the accumulation of cells harboring cancer-causing mutations. Since the identification of the Drosophila ortholog of the p53 gene by whole genome sequencing2,3, Drosophila has been used as a model organism to study the DNA damage response4. However, most studies in Drosophila and the mammalian system, have been performed using mitotically dividing somatic cells, and the DNA damage response in germline cells, including germline stem cells (GSCs), is relatively less characterized.
Drosophila adult females have a pair of ovaries, each containing approximately 16 ovarioles. Each ovariole consists of a germarium and several egg chambers with different developmental stages, up to 14. There are 2–3 GSCs in a germarium. GSCs undergo asymmetric cell division to produce a daughter GSC and a cystoblast that will undergo 4 synchronous cell divisions with incomplete cytokinesis to generate 16-cell cysts. Among the 16 cells, one cell is fated to become an oocyte and others become nurse cells, supplying proteins and RNAs to the oocyte at the end of oogenesis. Normal oogenesis is affected by stress, such as poor nutrition, resulting in cell death during early and mid-oogenesis and slowing of the cell cycle5. Previously, we have shown that high-dose irradiation induces cell cycle arrest of cystoblasts, cell death at early (germarium region 2) and mid-stage (stages 7–10), and morphological defects6. Most of these defects are recovered within a week after irradiation and the GSCs appear to survive high-dose irradiation and are able to produce viable oocytes. Since p53 is a major gene involved in irradiation-induced cell death in somatic cells, we aimed to investigate the function of p53 in oogenesis after irradiation.
Historically, three different Drosophila p53 isoforms, p53A (also reported as dp53, p53, DΔNp53, and p53A), p53B (Dp53, p53B), and p53n (p53ΔC), due to usage of alternative promoter and alternative splicing, were reported five years after the first isolation of the Drosophila p53 gene, p53A7. Currently, four p53 mRNA transcripts (A, B, C, and E) potentially encoding three different protein isoforms of 385, 495, and 334 amino acids (p53A, B, and E, respectively) are predicted in the Drosophila genome annotation8 (Fig. 1a). Except for the first 13, 123, and 10 amino acids of p53 A, B, and E, respectively, the amino acid sequences of the three isoforms are identical, generating a distinct N-terminal transactivation domain for each isoform with the same central DNA binding and C-terminal oligomerization domains (Fig. 1a).

Cell death increases after irradiation in Drosophila ovaries. Wild type adult females were irradiated and dying cells in the ovary were detected by TUNEL staining. (a) Schema of the Drosophila p53 mRNA and protein isoforms. Four p53 RNA transcripts (left) predicted by the current p53 gene annotation from Flybase are shown. Exons are indicated with boxes and the predicted open reading frames and 5′ or 3′ UTRs are indicated in orange and gray, respectively. The DNA regions deleted in p53 mutants are shown below. The transcriptional transactivation domains (TAD), DNA binding domain (DBD), and oligomerization domain (OD) of each p53 isoform are shown (right). The N-terminal regions with unique amino acid sequences are indicated with different colors. The amino acid regions recognized by the p53 antibodies (S1, d200, E5, and H3) are indicated above. (b) The percentages of TUNEL-positive germarium were counted 6 h after irradiation at the indicated doses. The values are the mean ± standard deviation (SD) of three independent experiments (*p < 0.05). At least 192 ovarioles in total were counted for each dose. (c) Representative images of TUNEL-positive (red) germarium 6 h after 3 Gy irradiation are shown. DAPI (blue) and Orb (green) staining shows DNA and germline cells in the germarium region 2 and 3, respectively. (d) The percentage of ovariole containing TUNEL-positive germline cells in the germarium or egg chambers at stage 7–10 were counted. In the case of follicle cells, the egg chambers containing more than 4 TUNEL-positive follicle cells at stages 2–6 (St. 2–6) or at stages 7–10 (St. 7–10) were counted. The values are the mean ± SD of three independent experiments (ns p > 0.05, **p < 0.01). At least 290 ovarioles or 330 egg chambers for follicle cell death in total were counted. (e) Ovarioles containing TUNEL-positive germline cells in the germarium or stage 7–10 egg chambers were counted at 0, 2, 4, 6, 8, 12, 24, and 48 h after 3 Gy irradiation. The values are the mean ± SD of three independent experiments. At least 160 ovarioles in total were counted for each time point.
Since the discovery of p53 isoforms, various studies have been performed to identify the specific functions of each isoform. When the p53A and p53B isoforms are overexpressed at the same protein level in the larval disc cells by targeted integration of the transgene, both p53A and p53B induce apoptosis, while a smaller isoform, p53E, inhibits irradiation-induced apoptosis9. When a BAC clone specifically affecting each isoform was used for rescue analysis, p53A, but not p53B, was primarily required for DNA damage-induced apoptosis by inducing hid gene expression in the larval disc cells9. In addition to the mitotically dividing somatic cells mentioned above, the role of the p53 isoforms in endocycling cells, where cell death is normally repressed, has been reported10. In this cell type, the overexpression of p53B, but not p53A, induces cell death. In male germline cells, p53 is required for programmed necrosis which occurs in mitotic germ cells in the absence of stress11. On the other hand, relatively less is known about the role of p53 isoforms in female germline cells.
In this study, we investigated irradiation-induced cell death during oogenesis and the role of p53 isoforms in the Drosophila germline cells after low- and high-dose irradiation. We found that IR-induced cell death occurs at two developmental stages of oogenesis, depending on the irradiation doses. Using transgenic flies expressing p53 isoforms, we found that the expression level of p53A, but not p53B, should be maintained at low levels to support normal oogenesis in the absence of stress. In response to DNA damage, both p53 isoforms restored IR-induced apoptosis during early oogenesis. Moreover, the p53A rather than the p53B isoform was responsible for maintaining the function of GSCs after high-dose irradiation.
Results
Low-dose irradiation induces cell death during early oogenesis in Drosophila female
Previously, we have shown that high-dose irradiation induces rapid and massive germline cell death during early oogenesis followed by a slow and lower amount of cell death during mid-oogenesis6. To understand radiation-induced cell death during Drosophila oogenesis in greater detail, adult females were treated with different doses of IR. TUNEL-positive dying cells in the germarium region 2 increased in a dose-dependent manner (Fig. 1b,c). Cell death was slightly but significantly increased 6 h after irradiation at as low as 0.3 Gy (14.9% compared to 7.3% of untreated sample, p < 0.05). To facilitate detection, low-dose irradiation was performed after 3 Gy irradiation, which induced cell death in half of the germarium (Fig. 1b,e). These results were compared with those of high-dose irradiation at 40 Gy. The maximum level of cell death was detected in the germarium 8 h after 3 Gy irradiation and was reduced to basal levels 48 h after irradiation. Conversely, cell death was not increased in stage 7–10 egg chambers until 72 h after irradiation (Fig. 1e and data for 72 h not shown), at which time significant cell death was observed after 40 Gy6. Similar to high-dose irradiation6, 3 Gy irradiation induced cell death in mitotically dividing somatic follicle cells at stages 2–6, but not in endocycling follicle cells at stages 7–10 (Fig. 1d). Therefore, low-dose irradiation induced rapid cell death in early oogenesis and mitotically dividing follicle cells as with high-dose irradiation but did not induce delayed degeneration of mid-stage egg chambers, suggesting that early oogenesis is more sensitive to DNA damage than mid-oogenesis.
lok, p53, and hid are required for IR-induced cell death in early oogenesis
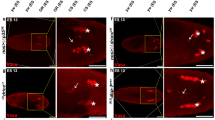
IR-induced apoptosis in Drosophila somatic cells requires the Drosophila ortholog of Chk2 (lok)12 and p53 (p53)13. Additionally, proapoptotic genes such as reaper and hid, which are induced by p53, are also required for cell death and the contribution of each gene depends on cell context. In our previous study, we investigated whether the same signaling pathway was induced in the germline cells and found that 40 Gy-induced cell death in the germarium required p53 and lok6. To further develop this finding, we investigated the cell death in the mutant germarium 6 h after 3 Gy. Cell death was increased in the reaper mutant but not in the lok, p53, and hid mutants, suggesting that lok, p53, and hid, but not reaper, are necessary for low-dose IR-induced cell death in the germarium (Fig. 2a). We also found that hid is required for IR-induced cell death in the germarium when irradiated at 40 Gy (Fig. S1). Therefore, lok, p53, and hid are required for cell death of the germline cells in the germarium after both low- and high-dose irradiation. To test if hid transcription is induced by p53 in the ovary after 40 Gy irradiation, RNA fluorescent in situ hybridization (FISH) and protein immunofluorescence (IF) double labeling was performed. When Dig-labeled anti-sense hid RNA was used as a probe, the hid transcript was increased in wild type germarium and egg chambers but not in p53 mutant after irradiation (Figs 2b and S2a). The signal was not detected before and after irradiation with a sense probe (Fig. S2b). These results suggest that irradiation-induced cell death in the germarium occurs through p53-dependent transcriptional activation of the hid gene. It is interesting to note that the hid transcript is induced in the entire germarium, suggesting that post-transcriptional regulation of hid is necessary for region 2-specific cell death after irradiation.

Genes involved in germline cell-death by IR and poor nutrition. (a) Females were irradiated at 3 Gy and TUNEL staining was performed with the ovaries 6 h after irradiation. The percentages of TUNEL-positive germline cells in the germarium region 2 in lok, p53, hid, and rpr mutant females were determined. The values are the mean ± SD of three independent experiments (ns p > 0.05, *p < 0.05, ***p < 0.001). At least 200 germarium in total were counted for each genotype. (b) Expression pattern of the hid transcript after irradiation. Wild type and p535A-1-4 mutant females were irradiated at 40 Gy and RNA fluorescent in situ hybridization and protein immunofluorescence double labeling was performed using Dig-labeled anti-sense hid RNA as a probe and antibody against Vasa. (c) Wild type and mutant females were grown in rich or poor nutrition conditions and TUNEL staining of the ovaries was performed. The percentages of ovarioles containing TUNEL-positive germline cells in the germarium region 2 (left panel) and TUNEL-positive egg chambers of stage 7–10 are shown (right panel). The values are the mean ± SD of at least two independent experiments (*p < 0.05, **p < 0.01, ***p < 0.001). At least 235 ovarioles in total were counted for each genotype. (d) Wild type and p535A-1-4 mutant females were irradiated with 40 Gy and ovarioles containing TUNEL-positive germline cells in the germarium region 2 or stage 7–10 egg chambers were counted at 6, 24, 48, 72, and 96 h after irradiation. The values are the mean ± SD of at least two independent experiments. At least 100 ovarioles in total were counted for each timepoint.
Poor nutrition also induces cell death in early and mid-oogenesis and p53 has been shown to be dispensable for cell death in mid-oogenesis14. To confirm and extend the genetic requirements of cell death induced by poor-nutrition, we tested whether cell death occurs in lok and p53 mutant ovaries. In contrast to irradiation, starvation of lok and p53 mutant females significantly increased cell death in the germarium (p < 0.001 and p < 0.01, respectively) and stage 7–10 egg chambers (p < 0.05 and p < 0.01, respectively) (Fig. 2c). This result suggests that different signaling pathways are utilized to induce cell death in the germarium depending on the nature of the stimuli: poor nutrition or DNA damage. We also found that the basal level of cell death in the germarium and stage 7–10 egg chambers was significantly increased in p53E4 (p < 0.001), rpr87 (p < 0.001), and lokp6 (p < 0.01), respectively. Increased cell death in the germarium in p53E4 does not appear to be induced by the lack of p53 protein since it was not observed in the null mutant, p535A-1–4, that disrupts all isoforms (Figs 1a and 2a). The role of rpr and lok in early and mid-stage cell death remains to be studied.
In the mitotically dividing somatic cells in Drosophila, cell death induced by high-dose irradiation occurs in two phases15: p53-dependent rapid cell death at high levels followed by slow lower-level cell death that occurs in a p53-independent manner. To test whether the delayed cell death in the germline cells by high-dose irradiation is also p53-independent, time-course analysis of TUNEL staining was performed in the wild type and p535A-1-4 mutant after 40 Gy irradiation. Similar to the finding in the somatic cells, the rapid high-level cell death that was observed in the wild type germarium at 6 h post-irradiation (Fig. 2d solid line with closed circle) was not detected, whereas low level delayed cell death at 24-48 h after irradiation was observed in the p535A-1-4 mutant germarium (Fig. 2d dotted line with open circle). Additionally, the slow and low-level cell death at the stage 7-10 egg chambers that was detected only after high-dose irradiation still occurred in the p535A-1-4 mutant (Fig. 2d dotted line with open triangle, p > 0.05 at 48 h and 72 h compared to wild type). These results suggest that three different signaling pathways are activated in the ovary during two developmental stages in response to high-dose irradiation to remove damaged germline cell: p53-dependent, rapid, and high-level cell death followed by p53-independent, slow, and lower-level death occurs in the germarium region 2 and p53-independent, slower, and lower-level death occurs at stage 7–10.
Role of p53 isoforms in IR-induced cell death
Although the RNA-seq data suggests that the p53B mRNA is expressed throughout Drosophila development8, endogenous p53B protein has not been reported. To determine the expression pattern of various p53 isoforms during development, we performed developmental western blotting using p53 E5 antibody, which recognizes the p53A, p53B, and p53E isoforms (Fig. 1a). The p53A isoform (with a molecular weight of 44 kDa) that was not detected in the p53 mutant ovaries was observed throughout the developmental stages with relatively higher levels in the early embryo and the adult ovaries (Fig. 3a). On the other hand, no specific band corresponding to the p53B or p53E (with a molecular weight of 56 kDa and 38 kDa, respectively) isoforms was detected, suggesting that the p53A is the major isoform expressed throughout development. To determine whether the p53B and p53E isoforms are expressed in the ovary, RT-PCR analysis was performed with total RNA isolated from the wild type and p535A-1-4 mutant ovaries. Using primers that specifically amplify each isoform (Fig. 3b lanes; 1, 2, and 3), the p53B transcript was detected in the wild type but not in the p535A-1-4 mutant ovaries after 30 cycles of amplification (Fig. 3b, upper panel). On the other hand, the p53E transcript was not detected even after 40 cycles of amplification (Fig. 3b, middle panel).

Expression of p53A and p53B can rescue IR-induced germline cell death in the p53 mutant germarium. (a) Lysates were generated from the embryos of the indicated ages, first, second, third instar larvae (1 L, 2 L, and 3 L), pupae, adult males and females, and testis and ovaries from the wild type or p53 mutants. The monoclonal antibody E5, which recognizes p53A, p53B and p53E was used. Bands corresponding to the p53A isoform are indicated by an arrow. Other bands, including the ones indicated by asterisks, appear to be a non-specific or degradation product of p53 protein. The membrane was stripped and reprobed with anti-α-Tubulin as a loading control. (b) To determine expression levels of the p53B and p53E transcripts, reverse-transcription PCR analysis was performed using total RNA preparation from ovaries of wild type and p53 mutant flies. UASp constructs containing the cDNA of each p53 isoform were used as controls. (c) Western blot of ovaries from wild type or transgenic flies expressing the indicated p53 isoform in the p535A-1-4 mutant. Two independent UASp-p53 insertion lines (446 and 450 for p53A, m6 and m63 for p53B) were used for each isoform. The blot was probed with anti-p53 H3 antibody followed by anti-α-Tubulin antibody. The band corresponding to each isoform is indicated. A signal indicated with * may be a degradation product of p53B isoform. (d) Transgenic lines expressing each p53 isoform in the p535A-1-4 mutant germline cells were tested for IR-induced cell death in the germarium by staining with antibody against cleaved Dcp-1 (cDcp-1). The values are the mean ± SD of three independent experiments (ns p > 0.05, *p < 0.05, ***p < 0.001). At least 200 germariums in total were counted for each sample except for p53A(450) rescued flies (48 and 76 germariums for 0 Gy and 40 Gy, respectively). Uncropped gels and blots are shown in Supplementary Fig. S5.
Since p53E was not detected by RT-PCR in the ovary, we focused on the functions of the p53A and p53B isoforms during oogenesis after irradiation. After expressing each p53 isoform in the ovary using germline-specific GAL4 (GAL4-NosVP16) in p535A-1-4 background, the expression level of each transgene from two independent transgenic lines was analyzed. Compared to the endogenous p53A protein in the ovary, the expression level of p53A and p53B transgene was approximately 14~23- and 5~6-fold higher, respectively (Fig. 3c). When p53A was expressed by GAL4-NosVP16 in a p53 mutant background and stained with antibody against Vasa, which detects germline cells, severe morphological defects were observed, most likely due to toxicity. We counted the cell death phenotype only in the germariums that showed an apparently normal Vasa staining pattern. In response to irradiation, lack of cell death in p535A-1-4 mutant germarium was rescued by the expression of p53A and p53B (Fig. 3d), suggesting that both p53A and p53B isoforms can support IR-induced germline cell death in the germarium.
Biochemical characterization of Drosophila p53 isoforms
In the mitotically dividing somatic cells, the mRNA and protein levels of Drosophila p53 are increased after irradiation, but protein stability is not increased due to lack of the MDM2 ortholog in Drosophila10. To test whether the p53A or p53B protein levels in the germline cells are changed after irradiation, we used p53 mutant flies expressing the p53A or p53B isoform in the germline cells by GAL4-NosVP16. Neither the p53A nor p53B protein levels were markedly increased after irradiation (Fig. 4a), suggesting that irradiation does not induce Drosophila p53 protein stabilization in the germline cells.

Biochemical characterization of p53 isoforms. (a) p53 mutant flies expressing p53A or p53B isoforms in the germline cells were irradiated at 40 Gy. Ovary extracts were generated 1 h after irradiation and western blotting was performed. The representative images are shown. After normalizing to the loading control α -Tubulin, the p53 protein levels after irradiation is indicated below the blot by setting each p53 isoform level from non-irradiated control as 1. (b) p53A or p53B protein was expressed individually or together in the germline cells of p53E4 mutant using GAL4-NosVP16 driver. The ovary extract was used to precipitate p53A isoform using the p53 S1 antibody, which recognizes p53A, not p53B. Immunoprecipitation with p53 d200 antibody, which recognizes both p53A and p53B, was used as a control. Western blotting was performed using the p53 E5 antibody. Lanes with “-“ indicate the input. The short and long exposures of the images are shown. Uncropped blots are shown in Supplementary Fig. S5.
In addition to protein stabilization, the tetramerization of p53 is also important for the transcriptional activation of p5316. Similar to human p53, the Drosophila p53 isoforms, both p53A and p53B, contain oligomerization domain, which has been shown to form tetramers17. To test whether p53A and p53B can form a heterotetramer, we developed a polyclonal antibody S1 that could specifically interact with p53A but not with p53B. We also generated p53 mutant flies that could express either p53A or p53B alone or the two isoforms together using GAL4-NosVP16 driver. Immunoprecipitation analysis of the ovary lysates confirmed that the p53A-specific antibody (S1) could precipitate p53A but not p53B (Fig. 4b lanes 2, and 5). When the S1 antibody was used to precipitate p53 in the flies expressing both p53A and p53B, the p53B isoforms were pulled down, confirming that p53A and p53B could form a complex (Fig. 4b lane 8).
Role of p53 isoforms in egg production of irradiated females
To test the role of p53 isoforms during oogenesis after irradiation, we compared the egg production of females after 3 and 40 Gy irradiation. The wild type females irradiated at 3 Gy showed slightly reduced egg production compared to the non-irradiated females until 11 days after irradiation (Fig. 5a). Interestingly, the large reduction of egg production observed in the females between 3–4 days after 40 Gy irradiation was not detected in the 3 Gy-irradiated females. This may be due to cell death in stage 7–10 egg chambers, which was induced after irradiation at 40 Gy but not at 3 Gy. In the 3 Gy-irradiated p535A-1-4 mutant females, daily egg production was not significantly different from that of the unirradiated females (p > 0.05) (Fig. 5b), suggesting that reduced egg production in the 3 Gy-irradiated wild type females may be due to p53-dependent cell death. After 40 Gy irradiation, the reduction in egg production 3 days after irradiation in the p535A-1-4 mutant females was never recovered and continued to decrease (Fig. 5b). In the p535A-1-4 mutant females expressing the p53A isoform by Gal4-NosVP16, we could not conclude whether p53A could restore the egg production after irradiation since the daily egg production was severely reduced even in the absence of irradiation (Fig. 5c). When p535A-1-4 mutant females were rescued by the NosVP16-driven p53B isoform, daily egg production after 3 Gy-irradiation was slightly reduced compared to unirradiated female similar to wild type females (Fig. 5d). In response to 40 Gy irradiation, the egg production that was decreased 3 days after irradiation remained low until day 7, but gradually increased by day 11 (Fig. 5d). These results indicate that the p53B isoform can rescue the changes in egg production in p53 mutant females after low- and high-dose irradiation.

Role of p53 isoforms in the egg production of females after low- and high-dose irradiation. Females of the indicated genotypes (a: wild type, b: p535A-1-4, c: GAL4-NosVP16/UASp-p53A(450); p535A-1-4, d: GAL4-NosVP16/UASp-p53B(m6); p535A-1-4) were irradiated at 0, 3, or 40 Gy and the eggs were collected every 24 h. Day 1 corresponds to the egg collection during the first 24 h after irradiation. The number of eggs per female were determined. The data are presented as the mean ± SD of the number of eggs per female of at least two independent experiments. Each experiment was performed using 5 cages for each treatment and each cage contained 3 pairs of females and wild type unirradiated males.
Moderate level expression of p53A and p53B isoform can restore loss of germline stem cells (GSCs) in the p53 mutant after high-dose irradiation
Since high levels of p53A isoforms were found to affect normal oogenesis, we attempted to reduce the expression level using another germline-specific driver, GAL4-NosNGTA. By altering transgene dosages, the expression level of the p53A isoform in the p53 mutant background was reduced to 5–49% of the endogenous p53A levels in the wild type ovary (Fig. 6a). Even though the expression level of p53A was reduced to 5% of that of endogenous p53A, NosNGTA-driven p53A was functional because it was able to restore the lack of cell death 6 h after irradiation in the germarium (Fig. 6b).

The moderate level expression of the p53A and p53B isoforms restores the loss of GSCs in the p535A-1-4 mutant after high-dose irradiation. (a) A representative image of the western blotting of the ovaries from wild type, p535A-1-4 mutant, and transgenic flies expressing p53A by Gal4-NosNGTA driver in p535A-1-4 mutant background [lane 3: UASp-p53A(446)/+; GAL4-NosNGTA, p535A-1-4/+, p535A-1-4, lane 4: UASp-p53A(450)/+; GAL4-NosNGTA, p535A-1-4/+, p535A-1-4, lane 5: UASp-p53A(446); GAL4-NosNGTA, p535A-1-4, and lane 6: UASp-p53A(450); GAL4-NosNGTA, p535A-1-4/+, p535A-1-4] is shown. The blot was probed with anti-p53 H3 antibody followed by anti-α-Tubulin antibody. The relative amount of p53A levels normalized by Tubulin is shown below the blot compared to that of wild type as 100. A signal indicated with * may be a degradation product of p53A isoform. (b) The wild type, p535A-1-4 mutant, and transgenic flies expressing p53A by Gal4-NosNGTA driver in p535A-1-4 mutant background were irradiated at 40 Gy and the germline cell death in the germarium was assayed by staining with antibodies against cleaved Caspase 3. The data represent the mean ± SD of two independent experiments (ns p > 0.05, **p < 0.01). At least 153 germariums in total were counted for each sample. (c) The wild type, p535A-1-4 mutant, and p535A-1-4 mutant flies expressing p53A or p53B were irradiated at 40 Gy. Six days later, the number of GSCs in the germarium was analyzed by staining with 1B1, Vasa, and Lamin C to detect fusome, germline cells, and cap cells, respectively. The values are the mean ± SD of two independent experiments (ns p > 0.05, *p < 0.05, **p < 0.01, ***p < 0.001). At least 51 germariums in total were counted for each sample. The transgenes were used as hemizygous (1×) or homozygous (2×) state. The number of GSCs after irradiation in each genotype compared to that of p535A-1-4 mutant is indicated below by setting the number of GSCs in irradiated p535A-1-4 mutant as 1. Uncropped blots are shown in Supplementary Fig. S5.
To confirm whether the changes in egg production represent loss of GSCs, we counted the number of GSCs after high-dose irradiation. The GSCs were identified as a germline cells at a close vicinity to the cap cell and were found to contain round fusomes by staining with VASA (germline cells), 1B1 (fusome), and Lamin C (cap cells) antibodies. In the absence of irradiation, the number of GSCs in the p535A-1-4 mutant, and the p535A-1-4 mutant expressing p53A or p53B was not significantly different from that of the wild type (Fig. 6c). Six days after 40 Gy irradiation, the number of GSCs in the wild type germarium was not significantly different from the untreated control (0 Gy: 2.0 vs 40 Gy: 1.7, Fig. 6c). On the other hand, the number of GSCs in the p535A-1-4 mutant was significantly decreased from 1.7 to 0.6 at 6 days after irradiation (p < 0.05) suggesting that reduced egg production in the irradiated p535A-1-4 mutant females is due to the loss of GSCs. To test which p53 isoform is involved in this process, we used two transgenic lines expressing p53B by GAL4-NosVP16 and four transgenic lines expressing p53A by GAL4-NosNTGA. The number of GSCs after irradiation in these flies were 1.9–2.9-fold higher (3.1-fold higher in wild type) than that of the p535A-1-4 mutant, suggesting that both p53A and p53B can partially rescue loss of GSCs after irradiation when expressed at moderate levels. It is interesting to note that the number of GSCs in the irradiated wild type and the p535A-1-4 mutant expressing p53B were restored to untreated level by day 6 after a slight reduction at 3 days post-irradiation (Fig. S3). On the other hand, the number of GSCs in the p535A-1-4 mutant continuously decreased by day 6 (Fig. S3), suggesting that p53 may be required for the self-renewal of GSCs.
Low level expression of p53A but not p53B isoform can restore loss of fertility in the p53 mutant after high-dose irradiation
To test whether the GSCs detected after high-dose irradiation were functional, the fertility of the adult female flies was tested by the production of pupae from the eggs laid by the female. In the absence of irradiation, the fertility of the wild type and p535A-1-4 mutant females was not significantly different at days 10-15 (Fig. 7 upper panel). The fertility of the irradiated wild type females was very low during the first 4 days after irradiation; however, it gradually increased up to that of the non-irradiated control at 10–15 days after irradiation (Fig. 7 lower panel). When p535A-1-4 mutant females were irradiated, the fertility was not increased at 4–15 days and remained low supporting the previous findings that high-dose irradiation reduces the egg production and the number of GSCs in the p535A-1-4 mutant (Figs 5b and 6c).

The expression of the p53A isoform at low levels restores the loss of fertility in the p535A-1-4 mutant after high-dose irradiation. The adult female flies of the indicated genotypes were irradiated at 0 or 40 Gy and incubated with non-irradiated males. The adults were transferred to new vials every 24 h. The fertility was scored by the number of pupae produced 7–8 days after the adults were removed. The data represent the mean ± SD of the number of pupae produced per day by a single female during the indicated period after irradiation of at least two independent experiments (ns p > 0.05, *p < 0.05, **p < 0.01). The transgenes were used as hemizygous (1×) or homozygous (2×) state. The fertility after irradiation at 10–15 days in each genotype compared to that of p535A-1-4 mutant is indicated below by setting the fertility of irradiated p535A-1-4 mutant as 1.
In the case of the p535A-1-4 mutant expressing p53A more than 30% of endogenous p53A, the fertility in the absence of irradiation was severely lower than that of the wild type (Fig. 7 last two genotypes in the upper panel). This result suggests that the GSCs in these flies are not functional and the amount of p53A protein should be maintained at low levels to support normal oogenesis. The expression of p53A at 5% of the endogenous p53A levels was able to support normal oogenesis in the absence of irradiation and could partially restore the fertility of the p535A-1-4 mutant female after irradiation (4.7 and 5.3-fold higher than p535A-1-4 mutant compared to 6.3-fold higher in the wild type, Fig. 7 5th and 6th genotypes). However, expression of p53B, which showed normal oogenesis without stress, was not able to restore the fertility (0.8- and 0.9-fold less than p535A-1-4 mutant, Fig. 7). These results suggest that both p53A and p53B isoforms could maintain the number of GSCs after high-dose irradiation, but the GSCs maintained by p53A, not by p53B, was functional to produce progeny.
Discussion
To understand the role of p53 isoforms in germline cells, we investigated the cellular response of germline cells during Drosophila oogenesis after low- and high-dose irradiation. We found that IR-induced apoptosis occurs at two developmental stages of oogenesis through different mechanisms. Rapid and high-level apoptosis is induced during early oogenesis in the germarium region 2 involving lok, p53A, p53B, and hid in response to both low- and high-dose irradiation. Mid-oogenesis was relatively resistant to irradiation and slow and low-level cell death was induced in a p53-independent manner only after high-dose irradiation. The expression levels of the p53 isoforms have distinct effects on oogenesis in the absence and presence of DNA damage. First, the expression level of p53A, but not p53B, should be maintained at a low level to support normal oogenesis in the absence of stress. Second, both p53 isoforms were able to induce IR-induced apoptosis during early oogenesis. Third, both p53A and p53B isoforms could restore the number of GSCs after high-dose irradiation. Lastly, the p53A rather than the p53B isoform was responsible for maintaining the function of GSCs after high-dose irradiation.
During normal development, cell death occurs in nurse cells at the late-stage of oogenesis (stages 12–14). Additionally, cell death can be induced in a stage-specific manner at early- and mid-oogenesis in response to developmental abnormalities or environmental stress such as poor nutrition18. We found that irradiation also induces death in germline cells in early- and mid-stage oogenesis. Cell death in the germarium occurs through apoptosis since both TUNEL and cleaved Dcp-1 positive cells were increased after irradiation (Figs 1b and 3d) and TUNEL and cleaved Dcp-1-positive cells appeared in close vicinity (Fig. S4). Cell death after irradiation in the germarium required lok and p53, while these genes were not necessary for poor nutrition-induced cell death (Fig. 2a,c). Irradiation-induced cell death in the germarium also requires hid, but not reaper, and hid transcription is induced by irradiation in a p53-dependent manner throughout the germarium (Fig. 2b) although cell death occurs only in region 2 (Fig. 1c). This is not surprising since hid is expressed both in cells that survive as well as in only a subset of cells that die19, suggesting that hid may be negatively regulated post-transcriptionally in the surviving cells. In support of this hypothesis, Hid is negatively regulated by EGFR signaling through MAPK-dependent phosphorylation20. The signaling pathway of cell death in mid-oogenesis by poor nutrition is unique in that it requires effector caspase Dcp-1 but not proapoptotic genes nor p5318. We also found that high-dose irradiation-induced cell death in mid-oogenesis does not require p53 (Fig. 2d). These results indicate that at least three distinct signaling pathways induce cell death in female germline cells in response to stress at two developmental stages: lok- and p53-dependent cell death in the germarium after irradiation, lok- and p53-independent cell death in the germarium in response to poor nutrition, and p53-independent cell death in mid-oogenesis in response to both irradiation and poor nutrition.
In response to DNA damage, the GSCs of adult Drosophila females immediately arrest the cell cycle and repair the damaged DNA faster than their differentiated progeny21. DNA damage-induced apoptotic cell death in the GSCs is prevented by the repression of hid transcription through the receptor tyrosine kinase Tie-mediated signaling pathway activated by the survival signal from dying daughter cells22. More severe DNA damage results in the loss of the GSCs and a progeny differentiation defect in a lok-dependent manner21. Drosophila ATM, ATR, and p53, but not Chk1, are required for preventing DNA damage-induced loss of GSCs21. We found that the Drosophila p53 isoforms exhibit two functions in the GSCs after irradiation: both p53A and p53B were able to restore the number of GSCs by 6 days after high-dose irradiation (Fig. 6c). Interestingly, irradiated GSCs maintained by p53A expression, but not by p53B, were functional, as observed in irradiated females with restored fertility (Fig. 7). Since it takes approximately 10 days for the GSCs to develop into mature eggs and oogenesis does not undergo significant delay after irradiation6, the fertility of the irradiated females 10–15 days after irradiation is likely to represent the function of the irradiated GSCs. The mechanism of GSC maintenance by p53 has not yet been fully elucidated, despite previous reports providing some clues. DNA repair does not appear to be responsible since the repair kinetics determined by phosphorylated histone H2Av staining were similar in the wild type and p53 mutant GSCs23. Delay in entry into the cell cycle in the irradiated p53 mutant GSCs was observed23, suggesting that the reduced self-renewal may result in the loss of GSCs. In support of this, self-renewal of irradiated human p53-deficient hematopoietic stem cells has been shown to be compromised in a serial transplantation experiment24. Since the number and function of GSCs were found to be affected in the p53 mutant, p53 may be required for the proper differentiation of stem cells. In addition, p53 reporter activity is increased in GSCs after irradiation23, suggesting that the identification of p53 target genes in irradiated GSCs could facilitate our understanding of the mechanism of p53 in GSCs maintenance after severe DNA damage.
Fluorescence correlation spectroscopy analysis showed that approximately 60% of p53 proteins are in dimeric form in living cells and DNA damage induces rapid assembly into tetramers16. Even when the p53 level was decreased by translation inhibition, p53 target genes were induced after DNA damage suggesting that the tetramerization in the absence of increased total p53 protein levels was sufficient to activate transcription16. We found that the Drosophila p53A and p53B proteins overexpressed in the germline cells are not stabilized after irradiation (Fig. 4a) and that they form a complex (Fig. 4b). These findings suggest that tetramerization may be the major mechanism of p53 activation in Drosophila and the relative protein levels of p53A and p53B will affect the relative levels of hetero- and homo-tetramers and resultant target gene expression. The identification of the oligomer-specific target genes will provide insights into the novel functions of p53 in vivo.
In summary, we have shown that early oogenesis is very sensitive to irradiation and induces cell death in a p53A- and p53B-dependent manner, whereas low-level apoptosis is induced in mid-oogenesis only after high-dose irradiation in a p53-independent manner. Moreover, the p53A rather than the p53B isoform is responsible for maintaining the function of GSCs after high-dose irradiation. The apoptotic function of p53 in early germline cells is similar to that of somatic cells, while the p53A isoform shows a unique function in adult female GSCs. The Drosophila ovary will serve as a great model system to further elucidate the novel functions of p53 in stem cell biology.
Materials and Methods
Drosophila strains
All Drosophila strains were raised at 25 °C. Canton S or w1118 were used as wild type controls. Drosophila strains were obtained from the Bloomington Drosophila Stock Center (BDSC; Bloomington, IN, USA) unless otherwise indicated. The lokp6, p53E4, hid05014, and rpr87 flies were provided by Drs. W. Theurkauf25, J. Chung13, H. Steller19, and K. White26, respectively. To generate flies expressing p53 isoforms, the cDNA for each isoform was cloned into a UASp vector and the transgenic flies were generated by germline mediated P-element transformation.
Sample preparation for immunoprecipitation and western blotting
Tissues from Drosophila at adult or various developmental stages were homogenized with buffer containing 50 mM Tris-HCl pH 7.4, 0.5% Nonidet P-40, 150 mM NaCl, 1 mM EDTA, 50 mM NaF, 20 mM NaVO3, 20 mM beta-glycerophosphate (Sigma-Aldrich; St. Louis, MO, USA), Protease Inhibitor Cocktail (Sigma-Aldrich), and 2 mM PMSF (Amresco; Solon, OH, USA). After centrifugation, the supernatants were used for immunoprecipitation or western blotting analysis following the standard procedures.
Immunofluorescence staining and antibodies
Adult females were either mock-treated or irradiated in a Cs137 gamma-irradiator and the ovaries were then subjected to immunofluorescence and TUNEL staining as previously described6. For nutrition study, two to three-day old females were incubated with rich or poor food for two days and the ovaries were dissected for staining. Plain molasses plates were used as a poor food source and molasses plates containing a layer of wet yeast served as a rich food source5. A polyclonal antibody (S1) that could recognize p53A but not p53B was generated by immunizing rabbits with a synthetic polypeptide (MYISQPMSWHKESTD) in the N-terminal region of p53A. Peptide synthesis and antibody generation were performed at AbFrontier (Seoul, Korea). Other primary antibodies used in this study included p53 H3 (Developmental Studies Hybridoma Bank; Iowa City, IA, USA), p53 E5 (Santa Cruz; Dallas, USA), p53 d200 (Santa Cruz), Vasa (Santa Cruz), Lamin C (Developmental Studies Hybridoma Bank), 1B1 (Developmental Studies Hybridoma Bank) and α-Tubulin (Sigma-Aldrich). The following secondary antibodies were used: goat anti-mouse Alexa568 (1:400, Molecular Probes; Eugene, OR, USA), goat anti-mouse Alexa647 (1:200, Molecular Probes), goat anti-rabbit Alexa488 (1:200, Molecular Probes), goat anti-rabbit Alexa568 (1:400, Molecular Probes) and donkey anti-goat Alexa488 (1:200, Molecular Probes). The ovaries were visualized using a confocal laser scanning microscope (LSM 700, Carl Zeiss, Oberkochen, Germany) or a fluorescence microscope (IX71, Olympus; Tokyo, Japan).
RNA fluorescent in situ hybridization (FISH) and protein immunofluorescence (IF) double labeling and RT-PCR
To generate RNA probes, part of the hid cDNA (375 bp) was cloned into pGEM-T easy vector (Promega; Madison, WI, USA) in sense and antisense orientation. The DNA plasmids were linearized by NdeI digestion and transcribed by T7 RNA polymerase using Digoxigenin (Dig) RNA labeling Mix (Roche Diagnostics; Germany) for 2 h at 37 °C. The reaction was stopped by adding 0.2 M EDTA (pH 8.0) and the RNA was precipitated by ethanol and dissolved in RNase free distilled water.
FISH/IF double labeling to detect the hid transcript and Vasa was performed as previously described with slight modifications27. Briefly, four- to six-day old females were irradiated at 40 Gy and ovaries were dissected in 1X PBS after 3–4 h. Samples were fixed in a 1:1 mixture of 4% formaldehyde (Sigma-Aldrich) and heptane (Sigma-Aldrich) for 20 min with rocking. Samples were permeabilized with 3 μg/ml Proteinase K for 13 min at room temperature and transferred to ice for 1 h before washing with 2 mg/ml glycine solution. After re-fixation and blocking, the samples were hybridized with 50 ng Dig-labeled RNA probe at 56 °C overnight. The ovaries were incubated with biotin conjugated mouse monoclonal anti-DIG (Jackson ImmunoResearch lab Inc., 1:400) and rat anti-Vasa (Developmental Studies Hybridoma Bank, 1:20) for 2 h at room temperature with shaking. Secondary antibody incubation was performed for 1 h with 1X streptavidin-HRP (Invitrogen) and goat anti-rat rhodamine (Molecular Probes, 1:200) at room temperature. Signal amplification was performed with Alexa 488 Tyramide conjugate (1:50) in 1X reaction buffer supplied with Alexa FlourTM 488 Tyramide SuperBoostTM kit (Invitrogen) for 1 h at room temperature. The reaction was terminated using stop solution for 10 min. The ovaries were mounted in 0.5% n-propyl gallate dissolved in glycerol.
RNA isolation from the ovary and the RT-PCR was performed as previously described28.
Fertility test
Four- to five-day-old adult females were irradiated at 0 or 40 Gy. Three vials containing four irradiated females and four unirradiated wild type males per vial were prepared for each genotype. The flies were transferred to new vials each day for 15 days. Fertility was scored according to the presence of pupae 7 to 8 days after the parents were removed from the vials. The daily egg production was determined as previously reported6.
Statistical analysis
The experiments were performed at least two times. The quantitative data are expressed as the mean ± SD. The significance of differences between two experimental samples was determined using two‐sided, unpaired Student’s t‐test. Differences were considered statistically significant at p < 0.05.
Data Availability
All data generated during this study are included in this published article. Additional raw data will be available on request.
References
Roos, W. P., Thomas, A. D. & Kaina, B. DNA damage and the balance between survival and death in cancer biology. Nat Rev Cancer 16, 20–33, https://doi.org/10.1038/nrc.2015.2 (2016).
Ollmann, M. et al. Drosophila p53 is a structural and functional homolog of the tumor suppressor p53. Cell 101, 91–101, https://doi.org/10.1016/S0092-8674(00)80626-1 (2000).
Brodsky, M. H. et al. Drosophila p53 binds a damage response element at the reaper locus. Cell 101, 103–113, https://doi.org/10.1016/S0092-8674(00)80627-3 (2000).
Song, Y. H. Drosophila melanogaster: a model for the study of DNA damage checkpoint response. Mol Cells 19, 167–179, doi: 828 (2005).
Drummond-Barbosa, D. & Spradling, A. C. Stem cells and their progeny respond to nutritional changes during Drosophila oogenesis. Dev Biol 231, 265–278, https://doi.org/10.1006/dbio.2000.0135 (2001).
Shim, H. J., Lee, E. M., Nguyen, L. D., Shim, J. & Song, Y. H. High-Dose Irradiation Induces Cell Cycle Arrest, Apoptosis, and Developmental Defects during Drosophila Oogenesis. PLoS One 9, e89009, https://doi.org/10.1371/journal.pone.0089009PONE-D-13-43342 (2014).
Bourdon, J. C. et al. p53 isoforms can regulate p53 transcriptional activity. Genes Dev 19, 2122–2137, https://doi.org/10.1101/gad.1339905 (2005).
Thurmond, J. et al. FlyBase 2.0: the next generation. Nucleic acids research 47, D759–D765, https://doi.org/10.1093/nar/gky1003 (2019).
Zhang, B., Rotelli, M., Dixon, M. & Calvi, B. R. The function of Drosophila p53 isoforms in apoptosis. Cell Death Differ 22, 2058–2067, https://doi.org/10.1038/cdd.2015.40 (2015).
Zhang, B., Mehrotra, S., Ng, W. L. & Calvi, B. R. Low levels of p53 protein and chromatin silencing of p53 target genes repress apoptosis in Drosophila endocycling cells. PLoS Genet 10, e1004581, https://doi.org/10.1371/journal.pgen.1004581 (2014).
Napoletano, F. et al. p53-dependent programmed necrosis controls germ cell homeostasis during spermatogenesis. PLoS Genet 13, e1007024, https://doi.org/10.1371/journal.pgen.1007024PGENETICS-D-17-00954 (2017).
Xu, J., Xin, S. & Du, W. Drosophila Chk2 is required for DNA damage-mediated cell cycle arrest and apoptosis. FEBS Lett 508, 394–398, doi:S0014-5793(01)03103-9 (2001).
Lee, J. H. et al. In vivo p53 function is indispensable for DNA damage-induced apoptotic signaling in Drosophila. FEBS Lett 550, 5–10, doi:S0014579303007713 (2003).
Peterson, J. S. et al. Noncanonical cell death pathways act during Drosophila oogenesis. Genesis 45, 396–404, https://doi.org/10.1002/dvg.20306 (2007).
Wichmann, A., Jaklevic, B. & Su, T. T. Ionizing radiation induces caspase-dependent but Chk2- and p53-independent cell death in Drosophila melanogaster. Proc Natl Acad Sci USA 103, 9952–9957, https://doi.org/10.1073/pnas.0510528103 (2006).
Gaglia, G., Guan, Y., Shah, J. V. & Lahav, G. Activation and control of p53 tetramerization in individual living cells. Proc Natl Acad Sci USA 110, 15497–15501, https://doi.org/10.1073/pnas.1311126110 (2013).
Ou, H. D., Lohr, F., Vogel, V., Mantele, W. & Dotsch, V. Structural evolution of C-terminal domains in the p53 family. Embo J 26, 3463–3473, https://doi.org/10.1038/sj.emboj.7601764 (2007).
Jenkins, V. K., Timmons, A. K. & McCall, K. Diversity of cell death pathways: insight from the fly ovary. Trends Cell Biol 23, 567–574, https://doi.org/10.1016/j.tcb.2013.07.005 (2013).
Grether, M. E., Abrams, J. M., Agapite, J., White, K. & Steller, H. The head involution defective gene of Drosophila melanogaster functions in programmed cell death. Genes Dev 9, 1694–1708 (1995).
Bergmann, A., Tugentman, M., Shilo, B. Z. & Steller, H. Regulation of cell number by MAPK-dependent control of apoptosis: a mechanism for trophic survival signaling. Developmental cell 2, 159–170 (2002).
Ma, X. et al. DNA damage-induced Lok/CHK2 activation compromises germline stem cell self-renewal and lineage differentiation. Development 143, 4312–4323, https://doi.org/10.1242/dev.141069 (2016).
Xing, Y., Su, T. T. & Ruohola-Baker, H. Tie-mediated signal from apoptotic cells protects stem cells in Drosophila melanogaster. Nat Commun 6, 7058, https://doi.org/10.1038/ncomms8058 (2015).
Wylie, A., Lu, W. J., D’Brot, A., Buszczak, M. & Abrams, J. M. p53 activity is selectively licensed in the Drosophila stem cell compartment. Elife 3, e01530, https://doi.org/10.7554/eLife.01530 (2014).
Milyavsky, M. et al. A distinctive DNA damage response in human hematopoietic stem cells reveals an apoptosis-independent role for p53 in self-renewal. Cell Stem Cell 7, 186–197, https://doi.org/10.1016/j.stem.2010.05.016 (2010).
Takada, S., Kwak, S., Koppetsch, B. S. & Theurkauf, W. E. grp (chk1) replication-checkpoint mutations and DNA damage trigger a Chk2-dependent block at the Drosophila midblastula transition. Development 134, 1737–1744, https://doi.org/10.1242/dev.02831 (2007).
Moon, N. S. et al. E2F and p53 induce apoptosis independently during Drosophila development but intersect in the context of DNA damage. PLoS Genet 4, e1000153, https://doi.org/10.1371/journal.pgen.1000153 (2008).
Lecuyer, E., Parthasarathy, N. & Krause, H. M. Fluorescent in situ hybridization protocols in Drosophila embryos and tissues. Methods Mol Biol 420, 289–302, https://doi.org/10.1007/978-1-59745-583-1_18 (2008).
Lee, E. M. et al. Drosophila Claspin is required for the G2 arrest that is induced by DNA replication stress but not by DNA double-strand breaks. DNA Repair (Amst) 11, 741–752, https://doi.org/10.1016/j.dnarep.2012.06.007 (2012).
Acknowledgements
This work was supported by the Mid-Career Researcher Program through the National Research Foundation (NRF) of Korea funded by the MEST (NRF-2007-0054123) and the Basic Science Research Program through the NRF of Korea funded by the Ministry of Education (NRF-2014R1A1A20543).
Author information
Authors and Affiliations
Contributions
Y.-H.S. designed the experiments and prepared the manuscript; J.-H.P., T.T.N.N., E.-M.L., V.C.-A., K.-S.L., R.W., and J.C. conducted the experiments; J.-H.P., T.T.N.N., E.-M.L., V.C.-A., K.-S.L., and R.W. analyzed the data. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Park, JH., Nguyen, T.T.N., Lee, EM. et al. Role of p53 isoforms in the DNA damage response during Drosophila oogenesis. Sci Rep 9, 11473 (2019). https://doi.org/10.1038/s41598-019-47913-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-47913-y
This article is cited by
-
Sensitive-stage embryo irradiation affects embryonic neuroblasts and adult motor function
Molecular & Cellular Toxicology (2022)
-
Myc plays an important role in Drosophila P-M hybrid dysgenesis to eliminate germline cells with genetic damage
Communications Biology (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
