
Abstract
Crop disease management often implies repeated application of fungicides. However, the increasing emergence of fungicide-resistant pathogens requires their rotation or combined use. Tank-mix combinations using fungicides with different modes of action are often hard to manage by farmers. An alternative and unexploited strategy are bifunctional fungicides, i.e. compounds resulting from conjugation of the pharmacophores of fungicides with different mechanisms of action. In this paper we describe a new approach to antifungal treatments based on the synthesis of dual agents, obtained by merging the strobilurin and succinate dehydrogenase inhibitor pharmacophores into a new entity. The compounds were tested against important fungal plant pathogens and showed good inhibition of Pyricularia oryzae and Sclerotinia sclerotiorum with activity comparable to commercial fungicides. The inhibition of the cytochrome bc1 and the succinate dehydrogenase enzyme activity confirmed that the new molecules are endowed with a dual mechanism of action. These results were further supported by molecular modelling which showed that selected compounds form stable complexes with both cytochrome b subunit and succinate dehydrogenase enzyme. This work can be considered an important first step towards the development of novel dual-action agents with optimized structure and improved interaction with the targets.
Similar content being viewed by others


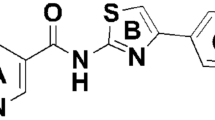
Introduction
As a constantly growing population is in demand of maximized agricultural yields, a precise pest management system is required, both to reduce the toxicity of pesticide application and minimize environmental hazards. Plant pathogenic fungi are among the major causes of crop losses1, and their efficient disease management often implies repeated application of fungicides. As a result, not only the cost of treatments increases, but such usage leads to unfavourable outcomes to the environment, including soil and water pollution. Furthermore, the number of fungal strains resistant to multiple antifungal compounds is dramatically increasing2. To overcome the cited drawbacks, a well-established approach is the use of tank-mix combination of molecules with different modes or sites of action. However, such combinations can be less effective than expected due to differential behaviour of each compound, difficulty to achieve optimal concentrations, unpredictable interactions and enhancement of adverse effects3.
The design of hybrid bifunctional compounds, i.e. conjugates resulting from merging the pharmacophores of active molecules with different mechanisms of action, appears to be a promising alternative to the combination approach, since it displays several advantages4,5. In particular, synergistic interaction of the two active components able to inhibit simultaneously multiple targets, improved bioactivity and lower risk of resistance are expected6. Even though the use of co-formulations or tank-mixes of fungicides with different modes of action is a well established strategy, their conjugation into a single molecule is a relatively underexplored approach. To the best of our knowledge, synthetic studies directed to find dual-action pesticides are very few7,8,9,10. In regard to antifungal compounds, Cheng et al.10 described 1,2,4-triazole-1,3-disulfonamides as dual inhibitors of mitochondrial complex II and complex III, whereas other groups reported examples of strobilurins functionalized with a 1,2,3-triazole moiety9 or with N-phenylpyrimidin-2-amines7.
Strobilurins, or quinone outside inhibitors (QoI), are an outstanding class of fungicides, whose discovery was inspired by a group of natural derivatives of β-methoxy acrylic acid, isolated mainly from basidiomycetes11. These compounds inhibit mitochondrial respiration by binding to a specific site in the mitochondria, the quinol oxidation (Qo) site (or ubiquinol site) of cytochrome b (Cyt b; subunit of the Cyt bc1 complex) and thereby hamper electron transfer between Cyt b and cytochrome c (Cyt c). This prevents oxidation of reduced nicotinamide adenine dinucleotide (NADH) and synthesis of adenosine triphosphate (ATP), thus leading to the inhibition of the energy production essential for survival12. At present, there are numerous synthetic analogues derived from natural strobilurins registered as fungicides in the world market and more are still being developed13. Since strobilurins have a single-site mode of action, they are prone to the development of resistance.
Succinate dehydrogenase inhibitors (SDHI) are the fastest growing class of fungicides in terms of new compounds launched into the market14. The SDH enzyme (also termed succinate ubiquinone oxidoreductase) is a mitochondrial heterotetramer composed of four nuclear-encoded subunits. In contrast to other dehydrogenases of the tricarboxylic acid (TCA) cycle, the SDH enzyme transfers succinate-derived electrons directly to the ubiquinone pool of the respiratory chain and not to soluble nicotinamide adenine dinucleotide (NAD+) intermediates. For this reason, SDH, named also complex II, is considered to be an essential component of the respiratory chain. All crop protection SDHI target the ubiquinone-binding pocket. Upon binding, they physically block the access to the substrate, which consequently prevents further cycling of succinate oxidation. Currently, the “overall” spectrum of SDHI fungicides is extremely broad, being comparable with the QoI spectrum. The most recent SDHI fungicides possess high level of activity against the most important pathogens causing diseases in crops15.
In this paper we report the results of our recent efforts to develop new hybrid fungicides, combining the pharmacophoric features of strobilurins and SDH inhibitors. This choice was mainly dictated by the following reasons: both classes of fungicides are already marketed and are characterised by highly specific mode of action on known and validated targets. Moreover, both strobilurins and SDHI are active at the same subcellular level, the mitochondrion. The design, synthesis and biological activity evaluation of the new compounds are described.
Results and Discussion
The design of dual inhibitors was based on SAR studies reported on both classes of fungicides14,16, which allowed to define the key moieties of the new hybrid molecules. A crucial step in the design was the identification of an appropriate combination and outdistancing between the pharmacophoric moieties of strobilurins and SDHI, to drive to compounds characterized by a suitable size and geometrical shape to fit the binding pockets of both enzymes.
The characteristic feature of all QoIs with excellent fungicidal activity is a methyl-(E)-β-methoxyacrylate pharmacophore, or its bioisosteric equivalent, attached to a phenyl ring with a side chain in an adjacent ortho-position. The side chains are usually represented by (hetero)aryloxy or (hetero)aryloxymethyl moieties17. On the other side, SDHIs are structurally very diverse, but they all display an essential common feature, which is the amide bond. The “core” moiety, an aromatic or heterocyclic ring, is attached to the carbonyl of the amide bond. The core moiety is essential for binding and in vivo potency, and it enters deeply into the active site of SDH. On the amine side of the amide bond, a “linker” is attached. The linker varies but it is usually an alkyl chain, or a substituted or unsubstituted (hetero) aromatic group. The ortho position usually carries a large hydrophobic group which is a further component of the molecule14.
Taking these findings into account, we conceived to use as prototypical strobilurin pharmacophore the β-methoxyacrylate moiety and to incorporate it into a properly substituted aromatic amide, considered the pharmacophoric group of SDHIs. On the basis of literature evidence, the phenyl ring attached to the β-methoxyacrylate should have a spacer linked to the ortho position (Fig. 1). Basically, two series of compounds were designed: 1) in the first series the aromatic ring of the SDHI moiety and the aromatic ring linked to the β-methoxyacrylate group are connected by a four-atom chain (including the carboxamide group) to maintain the distances between the rings comparable to the distances of the two aromatic rings in azoxystrobin and fluopyram (Fig. 2, compounds 6a–c); 2) the second series of compounds is characterized by a longer linker containing an aromatic ring (Fig. 3, compounds 12a–c), capable of giving additional hydrophobic interactions within the binding pockets of both target enzymes.

Design of hybrid compounds. The SDHI (red) and the strobilurine (blue) pharmacophoric groups are joined by a linker.

Synthesis of compounds 5 and 6a–c.

Synthesis of compounds 12a–c.
Synthesis
The α-iodo-β-methoxyacrylate 2 was prepared in good yield from the commercially available β-methoxyacrylate 1 following a literature method18. Next, compound 2 was coupled with the commercially available 2-methyl-1-phenylboronic acid in a Suzuki-Miyaura reaction in the presence of Pd(PPh3)4 (4 mol%) and K3PO419. Despite the good yield obtained, the relatively large amount of catalyst required made this protocol unsuitable for the scale up on gram scale. We observed that the amount of catalyst can be reduced up to 0.5 mol% without significant variations in yield and reaction time. Compound 3 was then converted into the benzyl bromide 4 by radical bromination in the presence of NBS20. Compound 4 was coupled with N-hydroxyphthalimide to obtain the derivative 5. The best yield was obtained in the presence of K2CO3 as a base21,22. Finally, 5 was deprotected using hydrazine in absolute ethanol and the unstable free O-alkyl hydroxylamine was subjected to acylation without purification. Acylation reactions were performed with acyl chlorides 7a–c in presence of pyridine as a base22 to obtain the final compounds 6a–c (Fig. 2).
The synthetic strategy to obtain compounds 12a–c is highlighted in Fig. 3. Protection of the commercially available 3-aminophenol with (Boc)2O gave compound 8 which was alkylated with benzyl bromide 4 in presence of K2CO3 as a base to afford compound 9 in 65% yield. Boc deprotection was easily performed with 10% v/v trifluoroacetic acid in absolute dichloromethane. The reaction afforded the trifluoroacetic salt, which that was converted in the free amine 10 during the work up by treatment with NaHCO3 saturated solution. Finally, the amine 10 was coupled with the three acids 11a–c to obtain the final compounds 12a–c in good yields. The coupling was performed with EDC·HCl and HOBt as coupling reagents and Hünig’s base.
Antifungal activity of dual compounds on mycelium growth of fungal plant pathogens
Due to their low solubility in water, the tested molecules were dissolved in acetone23. Therefore, two controls were included: MA (malt agar) medium (NTC) and MA supplemented with 1% acetone (ACT).
The antifungal activity of first-series compounds 6a–c was evaluated in terms of mycelium growth inhibition of 11 fungal strains belonging to 7 different fungal species. Compound 5 was tested as well, considering that it also possessed both strobilurin and SDH inhibitor pharmacophores. The activity of strobilurins on the growth of fungal pathogens is often evaluated in the presence of alternative oxidase (AOX) inhibitors such as salicyl hydroxamic acid (SHAM), which is based on the assumption that while AOX is active in vitro, it is not active in vivo. However, the use of SHAM in vitro is controversial. In fact, it was shown that in some pathogens AOX is active also in planta, therefore questioning the use of SHAM in vitro to suppress AOX, as it could lead to overestimation of QoI activity24. Moreover, inhibitory effect of SHAM on the growth of some fungal pathogens was observed25,26, and therefore we evaluated the effect of novel dual compounds and commercial fungicides without the addition of SHAM. The radial growth was measured at 7 DAI, except for fast-growing S. sclerotiorum, whose growth was evaluated at 3 DAI.
Among the first-series compounds, out of the four tested molecules, only 5 showed discrete inhibitory activity against selected fungi, while 6a, 6b and 6c showed no or low activity (Fig. 4a). In particular, 5 inhibited the growth of P. oryzae by ca. 35%, and its activity was comparable with the commercial fungicide kresoxim-methyl (QoI), and even better than that of fluopyram (SDHI). Moreover, 5 inhibited S. sclerotiorum and Curvularia sp. to the same extent of kresoxim-methyl, but less than fluopyram. P. ultimum and F. fujikuroi were not inhibited by any of the molecules, whereas they were well controlled by fluopyram.

Mycelium growth of seven fungal species on control media (NTC, ACT) or media supplemented with diverse fungicides and dual compounds at the concentration 25 mg/L. (a) First-series compounds (5, 6a–c). (b) Second-series compounds (12a–c). Number of replicates is shown in brackets. NTC – non treated control, ACT – 1% acetone, AZX – azoxystrobin, AZT – technical grade azoxystrobin, KM – kresoxim-methyl, FLP – fluopyram, FXP – fluxapyroxad. The measurements were done at 7 DAI, except for S. sclerotiorum, which were done at 3 DAI. The error bars represent standard error. The treatments with different letters for each fungal species are significantly different (Tukey HSD, P < 0.05).
Similarly, the antifungal activity of compounds 12a–c was evaluated in vitro following the same protocol used for compounds 5 and 6a–c. The results are highlighted in Fig. 4b. Compounds 12a–c showed similar activity, with minimal differences against individual pathogens. Moderate inhibitory activity (ca. 50%) was observed against Curvularia sp., P. ultimum, and R. solani, while F. fujikuroi and B. cinerea were only poorly inhibited. The best inhibitory activity for all compounds 12a–c was observed against P. oryzae and S. sclerotiorum. S. sclerotiorum was inhibited by compounds 12a–c in a range of ca. 70–80%. The activities were comparable to strobilurins, but inferior to SHDI. Significantly, 12a–c inhibited the growth of P. oryzae in a range of 60–70% and their activity was better than that of kresoxim-methyl and fluopyram and comparable to that of fluxapyroxad. Formulated azoxystrobin, which is the only commercial fungicide currently registered in the EU for the control of rice blast, showed the best inhibitory activity against P. oryzae. However, the technical grade azoxystrobin (AZT) showed activity comparable with the three tested dual molecules 12a–c. Interestingly, statistical differences were observed between technical grade and commercial azoxystrobin in S. sclerotiorum, P. oryzae and R. solani, but not in other fungi, which confirms that the formulations are specific for particular fungal pathogens and that they significantly influence the performance of the selected fungicide. This also indicates that the activity of compounds 12a–c can be further increased by proper formulation.
SDHI and QoI action of the designed compounds
To separately assess the action of the strobilurin-like (QoI) and SDHI pharmacophores in the putative dual compounds (compound 5 and compounds 12a–c), specific sets of experiments were carried out using the mitochondrial preparations of P. oryzae A2.5.2 in a cell-free system (Fig. 5).

Effect of compounds 5 and 12a–c on the P. oryzae A2.5.2 mitochondrial DBH2:Cyt c reductase and SDH activities. (a,b) Rate of the Cyt c-reduction mediated by the mitochondrial fraction was measured at 30 °C (λ = 550 nm), using decylubiquinol as electron-donor substrate, in the presence of 5, 12a–c and reference compounds AZX and FXP (20 µM) to evaluate the percent of inhibition due to the QoI action. (c,d) Rate of the succinate:2,3-dimethoxy-5-methyl-p-benzoquinone electron transfer mediated by the mitochondrial fraction was measured at 30 °C (λ = 595 nm) using DCPIP in the presence of 5, 12a–c and reference compounds AZX and FXP (50 µM – FXP, AZX, 5; or 80 µM – 12a–c) to evaluate the percent of inhibition due to the SDHI action. Data represent the mean ± standard deviation of at least three independent trials and different superscript letters indicate statistically significant differences (panels a and c: Tukey HSD, p ≤ 0.01; panels b and d: Tukey HSD, p ≤ 0.05).
The measurement of the DBH2:Cyt c reductase activity (i.e. the Cyt bc1 complex activity) was used to evaluate the action of the QoI pharmacophore of compound 5. To this aim, the reduction rate of Cyt c mediated by the mitochondrial fraction of P. oryzae A2.5.2 was measured, using decylubiquinol (DBH2) as electron-donor substrate (Fig. 5a). The Cyt c-reduction rate in the presence of the compound 5 (20 μM) was 68.2 ± 13.4% lower than that measured in the control containing DMSO. Interestingly, the inhibition with compound 5 at 100 μM (83.1 ± 10.9%) almost reached the saturation. Azoxystrobin (20 μM), used as positive control in the same assay conditions, showed an inhibition of the Cyt c-reduction rate (76.7 ± 11.2%) similar to that of compound 5. As expected, fluxapyroxad (20 μM), used as negative control, in the same assay conditions showed no clearly detectable inhibition of the Cyt c-reduction rate (6.7 ± 15%). Similarly, compound 5 was tested also for the ability to inhibit the SDH activity. For this test, the electron transfer rate mediated by the mitochondrial fraction of P. oryzae A2.5.2 in the presence of 2,3-dimethoxy-5-methyl-p-benzoquinone and succinate, was measured by a colorimetric method using the redox dye 2,6-dichlorophenolindophenol (DCPIP, Fig. 5c). Inhibition of the SDH activity (65.7 ± 17.4%) was observed for fluxapyroxad (50 μM). Conversely, no inhibition was observed for compound 5 and azoxystrobin. The obtained results for compound 5 support the hypothesis that this compound has only a strobilurin-like action.
Inhibition of DBH2:Cyt c reductase activity was also observed for second-series compounds 12a–c (Fig. 5b). Compounds 12a,b (20 μM) showed a complete inhibition, while for compound 12c a lower inhibition (54.6 ± 13.7%) was obtained. Compounds 12a–c were also tested for the ability to inhibit the SDH activity (Fig. 5d). Indeed, inhibition of the SDH activity was observed for all the tested compounds (80 µM). Out of three tested molecules, 12b (47.6 ± 16.6%) had an activity comparable with that of FXP (50 μM), while compounds 12a and 12c showed lower activity than FXP (50 μM). No inhibition was observed for azoxystrobin.
Thus, according to the overall inhibition data on the target mitochondrial enzyme activities, compounds 12a–c show the typical target actions associated to both QoI and SDHI pharmacophores.
Molecular modelling studies
With a view to rationalizing the dual activity, docking simulations were performed to explore the binding mode of the new molecules to both Cyt b subunit and succinate dehydrogenase enzyme from P. oryzae. In both cases, the proteins were modelled by homology modelling techniques and docking results were analyzed by comparison with known ligands.
The available sequence for Cyt b from P. oryzae comprises only a 170 residue-fragment, which is unsuitable for modelling the entire binding pocket. Thus, the Cyt b homology model was based on the primary sequence of the Cyt b protein from closely related P. grisea (Uniprot code: Q85KP9). The two sequences are substantially identical in the common region with only two conserved differences out of 170 residues, suggesting a very high identity degree between P. grisea and P. oryzae. Therefore, the structure of Cyt b from P. grisea was modelled by using the resolved Cyt b structure from S. cerevisiae in complex with the inhibitor Stigmatellin A (PDB ID: 3CX5) as the template.
The SDH structure is composed of four subunits which have to be modelled together, since the binding site is located at the interface among them. In detail, the homology model was based on three primary sequences from P. oryzae (i.e. Subunit A: G4NE44; Subunit B: L7JQS7; Subunit C: G4N2P5) while the sequence from P. grisea was used for subunit D (Q5G5B2), since the corresponding sequence from P. oryzae is unavailable. Then, the SDH structure was generated based on the resolved structure of the porcine enzyme in complex with N-[(4-tert-butylphenyl)methyl]-2-(trifluoromethyl)benzamide (E23).
With regard to the docking simulations with Cyt b, all tested compounds (5, 6a–c and 12a–c) showed a binding mode and an interaction pattern similar to that seen for azoxystrobin, which was included in the simulations for easy comparison. Fig. 6 compares the poses as computed for 6b, 12b and azoxystrobin, and reveals that all complexes are almost exclusively stabilized by hydrophobic contacts. In detail, the methyl (E)- β-methoxyacrylate moiety of the ligands is inserted within a highly hydrophobic subpocket, where it interacts with both alkyl (Val92, Leu96, Pro218, Leu222, Leu229) and aryl (Phe75, Tyr226) sidechains. The phenyl ring elicits π–π stacking with Phe225 plus S/π interactions with Met242. The variable portions linked to the phenyl ring are similarly accommodated within an apolar subpocket where they can contact Met71, Ala72, Phe75, Leu76, Ile93 and Phe243. In clear agreement with what was observed for the resolved complex between Bovine cytochrome bc1 and azoxystrobin (PDB ID: 1SQB), the computed complexes are completely devoid of polar interactions and there are no surrounding polar residues which might be at least involved in plausible water-bridged H-bonds. On these grounds, the difference in bioactivity seen for the here proposed compounds can be mostly ascribed to the capacity of the variable portion to stabilize π–π contacts with aromatic residues. Accordingly, the good activity of 5 can be explained by considering the enriched stacking interactions that the phthalimide moiety elicits with Phe24327 (Figure S1). Similarly, the notable activity of 12a–b can be explained by considering the reinforced hydrophobic contacts stabilized by these extended derivatives. The reduced inhibitory activity of 12c can be rationalized considering that the pyridine ring is less effective in stabilizing T-shape π–π stacking, as seen with Phe75 and to minor extent with Phe125.

Comparison of the putative poses as computed for (a) azoxystrobin, (b) 6b and (c) 12b within the Cyt b binding site.
Concerning the docking studies involving the SDH enzyme, Fig. 7 compares the computed poses for fluxapyroxad (used here as a reference) and 6b and 12b derivatives, revealing some common features between the reference compound and 12b which can clearly explain its good activity. In detail, both ligands similarly accommodate the carbonyl group, which elicits two key H-bonds with Tyr123 and Trp202, as well as the trifluoromethyl substituent which is engaged in halogen bonds with Asp122, Tyr123 and Ser199. While assuming a slightly different pose, the phenyl ring approaches Arg83 with which it can stabilize charge transfer interactions, similarly to what is observed for the pyrazole ring of fluxapyroxad. Greater differences are detected when comparing the pose of the anilino moieties of the two inhibitors. Due to the marked steric hindrance induced by the (E)-β-methoxyacrylate moiety group, the N-linked phenyl ring of 12b assumes a shifted pose (compared to fluxapyroxad) by which it is able to insert the phenyl (E)- β-methoxyacrylate moiety within a suitable and rather external subpocket. Here the phenyl ring approaches Tyr71 and Trp81 and the (E)-β-methoxyacrylate group is engaged in a H-bond with Tyr71 plus a set of hydrophobic contacts with Leu66, Leu76, Val80 and Ile84. The greater inhibition activity of fluxapyroxad can be ascribed to its capacity to better approach Trp202 with the N-linked phenyl ring thus reinforcing the key H-bond between Trp202 and the ligand carbonyl function. In contrast, the inactivity of the compounds of the first series (as here exemplified by the complex with 6b, Fig. 7b, and compound 5, Figure S2) can be justified by considering that their phenyl (E)-β-methoxyacrylate moiety, instead of being accommodated within the mentioned subpocket, assumes a pose comparable with that of the anilino ring of fluxapyroxad. Thus, the (E)-β-methoxyacrylate detrimentally bumps against Tyr123 and Trp202 and destabilizes their crucial interactions with the inhibitors.

Comparison of the putative poses as computed for (a) fluxapyroxad, (b) 6b and (c) 12b within the SDH binding cavity.
Conclusion
In this paper we report our first efforts to exploit an innovative approach based on the use of hybrid fungicides. In particular, we combined the pharmacophoric features of two classes of commercially available antifungal compounds: strobilurins and SDH inhibitors. The design of dual inhibitors was based on reported SAR studies on both classes of fungicides, which allowed to define the key moieties of the new molecules. Two series of hybrid compounds, characterized by a variously ortho-substituted aromatic carboxamide moiety connected to a β–methoxyphenylacrylate, were synthesized and tested to evaluate their inhibitory activity on mycelial growth of diverse fungal pathogens.
Out of the tested molecules belonging to the first series, compound 5 showed the highest inhibitory activity against a series of plant pathogenic fungi. In particular, 5 inhibited the growth of P. oryzae, one of the most detrimental pathogens worldwide28, with activity comparable to commercial fungicide kresoxim-methyl, and even better than that of fluopyram. The compound was tested to assess the inhibition of the Cyt bc1 complex and SDH enzyme activities. Inhibition was observed for the Cyt bc1 complex enzyme activity, while no inhibitory activity was observed for the SDH enzyme activity. These results indicate that compound 5 is able to express only a strobilurin-like activity, findings which were further confirmed by docking studies. The docking of compound 5 with the computational model of the Cyt bc1 complex subunit Cyt b showed a binding mode similar to that of the commercial fungicide azoxystrobin, whereas a binding mode different from the standard was observed for 5 in the active site of SDH enzyme. The observed differences were attributed to the steric hindrance of the (E)-β-methoxy acrylate group.
The second series of compounds (12a–c) were designed to increase the distance between the two moieties, which might improve the interaction with both binding pockets. This change led to increased biological activity against all tested fungal pathogens. In particular, good activities were observed against P. oryzae and S. sclerotiorum. All the tested compounds showed strong inhibition of the Cyt c reduction. More importantly, inhibition of SDH enzyme was observed as well. Therefore, these results strongly indicate that compounds 12a–c act as true dual target fungicides expressing both the activity of SDH inhibitors and strobilurins.
Molecular modelling studies further confirmed that the selected compounds are capable of forming stable complexes with both enzymes, providing a computational support to our objective to combine the strobilurin and SDHI-like motifs in a single molecule. Further work is required to develop analogues with optimized structure and improved interaction with the selected targets. Moreover, even if this work was focused on P. oryzae as a model organism to determine the SDHI and QoI action, the approach can be extended to other fungi causing disease on a wide range of economically important crops.
Materials and Methods
Chemistry
The detailed synthetic procedures for all the compounds are reported in supplementary material.
Antifungal activity evaluation
Fungal strains
The following fungal and oomycete strains, belonging to a collection maintained in the laboratory of Plant Pathology at DeFENS, University of Milan, Italy, were used in the study: Botrytis cinerea BC4A and BC-2F-2016, Curvularia sp. CURV3, Fusarium fujikuroi FFM2 and FFM4, Pyricularia oryzae A2.5.2 and TA102, Pythium ultimum FW407 and Sclerotinia sclerotiorum SW361 and SSP1. Rhizoctonia solani FW408 was obtained from Dr. Andrea Minuto (Centro di Sperimentazione e Assistenza Agricola, Albenga, Italy). The strains were maintained as single-spore isolates on malt-agar medium (MA: 20 g/L malt extract, 15 g/L agar) at 4 °C.
Fungicides
The following fungicides were used in the study: azoxystrobin (AZX, Amistar SC – suspension concentrate, 22.9% ai., Syngenta Crop Protection), technical grade azoxystrobin (AZT, Sigma-Aldrich), kresoxim-methyl (KM, Sovran WG – wettable granules, 50% ai., BASF Italia, S.p.A.), fluopyram (FLP, Luna Privilege SC, 50% ai., Bayer CropScience, S.r.l) and fluxapyroxad (FXP, Sercadis EC – emulsifiable concentrate, 30% ai., BASF Italia, S.p.A.).
Inhibition of mycelium growth on media supplemented with fungicides
The inhibitory activity of the first-series molecules 5 and 6a–c and commercial fungicides KM and FLP on mycelium growth of different fungal species was evaluated in a pilot study. A mycelium plug (0.5 cm in diameter) obtained from actively growing fungal colonies of B. cinerea BC4A and BC-2F-2016, Curvularia sp. CURV3, F. fujikuroi FFM2 and FFM4, P. oryzae A2.5.2 and TA102, P. ultimum FW407, R. solani FW408 and S. sclerotiorum SW361 and SSP1 was transferred to MA medium plates supplemented or not with commercial fungicides (KM, FLP) and compounds 5, 6a, 6b and 6c at the concentration 25 mg/L ai. Because the tested dual molecules had low solubility in water, they were dissolved in acetone. Therefore, two controls were included: MA medium (NTC, not treated control), and MA medium supplemented with acetone at the final concentration 1% v/v (ACT). The plates were incubated at 24 °C in the dark. The mycelium growth was measured 3–7 days after inoculation (DAI) and the inhibition of mycelium growth (%) was calculated by comparing the mycelium growth on control and fungicide-supplemented plates.
The activity of the second-series dual molecules 12a–c was evaluated against the same fungal pathogens as described above. Again, two controls were included: NTC and ACT. The commercial fungicides were AZX, KM, FLP and FXP at the concentration 25 mg/L ai. Moreover, technical grade azoxystrobin (AZT, 25 mg/L ai) was included for comparison. The plates were incubated at 24 °C in the dark and the mycelium growth was measured at 7 DAI.
All experiments were performed in at least three replicates. For statistical analysis, the mycelium growth data of the strains belonging to the same fungal species were pooled together and were submitted to ANOVA followed by a Tukey post hoc test for multiple comparison (P < 0.05), using the TukeyC package and R software, version R3.0.329,30.
SDHI and QoI activity of the designed compounds
Preparation of P. oryzae mitochondrial fraction
The mitochondrial fraction was prepared according to Ye and coworkers31 with some modifications. Mycelium liquid culture (200 mL, 20 g/L malt extract, in 1 L flask) was inoculated with 10 mycelium-MA plugs taken from the edge of actively-growing P. oryzae (A2.5.2) colony and incubated at 25 °C in an orbital shaker (125 rpm) in the dark for 6 days. Mycelium was cooled on ice and harvested by vacuum filtration through Miracloth (Calbiochem®, La Jolla, CA, USA). During vacuum filtration residual culture medium was removed by washing 3-times with 30 mL of ice pre-cooled water. The collected well-drained mycelium was chopped in small pieces by scalpel and thoroughly powdered in mortar and pestle in the presence of liquid nitrogen. The mycelium powder (∼ 6 g) was suspended in 10 volumes of ice-cooled mitochondrial buffer (0.5 M sucrose, 10 mM KH2PO4, 10 mM KCl, 10 mM MgCl2, 0.2 mM EDTA, pH 7.2), and gently mixed for 60 min by magnetic stirrer at 4 °C. The suspension was centrifuged two-times for 10 min at 4 °C, 5500 rpm (rotor JA20, Beckman). The cleared supernatant was further centrifuged for 20 min at 4 °C, 11250 rpm (rotor JA20, Beckman). The collected pellet (~ 33 mg) was suspended in mitochondrial buffer (2 mL), divided in 2 Eppendorf tubes and centrifuged again for 20 min at 4 °C, 10000 rpm (rotor F45-24-11, Eppendorf). Each pellet was resuspended in 0.2 mL of the same buffer, and stored at −80 °C until use.
Enzyme assays
The strobilurin-like (QoI) action was evaluated measuring the decylubiquinol:Cyt c reductase activity (i.e. the Cyt bc1 complex activity; EC 1.10.2.2) of the mitochondrial fraction in the presence of the synthesized molecules, using the method described by Zhu et al.7 with some modifications. The quinol enzyme substrate DBH2 was prepared using a NaBH4-driven reduction procedure of decylubiquinone32. At the end of the procedure, the residue containing DBH2 was vacuum-dried using a Savant SpeedVac concentrator and was dissolved in 10 mM HCl-ethanol degassed under nitrogen flow and stored at −80 °C as 0.3 M DBH2 aliquots. The enzyme assay was carried out at 30 °C using a spectrophotometer (Lambda 2, Perkin Elmer, USA) equipped with a Peltier system (PTP-6, Perkin Elmer, USA) temperature controller, in 0.5 mL total volume containing the enzyme assay buffer (50 mM KH2PO4, 3 mM NaN3 and 0.01% lauryl maltoside; pH 7.4; extensively degassed under vacuum) with the sequential addition of 20–100 µM tested molecule (25 µL from stocks, freshly prepared in DMSO) or DMSO (25 µL), 40 µM equine heart Cyt c (C2506, Sigma Aldrich; 40 µL from ice-cold 0.5 mM stock daily prepared in the enzyme assay buffer), 0.12 mM DBH2 (10 µL from 6 mM stock, freshly prepared in ice-cold 10 mM HCl-ethanol degassed under nitrogen flow) and 15 µL of mitochondrial fraction. The Cyt c reduction rate was monitored at 550 nm using the first 6-sec time window and was subtracted from the non-enzymatic Cyt c reduction rate measured during a 30-sec time interval before the addition of the mitochondrial fraction.
The SDHI action was evaluated measuring the succinate:quinone oxidoreductase (SQR) activity (i.e. the succinate dehydrogenase activity; EC 1.3.5.1) of the mitochondrial fraction in the presence of the synthesized molecules using a method based on the use of the redox dye, 2,6-dichlorophenolindophenol (DCPIP), and described by Ye et al.29, with some modifications. The enzyme assay was carried out at 30 °C in 96-well microplates (pre-equilibrated at 30 °C) in 0.2 mL total volume containing 5 µL of mitochondrial fraction (freshly pre-activated by a 30 min incubation, at 30 °C, in the presence of 10 mM sodium succinate), the enzyme assay buffer (50 mM NaH2PO4, 250 mM sucrose, 3 mM NaN3; pH 7.2), 50–80 µM tested molecule (10 µL from stocks, freshly prepared in DMSO) or DMSO (10 µL), 0.20 mM 2,3-dimethoxy-5-methyl-p-benzoquinone (10 µL from freshly prepared stock in the enzyme assay buffer; D9150, Sigma Aldrich), 0.14 mM DCPIP (10 µL from freshly prepared stock thoroughly dissolved in the enzyme assay buffer; D1878, Sigma Aldrich). The enzyme reaction was started by the addition of 10 mM sodium succinate (10 µL from freshly prepared 0.2 M stock in the enzyme assay buffer) and was monitored at 595 nm at different fixed times using a fixed wavelength microplate reader set. To calculate the SQR reaction rate, the absorbance decrease due to the DCPIP reduction during the 2–60 min interval time was considered.
In both enzyme assays, each enzyme rate in the presence of the tested molecule (ratemolecule) was compared to that achieved in the presence of DMSO (rateDMSO), in order to calculate the percent of inhibition (I %) as follows:
ANOVA followed by post-hoc Tukey’s HSD test for multiple comparison was used to determine the significance of the data.
Molecular modelling
The Cyt b homology model was based on the primary sequence of the Cyt b protein from P. grisea (Uniprot code: Q85KP9). The structure of Cyt b from P. grisea was modelled by using the resolved Cyt b structure from S. cerevisiae in complex with the inhibitor Stigmatellin A (PDB Id: 3CX5) as the template. Even though the resolved structure of the bovine Cyt b protein in complex with azoxystrobin was also available, the study involved the Cyt b structure from S. cerevisiae since the latter shows a higher sequence identity with the Cyt b protein from P. grisea (60% vs. 53% with S. cerevisiae and bovine cytochrome, respectively).
The SDH structure is composed of four subunits, which have to be modelled together since the binding site is located at the interface among them. In detail, the homology model was based on three primary sequences from P. oryzae (i.e. Subunit A: G4NE44; Subunit B: L7JQS7; Subunit C: G4N2P5) while the sequence from P. grisea was used for subunit D (Q5G5B2) since the corresponding sequence from P. oryzae is unavailable. Then, the SDH structure was generated based on the resolved structure of the porcine enzyme in complex with N-[(4-tert-butylphenyl)methyl]-2-(trifluoromethyl)benzamide (E23).
In both cases, the sequence alignment was performed by NIH Cobalt17 while the homology models were generated by Modeller9.1633. Among the proposed models, the best ones were selected based on DOPE and GA341 scores and optimized by keeping fixed the backbone atoms to preserve the predicted folding. Stigmatellin A and E23 were accommodated within the selected modelled binding sites by superimposing the key residues with those of the utilized templates and the complexes were minimized by keeping fixed all atoms outside a 10 Å radius sphere around the bound ligands. The optimized complexes revealed a set of stabilizing interactions very similar to those seen in the resolved structures, a finding which emphasizes the reliability of the generated homology models.
The conformational profile of the considered ligands was explored by Monte Carlo procedures as described elsewhere34.
The docking simulations were performed by PLANTS focusing the search on an 8.0 radius sphere around the bound ligands35. For each ligand, 10 poses were generated and scored by the ChemPLP score with a speed equal to 1. Along with the computed primary score, the best complexes were also selected based on the interaction similarity with the complexes obtained with Stigmatellin A and E23. The complexes were finally minimized by keeping fixed all atoms outside a 10 Å radius sphere around the bound ligand.
Data Availability
The datasets generated during the current study are available from the corresponding author on reasonable request.
References
Oerke, E. C. Crop losses to pests. J. Agric. Sci. 144, 31 (2006).
FRAC. FRAC List of plant pathogenic organisms resistant to disease control agents, www.frac.info (2018).
Sparks, T. C. & Lorsbach, B. A. Perspectives on the agrochemical industry and agrochemical discovery. Pest Manag. Sci. 73, 672–677 (2017).
Morphy, R., Kay, C. & Rankovic, Z. From magic bullets to designed multiple ligands. Drug Discov. Today 9, 641–651 (2004).
Cincinelli, R. et al. Camptothecin-psammaplin A hybrids as topoisomerase I and HDAC dual-action inhibitors. Eur. J. Med. Chem. 143, 2005–2014 (2018).
Müller-Schiffmann, A., Sticht, H. & Korth, C. Hybrid compounds. From simple combinations to nanomachines. BioDrugs 26, 21–31 (2012).
Li, H.-C. et al. Design, synthesis and fungicidal activity of novel strobilurin analogues containing substituted N-phenylpyrimidin-2-amines. Nat. Prod. Commun. 4, 1209–14 (2009).
Jiang, D., Zheng, X., Shao, G., Ling, Z. & Xu, H. Discovery of a novel series of phenyl pyrazole inner salts based on Fipronil as potential dual-target insecticides. J. Agric. Food Chem. 62, 3577–3583 (2014).
Li, Y., Lei, S. & Liu, Y. Design,synthesis and fungicidal activities of novel1,2,3-triazole functionalized Strobilurins. ChemistrySelect 4, 1015–1018 (2019).
Cheng, H. et al. Discovery of 1,2,4-triazole-1,3-disulfonamides as dual inhibitors of mitochondrial complex II and complex III. New J. Chem. 39, 7281–7292 (2015).
Lamberth, C. In Bioactive Carboxylic Compound Classes: Pharmaceuticals and Agrochemicals 371–384, https://doi.org/10.1002/9783527693931.ch28 (Wiley-VCH Verlag GmbH & Co. KGaA, 2016).
Bartlett, D. W. et al. The strobilurin fungicides. Pest Manag. Sci. 58, 649–662 (2002).
Balba, H. Review of strobilurin fungicide chemicals. J. Environ. Sci. Health. B. 42, 441–451 (2007).
Sierotzki, H. & Scalliet, G. A review of current knowledge of resistance aspects for the next-generation succinate dehydrogenase inhibitor fungicides. Phytopathology 103, 880–887 (2013).
Xiong, L. et al. In Discovery and synthesis of crop protection products, 175–194, https://doi.org/10.1021/bk-2015-1204.ch013 (2015).
Sauter, H., Steglich, W. & Anke, T. Strobilurins: evolution of a new class of active substances. Angew. Chemie Int. Ed. 38, 1328–1349 (1999).
Papadopoulos, J. S. & Agarwala, R. COBALT: constraint-based alignment tool for multiple protein sequences. Bioinformatics 23, 1073–1079 (2007).
Coleman, R. S. & Lu, X. Total synthesis of strobilurin B using a hetero-bis-metallated pentadiene linchpin. Chem. Commun. 0, 423–425 (2006).
Kim, H. H. et al. In Bulletin of the Korean Chemical Society. 191–192 (Korean Chemical Society, 2006).
Zhao, P.-L., Liu, C.-L., Huang, W., Wang, Y.-Z. & Yang, G.-F. Synthesis and fungicidal evaluation of novel chalcone-based strobilurin analogues. J. Agric. Food Chem. 55, 5697–5700 (2007).
Wang, M.-Z. et al. Design, synthesis and antifungal activities of novel pyrrole alkaloid analogs. Eur. J. Med. Chem. 46, 1463–1472 (2011).
Li, Y., Zhang, H.-Q., Liu, J., Yang, X.-P. & Liu, Z.-J. Stereoselective synthesis and antifungal activities of (E)-α-(Methoxyimino)benzeneacetate derivatives containing 1,3,5-substituted pyrazole ring. J. Agric. Food Chem. 54, 3636–3640 (2006).
Ma, B. & Uddin, W. Fitness and competitive ability of an azoxystrobin-resistant G143A mutant of Magnaporthe oryzae from perennial ryegrass. Plant Dis. 93, 1044–1049 (2009).
Liang, H.-J., Di, Y.-L., Li, J.-L., You, H. & Zhu, F.-X. Baseline sensitivity of pyraclostrobin and toxicity of SHAM to Sclerotinia sclerotiorum. Plant Dis. 99, 267–273 (2015).
Avila-Adame, C. & Köller, W. Disruption of the alternative oxidase gene in Magnaporthe grisea and its impact on host infection. Mol. Plant. Microbe. Interact. 15, 493–500 (2002).
Kunova, A., Pizzatti, C., Bonaldi, M. & Cortesi, P. Sensitivity of nonexposed and exposed populations of Magnaporthe oryzae from rice to tricyclazole and azoxystrobin. Plant Dis. 98, 512–518 (2014).
Hohenstein, E. G. & Sherrill, C. D. Effects of heteroatoms on aromatic π−π interactions: benzene−pyridine and pyridine dimer. J. Phys. Chem. A 113, 878–886 (2009).
Martin-Urdiroz, M., Oses-Ruiz, M., Ryder, L. S. & Talbot, N. J. Investigating the biology of plant infection by the rice blast fungus. Fungal Genet. Biol. 90, 61–68 (2016).
Faria, J. C., Jelihovschi, E. G. & Allaman, I. B. Conventinal Tukey Test. (Universidade Estadual de Santa Cruz, 2014).
R_Core_Team. R: A language and environment for statistical computing. (The R Foundation for Statistical Computing, 2013).
Ye, Y.-H. et al. Synthesis and antifungal activity of nicotinamide derivatives as succinate dehydrogenase inhibitors. J. Agric. Food Chem. 62, 4063–4071 (2014).
Gudz, T. I., Tserng, K. Y. & Hoppel, C. L. Direct inhibition of mitochondrial respiratory chain complex III by cell-permeable ceramide. J. Biol. Chem. 272, 24154–8 (1997).
Šali, A. & Blundell, T. L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 234, 779–815 (1993).
Vistoli, G., Mazzolari, A., Testa, B. & Pedretti, A. Binding space concept: a new approach to enhance the reliability of docking scores and its application to predicting butyrylcholinesterase hydrolytic activity. J. Chem. Inf. Model. 57, 1691–1702 (2017).
Korb, O., Stützle, T. & Exner, T. E. Empirical scoring functions for advanced protein-ligand docking with PLANTS. J. Chem. Inf. Model. 49, 84–96 (2009).
Acknowledgements
We thank Dr. Ilaria Teresa Di Gennaro for assistance in the enzyme assays experiments. The Department of Food, Environmental and Nutritional Sciences, Università degli Studi di Milano, partially covered the open access APC.
Author information
Authors and Affiliations
Contributions
M.Z. and L.M. performed the synthesis of dual-action compounds. A.K. assessed the activity of dual compounds against fungal plant pathogens and performed the statistical analysis of the corresponding data. F.F. conducted the SDHI and QoI enzyme inhibition assays on mitochondrial fractions and performed the statistical analysis of the corresponding data. A.P. participated on the synthesis and characterization of dual-action compounds. G.V. and S.G. conducted the modelling experiments. P.C. and S.D. conceived and designed the experiments. All authors participated on writing and reviewed the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zuccolo, M., Kunova, A., Musso, L. et al. Dual-active antifungal agents containing strobilurin and SDHI-based pharmacophores. Sci Rep 9, 11377 (2019). https://doi.org/10.1038/s41598-019-47752-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-47752-x
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
