
Abstract
Calcium (Ca2+) signals are decoded by the Ca2+-sensor protein calmodulin (CaM) and are transduced to Ca2+/CaM-binding transcription factors to directly regulate gene expression necessary for acclimation responses in plants. The molecular mechanisms of Ca2+/CaM signal transduction processes and their functional significance remains enigmatic. Here we report a novel Ca2+/CaM signal transduction mechanism that allosterically regulates DNA-binding activity of GT2-LIKE 1 (GTL1), a transrepressor of STOMATAL DENSITY AND DISTRIBUTION 1 (SDD1), to repress stomatal development in response to water stress. We demonstrated that Ca2+/CaM interaction with the 2nd helix of the GTL1 N-terminal trihelix DNA-binding domain (GTL1N) destabilizes a hydrophobic core of GTL1N and allosterically inhibits 3rd helix docking to the SDD1 promoter, leading to osmotic stress-induced Ca2+/CaM-dependent activation (de-repression) of SDD1 expression. This resulted in GTL1-dependent repression of stomatal development in response to water-deficit stress. Together, our results demonstrate that a Ca2+/CaM-regulated transcriptional switch on a trihelix transrepressor directly transduces osmotic stress to repress stomatal development to improve plant water-use efficiency as an acclimation response.
Similar content being viewed by others


Introduction
Plants sense and respond to external stimuli to acclimate and adapt to diverse environmental niches. In response to water deficit, plants limit transpirational water loss via reduced stomatal aperture as a rapid plant response1. As a longer-term response, plants may repress stomatal development, the result of which is reduced water loss to improve water-use efficiency (WUE) and/or drought tolerance2,3. Hyperosmotic stress induces signaling pathways that activate or repress genes necessary for water-deficit acclimation responses4 and transcriptional regulation is a key regulatory mechanism that governs stomatal development5. The abundance of SPEECHLESS, the master transcriptional regulator of stomatal cell identity, is decreased by osmotic stress via a mitogen-activated protein kinase (MAPK) cascade6,7. However, the earliest signal transduction mechanism from sensing water-deficit to the direct regulation of gene expression responsible for repression of stomatal development remains enigmatic.
Calcium ions (Ca2+) are versatile second messengers regulating plant growth and development. Environmental stimuli, and specifically hyperosmotic stress, induce rapid and transient changes in cytosolic and nuclear Ca2+ concentrations that differ in amplitude and duration (Ca2+ signatures)8,9,10,11,12. These stimuli-specific Ca2+ changes are decoded by Ca2+ sensor proteins, including the ubiquitous Ca2+-binding protein calmodulin (CaM). Ca2+ binding to CaM induces conformational change from a closed to an open state, facilitating hydrophobic and electrostatic interactions with basic amphipathic helices of target peptides to activate or repress the molecular activity of target proteins9,10,11. Among various CaM-binding targets, Ca2+/CaM-binding transcription factors, such as the calmodulin-binding transcriptional activator (CAMTA), myeloblastosis (MYB), and basic helix-loop-helix (bHLH) families, transduce signals to activate or repress gene expression necessary for inducing cellular responses12,13,14,15,16,17,18,19,20. However, the molecular and structural mechanisms of signal transduction by which Ca2+/CaM regulates transcriptional activity or DNA-binding activity of transcription factors are not fully understood.
Trihelix transcription factors are characterized by trihelical (α1-loop-α2-loop-α3) DNA-binding domains that specifically bind to the GT motif21,22. GT1 and GT2 sub-families are distinguished by the presence of one or two trihelical DNA-binding domains, respectively. The solution structure of the Arabidopsis GT-1 DNA-binding domain by NMR spectroscopy revealed that the trihelical tertiary structure is stabilized by hydrophobic residues forming a hydrophobic core that allows α3 docking to the GT1 box in target gene promoters23. GT2 factors have been implicated in developmental, light, and abiotic stress responses in Arabidopsis, poplar, soybean, and wheat24,25,26,27,28,29,30. We previously reported that the Arabidopsis GT2-LIKE 1 (GTL1) is a transrepressor of STOMATAL DENSITY AND DISTRIBUTION 1 (SDD1) that negatively regulates stomatal development and transpiration27,28. Ca2+/CaM-binding affinity has been demonstrated in AtGT2L and PtaGTL126,31. However, Ca2+/CaM signal transduction mechanisms of GTL1 and their biological implications are not known. Here, we show that Ca2+/CaM directly binds to the N-terminal DNA-binding domain of GTL1 and allosterically inhibits DNA-binding activity by disturbing the hydrophobic core. Hyperosmotic stress-induced Ca2+/CaM allosteric control on the GTL1 transrepressor leads to SDD1 derepression, both by inhibiting binding to and promoting release from the SDD1 promoter, leading to repression of stomatal development in response to water-deficit stress. We propose that this allosteric control of the GTL1 transcription factor by Ca2+/CaM is a transcriptional switch to modulate stomatal development, thereby conserving plant water loss as a long-term developmental adaptation during water stress.
Results
Ca2+/CaM binds to the GTL1 N-terminal trihelical DNA-binding domain
We hypothesized that Ca2+/CaM directly binds to AtGTL1, a negative regulator of stomatal development, to mediate water-deficit stress acclimation. The domain structure of GTL1 includes two trihelical DNA-binding domains in the N- and C-termini (Fig. 1a). A prediction algorithm identifies the α2 helix within the N-terminal DNA-binding domain (GTL1N) as a putative CaM-binding domain, but not within the C-terminal DNA-binding domain (GTL1C) (Supplementary Fig. S1)32. The helical projection of α2 indicates a canonical structure of a CaM-binding domain, which is a basic amphipathic helix with hydrophobic residues on one side and positively-charged basic residues on the other side (Fig. 1b). An in vitro pull-down assay using HA-Arabidopsis CaM2 (AtCaM2) showed that HA-AtCaM2 directly interacted with the GTL1N fused with maltose-binding protein (MBP) in the presence of Ca2+, but not with the MBP alone (Fig. 1c). The interaction between Ca2+/CaM and GTL1N was abolished by EGTA treatment, a chelator of Ca2+, indicating that Ca2+ is required for the interaction between CaM and GTL1N (Fig. 1d). In addition, the Ca2+-dependent CaM interaction only occurred with GTL1N, but not with GTL1C, indicating that the CaM-binding property is specific to the N-terminal trihelical DNA-binding domain (Fig. 1d). An in vitro CaM overlay assay using AtCaM2-conjugated with horseradish peroxidase (HRP) further confirms that the interaction of GTL1N and AtCAM2 occurred only in the presence of Ca2+ (Supplementary Fig. S2). Together, these results indicate that GTL1 is a Ca2+/CaM-binding protein and Ca2+/CaM directly interacts with the N-terminal DNA-binding domain, presumably via the α2 helix.

CaM binds to the N-terminal DNA-binding domain of GTL1 in a Ca2+-dependent manner. (a) Schematic illustration of GTL1 domain topology with N- and C-terminal trihelical domains. The primary and secondary structure of GTL1N (residue number 60~126) consists of three α-helices (α1 – grey, α2 – green, and α3 – orange) with α2 containing a predicted CaM-binding domain. (b) The helical wheel projection of the α2 helix shows a canonical amphipathic helix with hydrophobic (yellow) and hydrophilic (basic – blue and acidic – red) residues. (c) GTL1N interaction with Ca2+/CaM was performed by in vitro pull-down assay using Escherichia coli expressed MBP-GTL1N or MBP to pull down in vitro-translated HA-AtCaM2 that was detected by immunoblots using anti-HA antibodies (upper panel). Immobilized MBP and MBP-GTL1N fusion proteins are shown in the Coomassie blue-stained SDS-PAGE gel (lower panel). (d) Schematic illustration of the bait and prey proteins used in the pull-down assays. Ca2+-dependent CaM interaction with GTL1N or GTL1C were performed by pull-down assay using HA-AtCaM2. Bound and 10% of input HA-AtCaM2 fractions were detected by immunoblot using anti-HA antibodies. The MBP-GTL1N (Nt) and MBP-GTL1C (Ct) fusion protein bands are indicated by red arrows. Full-length blots and gels for c and d are presented in Supplementary Fig. S10.
To confirm that the α2 helix contains a CaM-binding domain, the GTL1N∆del fragment (without α2 and α3 helices) was tested for interaction with Ca2+/CaM. GTL1N∆del completely lost binding affinity to Ca2+/CaM (Fig. 2a), confirming the in silico prediction of α2 as a CaM-binding domain. To identify the critical residue important for CaM binding, the amino acid sequences of the α2 helix of the N- and C-terminal DNA-binding domains from CaM-binding and non-CaM binding groups of GT2 family proteins were aligned (Fig. 2b). The H94 residue in α2 of GTL1 has a similar basic property to the arginine (R501) in α2 of AtGT2L, another CaM-binding GT2 family protein31. The alternative residue for H94 of GTL1 and R501 of AtGT2L is glutamic acid (E) in the non-CaM binding group, suggesting that the H94 residue may determine the CaM-binding property of the α2 helix. To test this hypothesis, we introduced a site-directed mutation by substituting histidine for glutamic acid (H94E) in GTL1N and tested for CaM-binding activity. An H94E substitution resulted in the loss of CaM binding (Fig. 2c), presumably due to electrostatic repulsion with conserved E residues of CaM9,11. These results indicate that Ca2+/CaM binds to GTL1N α2, and that H94 is a critical residue for interaction with Ca2+/CaM.

Ca2+/CaM binds to the α2 helix of GTL1N. (a) Schematic illustration of two protein fragments (GTL1N and GTL1N∆del) fused to maltose-binding protein (MBP). In vitro pull-down assay was performed using MBP-fusion proteins with Ca2+/HA-AtCaM2. Bound and 10% of input HA-AtCaM2 fractions were detected by immunoblot using anti-HA antibodies. The MBP-GTL1N and MBP- GTL1N∆del fusion protein bands are indicated by red arrows. (b) Sequence alignment of the α2 helix of the N-terminal (Nt) or C-terminal (Ct) trihelical domain from GT2 family proteins, including GTL1. AtGTL1_Nt and AtGT2L_Ct have CaM-binding activity. *Indicates the residue that distinguishes CaM-binding and non-CaM-binding groups. (c) An in vitro pull-down assay was performed using MBP-GTL1N or MBP-GTL1N[H94E] fusion proteins with Ca2+/HA-AtCaM2. Bound and 10% of input HA-AtCaM2 fractions were detected by immunoblot using anti-HA antibodies. The MBP-GTL1N and MBP-GTL1N[H94E] fusion protein bands are indicated by red arrows. Full-length gels for a and c are presented in Supplementary Fig. S10.
The GTL1 N-terminal trihelical DNA-binding domain requires hydrophobic core formation for its DNA-binding activity
Ca2+/CaM binding to the α2 helix within the GTL1 N-terminal trihelical DNA-binding domain suggests a potential link between Ca2+/CaM binding and the regulation of DNA-binding activity. To understand the functional characteristics of the trihelical domain, the structure of GTL1N was constructed by comparative homology modeling with the NMR structure of the AtGT-1 trihelical DNA-binding domain as a template23. The primary and secondary structural comparison indicates that the key residues required for hydrophobic core formation in GT-1 are conserved in the GTL1N trihelical DNA-binding domain with 35.94% sequence identity (Supplementary Fig. S3a), which is above 30%, a good standard for accuracy of homology modeling33. The modeled GTL1N trihelical domain indicates that GTL1N α1 (residues 65–83) and α2 (residues 90–102) are oblique to each other, and that α3 (residues 107–125) is perpendicular to α1 (Fig. 3a). Ten residues (N terminus-W63; α1-L71, L72, and F82; α2-L91, W92, V95, and L99; and α3-F115 and V118) form a hydrophobic core to stabilize the GTL1N tertiary structure, which is conserved with hydrophobic residues important for the hydrophobic core of GT-1 (Fig. 3a and Supplementary Fig. S3b). Charged residues on the α3 helix, important for electrostatic interactions with the GT box cis-element, are also conserved between GT-1 and GTL1 (Supplementary Fig. S3c), suggesting that the functional structure of the trihelix DNA-binding domain is conserved between GT-1 and GTL1.

The GTL1 N-terminal DNA-binding domain forms a trihelical tertiary structure that is stabilized by hydrophobic residues. (a) Schematic illustration of GTL1 N-terminal trihelical domain. The primary and secondary structure of GTL1N (Residue Number 60–126) consists of three helices with α2 (green) and α3 (orange) helices containing CaM-binding and DNA-recognition sequence motifs, respectively. The three-dimensional GTL1N structure is predicted to be stabilized by a hydrophobic core (grey dot area) that is formed by hydrophobic residues shown in black, green, and orange distributed in the three helices. Blue residues indicate charged residues necessary for DNA recognition (α3) and CaM binding (α2). (b) DNA-binding activity of recombinant proteins (5 or 10 μg) of MBP, GTL1N (WT), or GTL1N mutations ([V95D], [W92R], and [L91R]) with biotin-labeled SDD1 promoter fragments was performed by electrophoretic mobility shift assay (EMSA). (c) GTL1N (WT) or H94E proteins were used to determine binding activity with a biotin-labeled SDD1 promoter fragment (left panel) or rice PHYA promoter fragment (right panel). Free promoter and protein-promoter complexes are indicated by an asterisk and arrows, respectively.
The GTL1N tertiary structure allows the α3 helix to interact with the major groove of the GT-3 box in the predicted docking structure between GTL1N and the SDD1 promoter (5′-gcttGGTAAAactt-3′) (Supplementary Fig. S4). The predicted model suggests that GTL1N-SDD1 promoter interactions occur via hydrogen bonds between α3 K109 (hydrogen donor) and the first guanine (Gua1) of the GT3 box (hydrogen acceptor), and α3 E116 (hydrogen acceptor) and the fourth adenine (Ade4) of the GT3 box (hydrogen donor) (Supplementary Fig. S4b). Other residues W63, α2-W92, and α3-K110, K112, K114, N117, Q119, and K120 interact with the negatively charged phosphate backbone of the GT3 box (Supplementary Fig. S4c), which stabilizes the GTL1N-SDD1 promoter interaction. Based on the docking model of GTL1N-SDD1 promoter, we hypothesize that a hydrophobic core in the trihelical GTL1N DNA-binding domain is essential for its DNA-binding activity. To validate the importance of the hydrophobic core in tertiary structure for DNA-binding activity, hydrophobic residues (L91, W92, and V95) were substituted to acidic or basic amino acids to disrupt the formation of a hydrophobic core. Consistent with the homology modeling and docking prediction, site-directed mutations (L91R, W92R, and V95D) of hydrophobic residues to charged residues results in the complete loss of binding to the SDD1 promoter fragment (Fig. 3b). These data support the model that the hydrophobic core formed by the hydrophobic residues in the GTL1N trihelical structure is required for native folding and its function as a DNA-binding transcription factor. However, the H94E mutation in the H94 residue required for Ca2+/CaM interaction did not result in the loss of DNA binding to the SDD1 promoter or rice PHYA promoter (Fig. 3c), suggesting that the α2 helix is not directly involved with DNA-recognition. Together, this result further supports the notion that the hydrophobic core formation results in a protein topology for surface exposure of the α2 (H94, R97, and K98) and α3 (K110, K112, K114, and K120) basic residues that are necessary for CaM and DNA interactions, respectively.
Ca2+/CaM binding to α2 of GTL1N inhibits its association with and facilitates dissociation from the SDD1 promoter
Ca2+/CaM binding to the α2 helix within the GTL1 N-terminal DNA-binding domain prompted us to postulate that Ca2+/CaM binding may allosterically inhibit GTL1N DNA binding to the SDD1 promoter. This hypothesis is based on the typical Ca2+/CaM-target peptide structure requiring hydrophobic residues9,34, which are important to form the hydrophobic core of GTL1N (Fig. 3a). We first tested this hypothesis with an in vitro electrophoretic mobility shift assay (EMSA) that was previously used to show GTL1 DNA binding to the SDD1 promoter27. GTL1N was able to interact with the SDD1 promoter fragment in the presence of CaM or CaM with EGTA (Fig. 4a). However, addition of Ca2+ substantially reduced the binding affinity with SDD1 (Fig. 4a). Interestingly, when GTL1N was pre-incubated with the SDD1 promoter fragment, the addition of CaM in the presence of Ca2+ was also able to dissociate GTL1N from the SDD1 promoter fragment (Fig. 4b). This result indicates that Ca2+/CaM is able to dissociate GTL1N from the SDD1 promoter. To further confirm the effect of Ca2+/CaM on GTL1N DNA-binding activity, the EMSA was performed using GTL1N[H94E] that is unable to bind Ca2+/CaM (Fig. 2c). The addition of CaM in the presence of Ca2+ was able to dissociate the GTL1N-SDD1 promoter complex, but not the GTL1N[H94E]-SDD1 promoter complex (Fig. 4c). These in vitro assays indicate that Ca2+/CaM binding to the α2 helix of GTL1N inhibits association and facilitates dissociation from the SDD1 promoter.

Hyperosmotic stress-induced Ca2+/CaM binding to the α2 helix inhibits DNA-binding activity of GTL1 to the SDD1 promoter. (a–c) EMSA was performed using GTL1N or GTL1N[H94E], and a biotin-labeled SDD1 promoter fragment that includes the GT3 box. (a) GTL1N (1 or 5 mg) was pre-incubated with human CaM (100 ng) without or with 2 mM CaCl2 or 10 mM EGTA, and then incubated with the biotin-labeled SDD1 promoter fragment (250 ng). (b) GTL1N (2.5 or 5 mg) was pre-incubated with the SDD1 promoter fragment without or with 2 mM CaCl2, and then with human CaM (100 ng). (c) GTL1N or GTL1N[H94E] was pre-incubated with the SDD1 promoter fragment without or with 2 mM CaCl2 and then with human CaM (50 or 100 ng). (d) ChIP assays using anti-HA antibody were performed using protoplasts isolated from gtl1-4 plants, transformed with HA-GTL1 or HA-GTL1[H94E], and incubated with 0 or 200 mM mannitol for 1 h. Input is the total isolated chromatin before immunoprecipitation. Mouse IgG was the negative control for immunoprecipitation. The SDD1 promoter region including the GT3 box was amplified by SDD1 promoter-specific primers.
To recapitulate these results in vivo using full-length GTL1 protein, a chromatin-immunoprecipitation (ChIP) assay was performed in gtl1-4 protoplasts expressing wild-type HA-GTL1 or mutant HA-GTL1[H94E]. Both GTL1 and GTL1[H94E] proteins associated with the SDD1 promoter region that contains the GT3 box, indicating again that the GTL1[H94E] protein is functional (Fig. 4d). To activate in planta Ca2+ signaling, 200 mM mannitol was used to induce hyperosmotic stress, which has been reported to induce cytosolic and nuclear Ca2+ transients in tobacco and Arabidopsis35,36,37,38. Mannitol reduced GTL1 association with the SDD1 promoter, but did not substantially reduce GTL1[H94E] association with the SDD1 promoter (Fig. 4d). Together, both in vitro and in vivo results indicate that hyperosmotic stress-induced Ca2+/CaM attenuates GTL1 docking to the SDD1 promoter.
Hyperosmotic stress induces SDD1 expression through Ca2+/CaM-dependent inhibition of the GTL1 transrepressor
To understand the biological significance of Ca2+/CaM interaction with GTL1 on the regulation of SDD1 promoter-binding activity, we hypothesized that Ca2+/CaM-dependent inhibition of the GTL1/SDD1 promoter complex interaction is a key signal transduction mechanism to activate (derepress) SDD1 expression in response to water-deficit stress. Indeed, transcript abundance of SDD1 mRNA is up-regulated in response to water-deficit stress when DREB2A, a drought-induced marker gene, was also up-regulated (Fig. 5a). Next, to determine if hyperosmotic stress regulates SDD1 transcription through GTL1 and Ca2+ and CaM signaling, we first established the 2 kb SDD1 promoter-reporter system (Fig. 5b). SDD1-LUC activity was higher in gtl1-4 than in wild-type (Col-0) protoplasts, whereas GTL1 promoter-driven GTL1-GFP expression in gtl1-4 (GTL1/gtl1-4) protoplasts repressed SDD1 expression to a level comparable to wild-type protoplasts (Fig. 5b), which is consistent with previous results in mature leaves26,27. The SDD1 promoter-reporter system established that the 2 kb promoter fragment of SDD1 including the GT3 box is sufficient for SDD1 expression and GTL1 transrepression.

Hyperosmotic stress induces SDD1 expression through Ca2+/CaM-dependent inhibition of the GTL1 transrepressor. (a) Transcript abundance of SDD1, DREB2A, and UBC was determined in total RNA extracted from 5-week-old Col-0 plants that were well-watered or water-deficit stressed. (b) Schematic diagram of the reporter and internal control constructs. The reporter construct included the 2 kb SDD1promoter fused with firefly luciferase (LUC) gene, and nopaline synthase termination signal (NOS-t). The internal control construct included the 35Spromoter, Renilla reniformis LUC (RrLUC), and NOS-t. SDD1-LUC activity relative to RrLUC activity (% of SDD1-LUC to RrLUC activity) was measured from protoplasts isolated from Col-0 (wild type), gtl1-4, and gtl1-4 expressing GTL1 (GTL1/gtl1-4). (c) Relative SDD1-LUC activities were determined in Col-0 and gtl1-4 protoplasts that were incubated with or without 1 mM GdCl3, 600 mM W7, or GdCl3 and W7 combined prior to addition of mannitol to a final concentration of 200 mM. (d) SDD1- LUC activities of Col-0, gtl1-4, or HA-GTL1 or HA-GTL1[H94E] protoplasts were determined after incubation with 0 or 200 mM mannitol for 1 h. All results shown are mean ± SEM (n = 3). Columns with the same letters above are not significantly different from each other based on Tukey’s Honestly Significant Difference (HSD) test (P < 0.05) (One-way ANOVA).
We next determined the involvement of hyperosmotic stress-induced Ca2+ and CaM signaling in the transcriptional regulation of SDD1. SDD1 expression in wild type protoplasts was induced by hyperosmotic stress (200 mM mannitol addition to the protoplast incubation solution), which was partially inhibited by pretreatment with either the Ca2+ channel blocker gadolinium ion (Gd3+) or the CaM antagonist W7 and abrogated when incubated with both (Fig. 5c). This result indicates that hyperosmotic stress-induced SDD1 expression is Ca2+- and CaM-dependent. Constitutive SDD1 expression was evident in gtl1-4 protoplasts without hyperosmotic treatment (Fig. 5c), confirming that GTL1 transrepresses SDD1 expression27. Hyperosmotic stress resulted in a slight increase in SDD1 expression in gtl1-4. W7, Gd3+, or Gd3+ + W7 pretreatment only marginally reduced SDD1 expression in gtl1-4 plants compared to the response in Col-0 (Fig. 5c). This could be due to involvement of a GTL1-independent pathway or other members of the GT-2 family in the regulation of SDD1 expression. A defect of Ca2+-dependent hyperosmotic SDD1 expression in gtl1-4 was restored in GTL1-GFP expressing lines (gtl1-4::GTL1promoter:GTL1:GFP) (Supplementary Fig. S5). These results indicate that GTL1 is required for Ca2+- and CaM-dependent hyperosmotic SDD1 expression. Together, these results indicate that hyperosmotic stress induces SDD1 expression through Ca2+/CaM signaling and by attenuating GTL1 transrepressor activity.
To determine the in vivo direct outcome of the Ca2+/CaM-binding function on GTL1 DNA-binding activity, SDD1 promoter-LUC reporter activity in response to hyperosmotic stress was compared by expressing wild-type GTL1 or GTL1[H94E]. Hyperosmolality increased SDD1 expression in wild-type and GTL1-expressing gtl1-4 protoplasts but not in GTL1[H94E]-expressing gtl1-4 protoplasts (Fig. 5d). Moreover, GTL1[H94E] expression suppressed constitutive SDD1 expression in gtl1-4 protoplasts (Fig. 5d). Together, these results indicate that SDD1 expression is activated or derepressed in response to hyperosmotic stress via allosteric inhibition of GTL1 docking to the SDD1 promoter by Ca2+/CaM.
Water-deficit stress represses stomatal development in a Ca2+/CaM- and GTL1-dependent manner
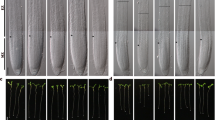
Hyperosmotic stress inhibits stomatal development in Arabidopsis6,39. Overexpression of SDD1 represses stomatal development in species including Arabidopsis, tomato, and maize40,41,42. We have shown that hyperosmotic stress results in up-regulated SDD1 expression in a Ca2+/CaM- and GTL1-dependent manner (Fig. 5). This suggests that water-deficit stress may repress stomatal development through Ca2+/CaM-dependent inhibition of the GTL1 transrepressor in plants. To test whether osmotic stress-induced repression of stomatal development is dependent on GTL1, we quantified changes in stomatal development in wild type and gtl1-4 plants in response to water-deficit stress (Supplementary Fig. S6). The presence of a significant water stress was confirmed by reductions in leaf area (Fig. 6a) and stomatal length (Fig. 6b), both of which are well-established indicators of water stress39.

Stomatal development is repressed under severe water-deficit conditions through Ca2+/CaM and the GTL1 transrepressor. (a) Leaf area of Col-0 and gtl1-4 plants grown under well-watered (WW), mild water-deficit (50% media water content, MWC), and severe water-deficit (30% MWC) conditions. These leaves were used to quantify stomatal length (b), stomatal index (c), and stomatal precursor index (d) in adaxial and abaxial leaf surfaces of Col-0 and gtl1-4 plants. Adaxial (upper case) and abaxial (lower case) leaves were analyzed separately for the statistical comparisons. (e) Stomatal index of the abaxial surface was quantified in Col-0 and gtl1-4 plants grown under 0 and 200 mM mannitol conditions with or without W7 (50 and 100 μM) and GdCl3 (85 and 170 μM). Col-0 (left panel) and gtl1-4 (right panel) were analyzed separately for the statistical comparisons. Data shown are the means with SD for 8–12 replicates (a–d) and 7 replicates (e). Columns with the same letters above are not significantly different from each other based on Tukey’s Honestly Significant Difference (HSD) test (P < 0.05) (Two-way ANOVA).
Stomatal index was reduced in gtl1-4 plants compared to wild type plants (Fig. 6c) as previously reported27. We also observed a reduction in stomatal development in severely water-stressed Col-0 plants, evidenced by a decrease of stomatal index in both adaxial and abaxial leaves (Fig. 6c). Mild water-deficit stress (50% MWC) resulted in reduced leaf area, but no change in stomatal index, suggesting that inhibition of stomatal development depends on the severity of the stress. In addition, the number of stomatal precursor cells produced by asymmetric divisions (meristemoids and guard mother cells) was increased in response to severe water-deficit stress (Fig. 6d). In contrast to the water-deficit-induced reduction in stomatal index in Col-0 plants, stomatal development in gtl1-4 was insensitive to severe water-deficit stress (Fig. 6c), indicating that GTL1 is required for water-deficit stress-induced repression of stomatal development.
To further test the hypothesis that water-deficit stress-repressed stomatal development is dependent on Ca2+/CaM signaling, plants were grown on media containing the CaM antagonist W7 alone or with GdCl3 with or without mannitol to induce osmotic stress. A lower concentration of W7 (50 and 100 µM), relative to protoplast assays, was used to determine the effects on stomatal development in seedlings without major effects on growth. W7, whether alone or in combination with GdCl3, did not significantly affect stomatal index of Col-0 under water-sufficient conditions. However, W7 completely rescued the water-deficit-induced repression of stomatal development regardless of GdCl3 treatment (Fig. 6e). There was no effect of W7 or mannitol on stomatal index of gtl1-4 plants (Fig. 6e). Leaf area was lower in both Col-0 and gtl1-4 when grown with mannitol, and this phenotype was not rescued by W7 (Supplementary Fig. S7). Together, these results indicate that water-deficit stress results in decreased stomatal development through Ca2+/CaM signaling, and that GTL1 is required for transducing the osmotic stress signal to the repression of stomatal development.
Discussion
Hyperosmotic stress-induced Ca2+/CaM signaling requires a rapid and efficient signal transduction mechanism to modulate global transcriptional regulation to cope with water deficit during drought stress. The widely accepted model of this signal transduction mechanism is that Ca2+/CaM directly interacts with transcription factors to modulate their activity and regulate transcription. However, molecular and structural mechanisms of this modulation specifically on a transcriptional repressor are largely unknown. In this set of experiments, we have demonstrated a mechanism of Ca2+/CaM-dependent allosteric control of the GTL1 transcriptional repressor that directly controls its DNA-binding activity in response to hyperosmotic stress (Fig. 7). Under water-sufficient conditions, GTL1 binds to the SDD1 promoter and represses SDD1 expression, resulting in maintained stomatal development. Water-deficit induces a transient increase of intracellular Ca2+ level36,38,43,44, which is transduced by CaM that directly binds to GTL1, destabilizes the trihelical structure of the DNA-binding domain, and facilitates dissociation of GTL1 from the SDD1 promoter. This novel signal transduction mechanism leads to transcriptional derepression (activation) of SDD1, a negative regulator of stomatal development, and thus inhibits stomatal development to reduce transpirational water loss under water-deficit conditions as an acclimation response (Fig. 7).

A Ca2+/CaM-regulated GTL1 transrepressor is a transcriptional switch to control stomatal development, transpiration, and water-use efficiency in plants. Proposed model for Ca2+/CaM-regulated transcriptional switch to repress stomatal development and to improve water-use efficiency and drought tolerance through GTL1.
A prototypical CaM-binding domain exists in the basic amphipathic region of GTL1N α2 (Fig. 1b and Supplementary Fig. S8). It is well known that Ca2+/CaM binds to target peptides via hydrophobic and electrostatic interactions9,34,45. Ca2+ binding to CaM causes a conformational change from a dumbbell shape to a globular shape, which exposes hydrophobic pockets and negatively charged conserved glutamate residues that can interact with protein target regions such as the basic amphipathic α2 helix of GTL1N (Supplementary Fig. S8). The prediction of the GTL1N α2-Ca2+/CaM complex suggests that α2 basic residues interact with conserved acidic glutamate residues of CaM through electrostatic interactions (Supplementary Fig. S8)9,11,45. The α2 hydrophobic residues interact with the hydrophobic pocket of Ca2+/CaM. Since α2 hydrophobic residues are also important for the formation of the hydrophobic core and GTL1N DNA-binding activity (Fig. 3), Ca2+/CaM binding to the α2 may interfere with the formation of the hydrophobic core and prevent binding to the SDD1 promoter. Our results demonstrate that Ca2+/CaM binds to the α2 inner-core hydrophobic residues, which destabilizes GTL1N tertiary structure by conformational change, preventing SDD1 promoter docking, which results in transcriptional derepression of SDD1 expression.
This allostery is in contrast to the Ca2+/CaM steric hindrance mechanism of E proteins in which Ca2+/CaM blocks promoter binding of E proteins but does not dissociate the E protein-promoter complex in animals12. Because GTL1 is associated with the SDD1 promoter, resulting in repression under well-watered conditions, this allosteric mechanism allows for dissociation of GTL1 from the promoter and the prevention of free GTL1 binding to the promoter, thereby maintaining SDD1 activation through both mechanisms. Allostery can involve large or subtle conformational change or can function without conformational change by redistributing electrostatic interactions necessary for binding interface46. Whether Ca2+/CaM-mediated allosteric regulation of GTL1 DNA-binding activity requires conformational change of trihelical tertiary structure is unknown without analyzing the solution structure of the Ca2+/CaM-GTL1 complex. It is, however, well-accepted that allostery is one of the fundamental molecular mechanisms in cellular signaling pathways of all living organisms47. We posit that the allosteric mechanism may fine-tune transcriptional regulation that is necessary for precise gene expression required when an organism must decode different signatures encoded by environmental changes necessary for adaptive fitness48.
Water deficit in soil results in decreased cell turgor pressure and imposes osmotic stress on plant cells, which activates various acclimation responses, including physiological and developmental changes49. Mannitol-induced hyperosmotic stress has been reported to induce cytosolic and nuclear Ca2+ transients in tobacco and Arabidopsis36,38,43,44. Hyperosmotic stress also results in decreased stomatal development in Arabidopsis6,39. Consistent with these reports, our study also showed that water-deficit stress repressed stomatal development as evidenced by a reduction of stomatal index (Fig. 7). However, in gtl1-4 plants, stomatal index was not further decreased by mannitol, indicating that GTL1 is required for transducing the signal from the osmotic stress to the modulation of stomatal development. We showed that the Ca2+/CaM-GTL1-SDD1 modular relay system allows for transduction of water-deficit (e.g. turgor reduction or low water potential) sensing. When water is sufficient, GTL1 transrepression of SDD1 (a negative regulator of guard cell lineage) is necessary to facilitate stomatal development27,28. The Ca2+/CaM-GTL1-SDD1 module appears to be a transcriptional switch mechanism through which plants can acclimate to reduced water availability via a repression of stomatal development (Fig. 7). Since YODA (a MAPKKK) is known to act downstream of SDD1, we posit that the Ca2+/CaM-regulated transcriptional switch through the GTL1-SDD1 module is the most upstream signal transduction pathway responsible for this response identified to date. Whether the function of SDD1 is to send a signal in response to water deficit stress remains to be tested.
A Ca2+-dependent transcriptional derepression mechanism can be considered to be an efficient signal transduction mechanism for rapid activation of acclimation genes17,19. Recently, a similar signal transduction mechanism through the SR1/CAMTA3 transrepressor has been reported in Arabidopsis. SR1/CAMTA3 is a Ca2+/CaM-binding transcriptional repressor that suppresses the expression of salicylic acid (SA)-related genes involved in plant immunity14. Ca2+/CaM binding to AtSR1/CAMTA3 is required for repression of the immune response. Low temperature induces expression of SA-related genes through de-repression of SR1/CAMTA3 activity by Ca2+/CaM17,20. However, the CaM-binding domain is located in the C-terminus, while the DNA-binding domain is located in the N-terminus. It is not known how Ca2+/CaM activates CAMTA transcriptional activity or DNA-binding activity. This report together with our results suggest that Ca2+/CaM-regulated transcriptional derepression is a common signal transduction mechanism in response to various environmental stresses.
The capacity for plants to sense water-deficit through the Ca2+/CaM-GTL1-SDD1 module and to transduce this signal to a developmental program may be an important mechanism to modulate plant water use. The presence of GTL1 orthologs in a large number of diverse species suggests that this acclimation mechanism is conserved among higher plants27,28. Supporting this notion, TaGT2L1D, a GTL1 ortholog in wheat, is also a transrepressor of SDD1 to negatively regulate stomatal development30. We reported that the poplar GTL1 ortholog interacts with Ca2+/CaM through the C-terminal DNA-binding domain that binds to the GT2 box of the poplar SDD1 promoter26. It is interesting that the Ca2+/CaM-binding domain of AtGT2L is located in the α2 helix of the C-terminal trihelical domain, whereas in PtaGTL1 it is located in the α1 of the C-terminal trihelical domain. Some of the GT2 family do not have Ca2+/CaM-binding domains within their trihelical domain due to a change of residue to an acidic residue (Fig. 2b). This suggests that the Ca2+/CaM-binding property might have evolved in specific members of the GT2 family, such as GTL1. The Ca2+/CaM-binding property of the trihelical DNA-binding domain in GTL1 may have evolved to activate SDD1 expression by disrupting DNA-binding activity under selective pressure as a drought adaptation mechanism. Future work will reveal the evolutionary/adaptive significance of the allosteric inhibition mechanism in GTL1 orthologs to improve crop production under limited water availability.
Methods
Plant materials and growth conditions
The Arabidopsis thaliana genetic resources used in this study were wild type Columbia-0 (Col-0), gtl1-4 (SALK_005972), and transgenic plants expressing GTL1promoter:GTL1:GFP in gtl1-427. Plants were grown on an MS-based medium (1xMS, 2% sucrose, 2.5 mM MES, pH 5.7, 0.5% agar) under fluorescent lights with a light level of ca. 60 µmol quanta m−2 s−1 for 4 weeks (temperature: 22 °C [light]/18 °C [dark]; photoperiod: 16 h [light]/8 h [dark]). For stomatal development analysis in soil-grown plants, Col-0 and gtl1-4 seeds were germinated in a 1:2 soil mix of Turface (PROFILE Products LLC) and Fafard F2 soilless media (Sungro Horticulture) in SC7 Ray Leach ‘Cone-Tainers’ (Stuewe & Sons Inc.). Plants were grown in the greenhouse under an 8 hour photoperiod, with light intensity of 150 µmol m−2 s−1, day/night temperature of 23/18 °C, and relative humidity of 65%. For stomatal development analysis in plate-grown plants, Col-0 and gtl1-4 seeds were plated (8–12 plants per genotype per plate) in quadrants on agar plates containing one or more of 200 mM mannitol, 50 or 100 µM W7, and 85 or 170 µM GdCl3. Seedlings were grown under a 16 hour photoperiod at 150 µmol m−2 s−1 for 6 weeks.
Plasmid construction
SDD1promoter:LUC was constructed in the 5′GAL4:LUC vector by replacing the GAL4 binding site with the 2 kb SDD1 promoter50. The 2 kb SDD1 promoter including the GT3 box and the 5′ untranslated region was amplified with designated primers (SDD1p-F-EcoRV and SDD1p-R-NcoI) and was fused to the firefly luciferase gene (LUC) and nopaline synthase (NOS) termination signal (NOS-t) to produce an SDD1 reporter (SDD1-LUC) for protoplast transactivation assays. The promoter containing 5′GAL4 from 5′GAL4:LUC vector was removed by Hind III (with blunt-end generation by Klenow) and NcoI. The amplified SDD1 promoter digested by EcoRV (blunt-end) and NcoI was inserted to the HindIII (blunt-end)/NcoI site.
Two GTL1 coding fragments (GTL1N and GTL1Δdel) were amplified by designated primers (GTL1N, N-F-EcoRI and N-R-PstI; GTL1Δdel, N-F-EcoRI and del-R-PstI). Two site-specific mutations (V95D and H94E) were amplified by designated primers (V95D-F, V95D-R, H94E-F, and H94E-R with N-F-EcoRI and N-R-PstI) by PCR-based site-directed mutagenesis. The amplified fragments and mutations were inserted into the EcoRI and PstI sites in the pMAL-C2 vector to produce MBP-fusion proteins. For the transient protoplast assay, HA:GTL1 and HA:GTL1[H94E], driven by the 35S promoter, were constructed in the p326:HAN vector51. GTL1 or GTL1[H94E] coding fragment was amplified by designated primers (GTL1, GTL1-F-XmaI and GTL1-R-EcoRI; GTL1[H94E], GTL1-F-XmaI and H94E-R, and H94E-F and GTL1-R-EcoRI). p326:HAN vector was digested with KpnI (with blunt-end generation by Klenow) and XmaI. Amplified GTL1 and GTL1[H94E] products were digested with EcoRI (with blunt-end generation by Klenow) and XmaI, which was inserted into KpnI (blunt end)/XmaI sites.
Arabidopsis CaM2 was amplified by designated primers (AtCaM2-F and AtCaM2-R). The amplified fragments were inserted into the BamHI and XhoI sites of pCMX-PL2-NterHA using HiFi Assembly (NEB). Sequence information for all designated primers used for plasmid construction is provided in Supplementary Table S1.
Arabidopsis protoplast transient expression and luciferase reporter assay
Protoplasts were isolated from 4-week-old plants as described previously52 with modifications. Shoots from 150 seedlings were incubated in 30 mL of 1 M mannitol solution for 30 min and then incubated in 20 mL of enzyme solution (1% cellulose R-10, 0.25% macerozyme R-10, 500 mM mannitol, 1 mM CaCl2, 10 mM MES, 20 mg BSA) for 12 h to digest cell walls. The protoplast mixture was filtered through 100 mm mesh and transferred onto 20 mL of 21% sucrose solution, which was centrifuged at 57 g for 10 min in a swinging bucket rotor. Approximately 10 mL of protoplasts at the interface were transferred to 30 mL of W5 protoplast stabilization solution (154 mM NaCl, 125 mM CaCl2, 5 mM KCl, 2 mM MES, pH 5.7) and centrifuged at 43 g for 6 min. Supernatant was removed, replaced by 30 mL of W5 solution, and incubated on ice for 6 h to stabilize protoplast.
SDD1promoter:LUC (29 mg), 35Spromoter:LUC (1 mg), HA:GTL1 (5 mg), or HA:GTL1[H94E] (5 mg) was transformed into protoplasts using the polyethylene glycol (PEG)-mediated transformation method52. First, W5 solution was replaced by mannitol solution (400 mM mannitol, 30 mM MgCl2, and 5 mM MES, pH 5.6). Plasmids (30 ml) were added to 300 ml of protoplast MaMg solution. Then, 300 ml of PEG solution (400 mM mannitol, 100 mM Ca(NO3)2, 40% PEG6000) was added, mixed by inverting for 1.5 min, and incubated at room temperature for 30 min. PEG protoplast solution was slowly diluted with 5 ml of W5 solution by adding 1 ml of solution every 2 min. The protoplast solution was centrifuged at 43 g for 4 min. Supernatant was completely removed, replaced by 2 ml of W5 solution, and incubated at room temperature for 12 h. To induce hyperosmotic stress, 200 mM mannitol was added and protoplasts were incubated for 1 h. To inhibit Ca2+ or CaM signaling, protoplasts were pre-incubated in 1 mM GdCl3, 0.5 mM LaCl3, or 600 mM W7 (N-(6-aminohexyl)-5-chloro-1-naphthelenesulfon-amide-hydrochloride) for 10 min prior to mannitol treatment. After incubation with mannitol, protoplasts were harvested by centrifugation at 57 g for 2 min, then frozen in liquid nitrogen. Frozen protoplasts were lysed with passive lysis buffer (Promega) and the SDD1-LUC and RrLUC activities were measured using a dual-luciferase assay kit according to the manufacturer’s instructions (Promega) using a luminometer (TD20/20). Relative SDD1-LUC activity was shown as 100× (SDD1-LUC activity/RrLUC activity). Osmotic potential (ψs) of the osmotic stabilization solution (W5; (ψs: −1.19 MPa) and hyperosmotic solution (W5 with 200 mM mannitol; ψs: −1.73 MPa) was calculated by the equation (ψs = −CRT; C = osmolality, R = gas constant, T = Kelvin temperature). Osmolality of the solution was measured using a vapor pressure osmometer (Wescor 5200).
Purification of recombinant proteins
MBP and MBP-fusion proteins (GTL1N, GTL1C, GTL1Δdel, GTL1N[L91R], GTL1N[W92R], GTL1N[H94E], and GTL1N[V95D]) were purified by amylose resin (NEB) as described previously27.
CaM-binding assays
CaM-binding assays were performed using bacteria lysates expressing MBP-fusion proteins and in vitro-translated HA-AtCaM2 as described previously53 with the following the modifications. Briefly, MBP fusion proteins were expressed in Escherichia coli strain Rosetta (DE3) carrying pMAL-c2 vectors. After harvesting by centrifugation, the cells were lysed by French press in lysis buffer containing 50 mM Tris-HCl, pH 7.5, 100 mM NaCl, 1% DMSO, 2 mM DTT, and protease inhibitor cocktail (Sigma-Aldrich). The cell extract was prepared by centrifugation at 10,000 g for 20 min at 4 °C and cleared by filtration. The clear lysates were incubated with amylose resin at 4 °C for 2 h. The beads with immobilized MBP fusion proteins were washed with wash buffer containing 50 mM Tris-HCl, pH 7.5, 100 mM NaCl, 1% DMSO, 2 mM DTT, and 0.1% Nonidet P-40. AtCaM2 with an N-terminal HA tag was synthesized using plasmid pCMX-PL2 and the TNT T7 Coupled Reticulocyte Lysate System (Promega) according to the manufacturer’s protocol. The in vitro-translated HA-AtCaM2 was diluted with CaM-binding buffer (50 mM Tris-HCl, pH 7.5, 100 mM NaCl, 1% DMSO, 2 mM DTT, and 2 mM CaCl2) and incubated with the affinity-purified MBP fusion proteins immobilized on the beads at 4 °C for 2 h. The beads were then washed four times with wash buffer. Bound HA-AtCaM2 was eluted by boiling in 1x SDS sample buffer and subjected to 12% SDS-PAGE. Input and immunoprecipitated HA-AtCaM2 proteins were detected by immunoblots using rabbit polyclonal anti-HA antibodies (Abcam) and the Enhanced Chemiluminescence Plus protein gel blotting detection system (GE Healthcare). The amount of MBP fusion proteins used in each CaM-binding assay was separated by another 8% SDS-PAGE gel and visualized by staining with Coomassie Brilliant Blue.
CaM-overlay assays using HRP-conjugated AtCaM2 proteins were performed as described previously54. Briefly, MBP fusion GTL1 proteins were separated by SDS-PAGE and transferred to two PVDF membranes. After blocking in TBST buffer containing 7% non-fat dry milk, membranes were incubated in the overlay buffer (50 mM imidazole-HCl, pH 7.5 and 150 mM NaCl) containing 1 µg/ml HRP-AtCaM2 in the presence of Ca2+ or EGTA for 1 h. The bound HRP-AtCaM2 signal was detected by the Enhanced Chemiluminescence Plus protein gel blotting detection system.
Electrophoretic mobility shift assay (EMSA)
Purified recombinant proteins were used to determine DNA-binding activity based on interaction with biotin-labeled Arabidopsis SDD1 promoter fragments that contain the GT3 box. EMSA procedures were as described previously27, with the following modifications to determine the effect of Ca2+/CaM on DNA-binding activity of recombinant proteins. To determine the effect of CaM on the association between GTL1N and the SDD1 promoter, human CaM protein (50 or 100 ng) was incubated with recombinant proteins with 2 mM CaCl2 or 10 mM EGTA for 1 h. Then, biotin-labeled SDD1 promoter was added and incubated for 20 min. To determine the CaM effect on the dissociation of GTL1N from the SDD1 promoter, biotin-labeled SDD1 promoter was incubated with GTL1N or GTL1N[H94E] for 20 min. Then, CaM protein (100 ng) was added to the GTL1-SDD1 promoter complex mixture with 2 mM CaCl2 and incubated for 1 h. The mixture was separated in a 6% PAGE gel and transferred to a Biodyne B Nylon membrane (Thermo Fisher Scientific). Migration of biotin-labeled SDD1 promoter was detected by LightShift Chemiluminescent EMSA kit (Thermo Fisher Scientific) according to the manufacturer’s protocol.
Chromatin immunoprecipitation
ChIP assays were performed using chromatin isolated from protoplasts of 4-week-old gtl1-4 plants using an EZ ChIP chromatin immunoprecipitation kit (Millipore) as described previously27 with the following modifications. Protoplasts were transformed with HA-GTL1 or HA-GTL1[H94E] and incubated for 12 h at room temperature. 200 mM mannitol was added to one group (each group includes 3 replicates) to induce hyperosmotic stress for 1 h. Then, formaldehyde was added for crosslinking of chromatin and 0.1 M glycine to quench crosslinking. After centrifuging briefly, protoplasts were resuspended in lysis buffer. Chromatin was sheared to 200~1000 bp fragments by sonication. Isolated and pre-cleared chromatin was incubated with mouse monoclonal anti-HA antibody or mouse IgG for 4 h at 4 °C to immunoprecipitate the chromatin associated with HA-GTL1 or HA-GTL1[H94E]. Immunoprecipitated chromatin was reverse-crosslinked and purified according to the manufacturer’s protocol. Input chromatin (immunoprecipitated by anti-HA antibody or IgG antibody) was used for PCR analysis by the SDD1 promoter-specific primers (SDD1ChIP-3F and SDD1ChIP-3R)27.
Stomatal development analysis
Following irrigation and drainage of excess water, containers were weighed to obtain media saturated weight (MSW). Plants were irrigated to saturation (well-watered) or water was withheld to 50 or 30% media water content (MWC), calculated as [media fresh weight (MFW) − media dry weight (MDW)]/[media saturated weight (MSW) − MDW] × 100. During the experiment, a MDW of 49 g was used to determine the appropriate amount of water to add to containers, based on prior experiments. After harvesting, containers were dried in a forced-air oven and MDW was obtained for each plant. Final average MWC of the 50 and 30% MWC treatments was Final average MWC of the 50 and 30% MWC treatments was between 38% (prior to watering) and 53% (post-watering), and 25% (prior to watering) and 33% (post-watering), respectively (Supplementary Fig. S6). Once the treatment MWC was reached, containers were wrapped with plastic film to prevent water loss by evaporation. Containers were weighed every two days and re-watered to bring the MWC to target levels (Supplementary Fig. S6). This treatment was maintained for 30 days after the emergence of the target leaf, enabling the leaf to complete development under a given water deficit treatment.
Leaf appearance was tracked with photographs to determine leaf number. Once the target leaves had fully expanded, leaf adaxial and abaxial surfaces were pressed onto cyanoacrylate droplets (Henkel Manufacturing) on glass slides. Impressions were viewed under a BH-2 light microscope (Olympus) at 200× magnification, with a field of view of 0.113 mm2. Four images were collected from each surface impression, and cell types were counted in ImageJ. Cell types were distinguished by shape; meristemoids were roughly triangular cells, while long oval cells were identified as guard mother cells (GMCs)55. Stomatal index was calculated as the number of stomata/(number of stomata + epidermal cells). A diagonal line was drawn through each image and the 5 stomata closest to this line were used for stomatal length measurements. Length was measured from end to end of the outer cell wall.
To test the necessity of calcium-CaM binding for the inhibition of GTL1-dependent stomatal development under osmotic stress conditions, a pharmacological disruption experiment was performed in vivo. Seedlings were grown for 6 weeks, at which point the first true leaf was collected and fixed in a 9:1 ethanol:acetic acid solution. Following clearing, leaf tissue was stored in 85% ethanol. Tissue was rehydrated by transferring to a 70% ethanol solution, then to 70, 50, and 30% methanol solutions, each for ca. one hour. The tissue was then stained with 0.2% toluidine blue and mounted for DIC microscopy in 50% glycerol. All microscopy was done at a 400× magnification. Leaf tissue used for microscopy was also photographed for measurements of leaf area in ImageJ.
Homology modeling, model validation, and computational docking
Homology modeling of the GTL1 N-terminal trihelix DNA-binding domain (residues 63 to 126) was modeled using the RaptorX web-based server56. Structure refinement of the predicted models was performed using ModRefiner57. The predicted model of GTL1N was validated by PROCHECK58 to check stereochemical quality of a native structure. Ramachandran plot (Supplementary Fig. S9a) and plot statistics (Supplementary Table S2) indicate 95.3% residues in most favored regions, showing a good quality of GTL1N structure. The GTL1N model was further evaluated using the ProSA web server59. ProSA calculates the Z-score (−4.13) for the model to show the overall model quality (Supplementary Fig. S9b) and knowledge-based energy plot of residue scores (Supplementary Fig. S9c).
The structure of the GTL1N-SDD1 promoter complex was determined using the High Ambiguity Driven biomolecular DOCKing (HADDOCK) web server60. The active residues of GTL1N involved in docking are chosen based on the NMR structure of GT1 and GT-1 box element and are N61, R62, K109, E113, E116, N117, K120, and K126. The residues around active residues were automatically defined as passive residues. 3D structure of the SDD1 promoter fragment (5¢-gcttGGTAAAactt-3¢) including a GT3-box was generated by 3D-DART, a DNA structure-modeling server61. The GT2 box residues in the SDD1 promoter fragment (Gua5, Gua6, Thy7, Ade8, Ade9, Ade10) and their complementary residues were considered active residues. A total of 20,000 structures were generated using rigid body energy minimization. The 200 lowest-energy structures were refined by semiflexible refinement and explicit solvent calculations. The model with the best (lowest) HADDOCK score (−175.6 ± 6.2) and root mean square deviation (2.0 ± 1.2) was selected and used for the analysis. All 3D structures were analyzed and visualized by 3D structure viewer UCSF Chimera62.
Statistical analysis
All column graphs were analyzed by Tukey’s honestly significant difference (HSD) test (one-way ANOVA) using SAS 9.2 software (SAS Institute).
References
Lawson, T. & Blatt, M. R. Stomatal size, speed, and responsiveness impact on photosynthesis and water use efficiency. Plant Physiol. 164, 1556–1570 (2014).
Yoo, C. Y., Pence, H. E., Hasegawa, P. M. & Mickelbart, M. V. Regulation of transpiration to improve crop water use. CRC Crit. Rev. Plant Sci. 28, 410–431 (2009).
Franks, P. J. W., Doheny-Adams, T., Britton-Harper, Z. J. & Gray, J. E. Increasing water-use efficiency directly through genetic manipulation of stomatal density. New Phytol. 207, 188–195 (2015).
Yoshida, T., Mogami, J. & Yamaguchi-Shinozaki, K. ABA-dependent and ABA-independent signaling in response to osmotic stress in plants. Curr. Opin. Plant Biol. 21, 133–139 (2014).
Simmons, A. R. & Bergmann, D. C. Transcriptional control of cell fate in the stomatal lineage. Curr. Opin. Plant Biol. 29, 1–8 (2016).
Kumari, A., Jewaria, P. K., Bergmann, D. C. & Kakimoto, T. Arabidopsis reduces growth under osmotic stress by decreasing SPEECHLESS protein. Plant Cell Physiol. 55, 2037–2046 (2014).
Wang, H., Ngwenyama, N., Liu, Y., Walker, J. C. & Zhang, S. Stomatal development and patterning are regulated by environmentally responsive mitogen-activated protein kinases in Arabidopsis. Plant Cell 19, 63–73 (2007).
Dodd, A. N., Kudla, J. & Sanders, D. The language of calcium signaling. Annu. Rev. Plant Biol. 61, 593–620 (2010).
Clore, G. M., Bax, A., Ikura, M. & Gronenborn, A. M. Structure of calmodulin-target peptide complexes. Curr. Opin. Struc. Biol. 3, 760–764 (1993).
Park, H. Y. et al. Conformational changes of calmodulin upon Ca2+ binding studied with a microfluidic mixer. Proc. Natl. Acad. Sci. USA 105, 542–547 (2008).
Crivici, A. & Ikura, M. Molecular and structural basis of target recognition by calmodulin. Annu. Rev. Biophys. Biomol. Struct. 24, 85–116 (1995).
Corneliussen, B. et al. Calcium/calmodulin inhibition of basic-helix-loop-helix transcription factor domains. Nature 368, 760–764 (1994).
Yoo, J. H. et al. Direct interaction of a divergent CaM isoform and the transcription factor, MYB2, enhances salt tolerance in Arabidopsis. J. Biol. Chem. 280, 3697–3706 (2005).
Du, L. et al. Ca2+/calmodulin regulates salicylic-acid-mediated plant immunity. Nature 457, 1154–1158 (2009).
Du, L., Yang, T., Puthanveettil, S. V. & Poovaiah, B. W. Decoding of Calcium Signal Through Calmodulin: Calmodulin-Binding Proteins in Plants. In Coding and Decoding of Calcium Signals in Plants (ed. Luan, S.) 177–233 (Springer Berlin Heidelberg, 2011).
Han, H. J. et al. The transcriptional repressor activity of Asymmetric Leaves 1 is inhibited by direct interaction with calmodulin in Arabidopsis. Plant Cell Environ. 35, 1969–1982 (2012).
Fromm, H. & Finkler, A. Repression and de-repression of gene expression in the plant immune response: The complexity of modulation by Ca2+ and calmodulin. Mol. Plant 8, 671–673 (2015).
Virdi, A. S., Singh, S. & Singh, P. Abiotic stress responses in plants: roles of calmodulin-regulated proteins. Front. Plant Sci. 6, 809 (2015).
Reddy, A. S. N., Ali, G. S., Celesnik, H. & Day, I. S. Coping with stresses: roles of calcium- and calcium/calmodulin-regulated gene expression. Plant Cell 23, 2010–2032 (2011).
Kim, Y. S. et al. CAMTA-mediated regulation of salicylic acid immunity pathway genes in Arabidopsis exposed to low temperature and pathogen infection. Plant Cell 29, 2465–2477 (2017).
Zhou, D. X. Regulatory mechanism of plant gene transcription by GT-elements and GT-factors. Trends Plant Sci. 4, 210–214 (1999).
Kaplan-Levy, R. N., Brewer, P. B., Quon, T. & Smyth, D. R. The trihelix family of transcription factors–light, stress and development. Trends Plant Sci. 17, 163–171 (2012).
Nagata, T. et al. Solution structures of the trihelix DNA-binding domains of the wild-type and a phosphomimetic mutant of Arabidopsis GT-1: Mechanism for an increase in DNA-binding affinity through phosphorylation. Proteins 78, 3033–3047 (2010).
Breuer, C. et al. The trihelix transcription factor GTL1 regulates ploidy-dependent cell growth in the Arabidopsis trichome. Plant Cell 21, 2307–2322 (2009).
Kaplan-Levy, R. N., Quon, T., O’brien, M., Sappl, P. G. & Smyth, D. R. Functional domains of the PETAL LOSS protein, a trihelix transcription factor that represses regional growth in Arabidopsis thaliana. Plant J. 79, 477–491 (2014).
Weng, H., Yoo, C. Y., Gosney, M. J., Hasegawa, P. M. & Mickelbart, M. V. Poplar GTL1 is a Ca2+/calmodulin-binding transcription factor that functions in plant water use efficiency and drought tolerance. PLoS One 7, e32925 (2012).
Yoo, C. Y. et al. The Arabidopsis GTL1 transcription factor regulates water use efficiency and drought tolerance by modulating stomatal density via transrepression of SDD1. Plant Cell 22, 4128–4141 (2010).
Yoo, C. Y., Hasegawa, P. M. & Mickelbart, M. V. Regulation of stomatal density by the GTL1 transcription factor for improving water use efficiency. Plant Signal. Behav. 6, 1069–1071 (2011).
Xie, Z.-M. et al. Soybean trihelix transcription factors GmGT-2A and GmGT-2B improve plant tolerance to abiotic stresses in transgenic Arabidopsis. PLoS One 4, e6898 (2009).
Zheng, X. et al. The wheat GT factor TaGT2L1D negatively regulates drought tolerance and plant development. Sci. Rep. 6, 27042 (2016).
Xi, J., Qiu, Y., Du, L. & Poovaiah, B. W. Plant-specific trihelix transcription factor AtGT2L interacts with calcium/calmodulin and responds to cold and salt stresses. Plant Sci. 185–186, 274–280 (2012).
Yap, K. L. et al. Calmodulin target database. J. Struct. Funct. Genomics 1, 8–14 (2000).
Martí-Renom, M. A. et al. Comparative protein structure modeling of genes and genomes. Annu. Rev. Biophys. Biomol. Struct. 29, 291–325 (2000).
O’Neil, K. T. & DeGrado, W. F. How calmodulin binds its targets: sequence independent recognition of amphiphilic alpha-helices. Trends Biochem. Sci. 15, 59–64 (1990).
Yuan, F. et al. OSCA1 mediates osmotic-stress-evoked Ca2+ increases vital for osmosensing in Arabidopsis. Nature 514, 367–371 (2014).
Zhu, X., Feng, Y., Liang, G., Liu, N. & Zhu, J.-K. Aequorin-based luminescence imaging reveals stimulus- and tissue-specific Ca2+ dynamics in Arabidopsis plants. Mol. Plant 6, 444–455 (2013).
Kiegle, E., Moore, C. A., Haseloff, J., Tester, M. A. & Knight, M. R. Cell-type-specific calcium responses to drought, salt and cold in the Arabidopsis root. Plant J. 23, 267–278 (2000).
Pauly, N. et al. The nucleus together with the cytosol generates patterns of specific cellular calcium signatures in tobacco suspension culture cells. Cell Calcium 30, 413–421 (2001).
Skirycz, A. et al. Pause-and-stop: the effects of osmotic stress on cell proliferation during early leaf development in Arabidopsis and a role for ethylene signaling in cell cycle arrest. Plant Cell 23, 1876–1888 (2011).
Liu, Y., Qin, L., Han, L., Xiang, Y. & Zhao, D. Overexpression of maize SDD1 (ZmSDD1) improves drought resistance in Zea mays L. by reducing stomatal density. Plant Cell Tissue Organ Cult. 122, 147–159 (2015).
Von Groll, U., Berger, D. & Altmann, T. The subtilisin-like serine protease SDD1 mediates cell-to-cell signaling during Arabidopsis stomatal development. Plant Cell 14, 1527–1539 (2002).
Morales-Navarro, S. et al. Overexpression of a SDD1-like gene from wild tomato decreases stomatal density and enhances dehydration avoidance in Arabidopsis and cultivated tomato. Front. Plant Sci. 9, 940 (2018).
Knight, H., Trewavas, A. J. & Knight, M. R. Calcium signalling in Arabidopsis thaliana responding to drought and salinity. Plant J. 12, 1067–1078 (1997).
Huang, F. et al. Cytosolic and nucleosolic calcium signaling in response to osmotic and salt stresses are independent of each other in roots of Arabidopsis seedlings. Front. Plant Sci. 8, 1648 (2017).
Villarroel, A. et al. The ever changing moods of calmodulin: how structural plasticity entails transductional adaptability. J. Mol. Biol. 426, 2717–2735 (2014).
Liu, J. & Nussinov, R. Energetic redistribution in allostery to execute protein function. Proc. Natl. Acad. Sci. USA 114, 7480–7482 (2017).
Nussinov, R., Tsai, C.-J. & Liu, J. Principles of allosteric interactions in cell signaling. J. Am. Chem. Soc. 136, 17692–17701 (2014).
Changeux, J.-P. & Edelstein, S. J. Allosteric mechanisms of signal transduction. Science 308, 1424–1428 (2005).
Xiong, L. & Zhu, J. K. Molecular and genetic aspects of plant responses to osmotic stress. Plant Cell Environ. 25, 131–139 (2002).
Miura, K. et al. SIZ1-mediated sumoylation of ICE1 controls CBF3/DREB1A expression and freezing tolerance in Arabidopsis. Plant Cell 19, 1403–1414 (2007).
Jin, J. B. et al. The SUMO E3 ligase, AtSIZ1, regulates flowering by controlling a salicylic acid-mediated floral promotion pathway and through affects on FLC chromatin structure. Plant J. 53, 530–540 (2008).
Yoo, S. D., Cho, Y. H. & Sheen, J. Arabidopsis mesophyll protoplasts: a versatile cell system for transient gene expression analysis. Nat. Protoc. 2, 1565–1572 (2007).
Trost, C., Bergs, C., Himmerkus, N. & Flockerzi, V. The transient receptor potential, TRP4, cation channel is a novel member of the family of calmodulin binding proteins. Biochem. J 355, 663–670 (2001).
Reddy, V. S. & Reddy, A. S. N. The calmodulin-binding domain from a plant kinesin functions as a modular domain in conferring Ca2+-calmodulin regulation to animal plus- and minus-end kinesins. J. Biol. Chem. 277, 48058–48065 (2002).
Vatén, A. & Bergmann, D. C. Mechanisms of stomatal development: an evolutionary view. Evodevo 3, 11 (2012).
Källberg, M. et al. Template-based protein structure modeling using the RaptorX web server. Nat. Protoc. 7, 1511–1522 (2012).
Xu, D. & Zhang, Y. Improving the physical realism and structural accuracy of protein models by a two-step atomic-level energy minimization. Biophys. J. 101, 2525–2534 (2011).
Laskowski, R. A., MacArthur, M. W., Moss, D. S. & Thornton, J. M. PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 26, 283–291 (1993).
Wiederstein, M. & Sippl, M. J. ProSA-web: interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 35, W407–10 (2007).
de Vries, S. J., van Dijk, M. & Bonvin, A. M. J. J. The HADDOCK web server for data-driven biomolecular docking. Nat. Protoc. 5, 883–897 (2010).
van Dijk, M. & Bonvin, A. M. J. J. 3D-DART: a DNA structure modelling server. Nucleic Acids Res. 37, W235–9 (2009).
Pettersen, E. F., Goddard, T. D. & Huang, C. C. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 (2004).
Acknowledgements
We thank Dr. Sharon Kessler, Thomas Davis, Rachel Flynn, Michael J. Gosney, and Huangai Bi for experimental assistance, and Nathan Deppe and Daniel Little for greenhouse assistance. Research was supported by a grant from the National Science Foundation Award (MCB-0424850 to P.M.H., MCB-5333470 to A.S.N.R., IOS-1021344 to B.W.P.), the United States Department of Agriculture - National Institute of Food and Agriculture (WNP00321) to B.W.P., the Agriculture and Food Research Initiative Competitive Grant (2019-67013-29239 to A.S.N.R., the United States-Israel Binational Agricultural Research and Development Award (IS-4243-09 to H.F. and P.M.H. and IS-4749-14CR to H.F., M.V.M. and P.M.H.), and the Department of Energy Office of Science, Office of Biological and Environmental Research (DE-SC0010733) to A.S.N.R.
Author information
Authors and Affiliations
Contributions
C.Y.Y., P.M.H. and M.V.M. conceived and designed the experiments. C.Y.Y., N.M., A.F., H.W. and I.S.D. performed the experiments. C.Y.Y., N.M., A.F., H.W., H.F., P.M.H. and M.V.M. analyzed and discussed the data. A.S.N.R. and B.W.P. originally identified GTL1 as a Ca2+/CaM-binding protein, analyzed and discussed data. C.Y.Y., P.M.H. and M.V.M., wrote the manuscript. All authors read and approve the final manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yoo, C.Y., Mano, N., Finkler, A. et al. A Ca2+/CaM-regulated transcriptional switch modulates stomatal development in response to water deficit. Sci Rep 9, 12282 (2019). https://doi.org/10.1038/s41598-019-47529-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-47529-2
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
