
Abstract
The emergence of cost-effective and rapid sequencing approaches has resulted in an exponential rise in the number of mitogenomes on public databases in recent years, providing greater opportunity for undertaking large-scale comparative genomic and systematic research. Nonetheless, current datasets predominately come from small and disconnected studies on a limited number of related species, introducing sampling biases and impeding research of broad taxonomic relevance. This study contributes 21 crustacean mitogenomes from several under-represented decapod infraorders including Polychelida and Stenopodidea, which are used in combination with 225 mitogenomes available on NCBI to investigate decapod mitogenome diversity and phylogeny. An overview of mitochondrial gene orders (MGOs) reveals a high level of genomic variability within the Decapoda, with a large number of MGOs deviating from the ancestral arthropod ground pattern and unevenly distributed among infraorders. Despite the substantial morphological and ecological variation among decapods, there was limited evidence for correlations between gene rearrangement events and species ecology or lineage specific nucleotide substitution rates. Within a phylogenetic context, predicted scenarios of rearrangements show some MGOs to be informative synapomorphies for some taxonomic groups providing strong independent support for phylogenetic relationships. Additional comparisons for a range of mitogenomic features including nucleotide composition, strand asymmetry, unassigned regions and codon usage indicate several clade-specific trends that are of evolutionary and ecological interest.
Similar content being viewed by others

Introduction
Traditional molecular systematic research has been largely dependent on phylogenetic reconstructions based on few molecular markers obtained via individual targeted polymerase chain reaction (PCR) and Sanger sequencing approaches1,2,3,4,5,6,7. More recently, the advent of second-generation sequencing (SGS), coupled with the development of simplified and streamlined laboratory and bioinformatic pipelines8,9,10,11,12, has seen a proliferation of eukaryotic and prokaryotic genomic resources. Notably, advances in genomic approaches have greatly increased the efficiency of recovering complete and near-complete mitochondrial genomes from species across the animal and plant kingdoms, making it a popular candidate for phylogenetic and comparative genomic studies, in addition to its role as a useful barcoding locus for species identification13. Despite the rapid growth in mitogenome resources, current datasets predominately come from small and disconnected studies on single or a limited number of related species, introducing sampling biases and impeding research of broad taxonomic relevance14,15,16. While there is a general consensus that new mitogenome resources remain valuable for comparative genomic and systematic research, improved representation of mitogenome resources for poorly-represented biological lineages is still required for studies aimed at addressing fundamental evolutionary, genomic and systematic questions14. More specifically, within the context of phylogenetics, inadequate taxon sampling and taxon biases can lead to topological distortions due to artefactual sources of error such as long-branch attraction17. This highlights the need for reviews of sampling biases and more exhaustive sampling to overcome such limitations.
Decapods are a highly speciose and diverse group of crustaceans, including 11 infraorders and almost 15,000 species18, which provide critical ecosystem services across marine, freshwater and terrestrial environments, and are of significant commercial and/or medical importance19,20,21,22. The first decapod mitogenome sequences emerged in the early 2000s based on tedious Sanger sequencing approaches but were biased primarily toward species of commercial importance23,24,25,26,27,28. While these studies provide an early hint on the heterogeneous nature of decapod mitogenomes, reporting novel gene orders in multiple lineages, phylogenetic and comparative genomic studies remained severely constrained by limited taxon sampling for almost a decade23,24,25,26,27,28. However, since the emergence of modern genomic sequencing approaches, the availability of mitogenome resources for the Decapoda has rapidly expanded. After the study by Miller and colleagues24,25, the number of decapod mitogenomes has expanded rapidly (now numbering in the hundreds), proving a valuable resource for studies that have provided new insights into decapod evolutionary relationships and comparative mitogenomics at various taxonomic levels11,29,30,31,32,33,34,35,36. Despite this progress, sampling within this large and diverse group has generally been skewed towards commercial or easily-sampled species, highlighting the need for more balanced taxonomic sampling17,36,37,38. At the time this study was initiated, mitogenomic resources for the less diverse and rare groups such as Polychelida, Stenopodidea, Axiidea and Gebiidea were scarce, which impeded the resolution of their relative positions within previous mitogenome phylogenetic reconstructions35,36. To gain a more comprehensive understanding of evolutionary relationships and genomic compositions within Decapoda, mitogenome resources for more species across a breadth of taxonomic groups are much needed.
Though the value of mitogenomes as the sole genetic marker for phylogenetic research is often contested6,39,40, mitochondrial DNA sequences remain the most abundant publicly-available molecular genetic resources, which continue to facilitate fundamental evolutionary research11,35,36. As mitogenomic resources continue to expand and bioinformatic tools capable of processing and analysing increasingly immense datasets are further developed, opportunities for more comprehensive analyses of mitogenome structural architecture and systematic relationships across broad taxonomic groups are emerging14,15. This is giving rise to increasing numbers of studies on the evolution of mitogenome features across various animal groups35,41,42. Specifically, these new resources are allowing transcriptional and translational features such as nucleotide and amino acid composition43,44, substitution rates34,45, codon usage32,46, unassigned and intergenic regions47,48 and strand asymmetry49,50 to be contrasted within and among a range of taxonomic groups.
As a whole, lineages within the Decapoda have successfully conquered a very broad range of environments (e.g. freshwater, terrestrial, marine) as well as adopted a wide variety of lifestyles, body forms and sizes. Considering the varied metabolic demands associated with this range of adaptations, it is expected that respiratory functions of the mitochondrion are subject to substantial selective pressures51,52. Thus, heterogeneity in mitogenomic features may provide insights into possible correlations between the evolution of the molecule and species diversification at a range of evolutionary scales41,53. The ease of recovering complete mitogenomes with modern genomic approaches is now allowing for the analysis of genomic structural diversification in unprecedented detail42,47,54,55,56. In the Decapoda, novel mitochondrial gene orders (MGOs) have been reported across a range of lineages57,58,59,60,61 and at an unusually high frequency compared with other higher-level taxa (e.g. several insect orders62, frogs63, birds49, batoids64, elasmobranchs65). Several studies have suggested possible correlations of certain highly-rearranged MGOs to accelerated nucleotide substitution rates in selected lineages or to the adaptations to specialised lifestyles and extreme ecological niches34,45,62,66. This may potentially be multifactorial, occurring in association with the evolution of various biochemical and metabolic traits (e.g. DNA repair, metabolic rate, generation time, body size)45. Similar to insects62, MGO patterns have been identified as potential synapomorphies for various decapod groups at a range of taxonomic levels, in some instances providing unequivocal resolution of contentious evolutionary relationships31,32,34,66,67,68,69.
In this study, we contribute 21 new mitogenomes from 7 decapod infraorders including a number from pre-identified taxonomic groups with limited sampling. We provide an overview of the distribution of mitochondrial gene orders (MGO) within the context of our inferred phylogeny and show that these rearrangements are unevenly distributed throughout Decapoda. Though noting a lack of correlation of these heightened rearrangements with variations in general ecology, lifestyles or nucleotide substitution rates, we point out instances of MGOs as likely synapomorphies at various taxonomic levels and also included comparisons of several mitogenomic features to gain further insights into decapod mitogenome evolution and phylogeny.
Results
New mitogenomes for under-represented infraorders
This work contributes mitogenomes for 21 species from 7 decapod infraorders (Table 1), substantially increasing complete mitogenome resources for infraorders that were most under-represented in previous mitogenome studies. All but one of the mitogenomes were successfully assembled into complete circular sequences with sizes ranging from 15.5 to 17.6 kbp. The assembled mitogenome of Parastacus brasiliensis contains a gap in the control region. All mitogenomes contain the typical 13 protein-coding genes and 2 ribosomal RNAs (rRNA), but with varying numbers (18 to 23) of identified transfer RNAs (tRNA) (Supplementary Data S1).
Mitogenome-based phylogenetics provide insights into infra-ordinal relationships
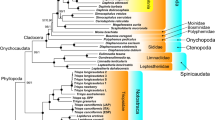
Concatenation of the mitochondrial genes resulted in 10 359, 11 694 and 3 453 alignment sites for Dataset I (nucleotide-based, 13 PCG), Dataset II (nucleotide-based, 13 PCG + 2 rRNA) and Dataset III (amino acid-based, 13 PCG), respectively. Out of the 21 new species for which mitogenomes were contributed in this study, eight species are observed in the trees to share sister relationships with species from the same genus. The remaining 13 species represent new lineages, providing new resources for their respective genera (Spongicola, Globospongicola, Remiarctus, Puerulus, Cardus, Pentacheles, Stereomastis, Parastacus, Ombrastacoides, Strahlaxius, Axianassa, Laomedia, Gebiacantha). These lineages also cluster with other members from their respective families or superfamilies supporting existing taxonomic arrangements (Fig. 1, trees are available as Supplementary Data S2). Relationships among all decapod infraorders are represented in this analysis, with the exception of Procarididea that lacks available complete mitogenome data at the time of this study.

Infraorder-level topology inferred from Maximum likelihood (ML) and Bayesian (BI) methods, based on three datasets. Red branches indicate nodes with weak support (ML: ultrafast bootstrap values of < 95%, BI: posterior probabilities of <0.90).
All three datasets recover monophyletic groups for the 10 Pleocyemata infraorders, with a consistent basal position for the shrimps (Caridea, Stenopodidea), followed by the burrowing shrimps (Axiidea, Gebiidea), with the ‘crabs’ (Anomura, Brachyura) in the most derived positions. The lobsters and crayfish and allies, mostly represented by Achelata, Astacidea, Glypheidea and Polychelida, see some discrepancies between topologies reconstructed from nucleotide (Dataset I and II) and amino acid (Dataset III) datasets. Nucleotide-based phylogenies place Achelata at a relatively basal position whereas the topology based on the amino acid alignment has Achelata nested within the larger lobster/crayfish clade. This, in turn, affects the positions of the other lobster/crayfish infraorders as well. On the other hand, the positions of several infraorders are variable in the Bayesian-inferred tree (Fig. 1), resulting in a clade of Caridea + Stenopodidea sharing a sister relationship. Also in this tree, while Axiidea and Gebiidea are still recovered as separate infraorders, the latter is placed at a more basal position, an observation that contrasts with the topologies shown in the other three trees.
Gene order rearrangements are unevenly distributed across decapod infraorders
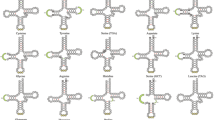
In general, the level of diversity in MGO patterns varies widely within and between infraorders (Fig. 2). Excluding the infraorders with less than 10 species sampled (Glypheidea, Stenopodidea, Polychelida, Axiidea), decapod groups ordered from most to least diversity of MGOs are as follows: Anomura (13 MGO patterns among 22 species), Gebiidea, Astacidea, Achelata, Brachyura, Dendrobranchiata and Caridea (4 MGO patterns among 37 species). Several MGO ‘hot-spots’ are identified from this analysis. For instance, almost every family in Anomura (An) and Gebiidea (Ge) has a unique MGO pattern. Within the Astacidea (As), species in the Parastacidae exhibit 12 different MGOs compared to its sister family group Astacidae with only one MGO pattern shared by all species. In addition, almost every genus in the brachyuran (Br) superfamilies Gecarcinucoidea and Potamoidea have unique MGOs (see Supplementary Data S3 for additional information).

Decapod phylogenetic tree. This cladogram was inferred using the maximum-likelihood method based on Dataset I (13 mitochondrial PCGs, 10 359 nucleotide alignment). Clades are coloured according to the different infraorders. The outer colour strip in the phylogenetic tree represents the distribution of mitochondrial gene orders (MGO) in various infraorders and summarises a total of 59 different MGOs across the 246 different decapod species analysed, labelled for each infraorder in the panels below. Orange-coloured MGO labelled with ‘Gr’ refers to the pancrustacean ground pattern; other derived MGOs are numerically labelled and attached with a 2-letter infraorder prefix. MGOs that differ from the ground pattern are a result of a series of CREx-predicted gene rearrangement events: transposition (T), reversal (r), reverse transposition (rT), duplication (d), deletion (x) and tandem duplication-random loss (tdrl). Yellow- or red-coloured circles on some nodes reflect the level of uncertainty for the TreeREx reconstruction of each ancestral MGO, with red exhibiting highest level of uncertainty, yellow for mid-level and no circle for consistent reconstruction (see Babbucci, et al.42 for details). Subsequent outer rings indicate, to the best of our knowledge, the possible environments (terrestrial, freshwater, marine, vents/seeps) inhabited by each decapod species.
Established as a gene order common to insects and crustaceans67, the pancrustacean ground pattern (coloured orange and labelled Gr) is predominantly observed for basal decapod groups such as the suborder Dendrobranchiata and infraorders Caridea, Achelata and Glypheidea, appears in a limited number of species within the Astacidea and Gebiidea, and is absent in all species sequenced so far in the other infraorders. Within the context of the inferred phylogeny, gene rearrangement events shared by members of specific clades provide examples of unifying evolutionary signatures (synapomorphies) from higher levels such as the infraorder-specific St1 pattern shared by the four analysed Stenopodidea species, through the superfamily-specific As1 for species in the Astacoidea, and family-specific rearrangements at the base of the Upogebiidae clade illustrated by the Ge1 pattern.
Lack of correlation between MGO variation and substitution rates
Overall, evidence of episodic positive selection is observed in 10 out of 13 mitochondrial PCGs (ATP6, COX1, COX2, COX3, CYTB, ND1, ND2, ND4, ND5, ND6) and for main branches of most infraorders/suborders, with the exception of Dendrobranchiata, Glypheidea and Axiidea (Supplementary Data S4). Notably, a higher frequency of statistically significant selection is reported in Caridea mostly detected along branches leading to the genera Alpheus, Palaemon and Macrobrachium. On the other hand, accelerated evolutionary rates are observed for multiple decapod lineages across all infraorders (Supplementary Data S4). Based on observation, while there appears to be a link between substitution rates and heightened gene rearrangements in some taxonomic clades, for example in Parastacoidea (southern hemisphere crayfish) or Caridea, there are also instances where accelerated rates are observed for lineages with highly conserved MGOs as well. However, across Decapoda as a whole, the Spearman correlation analysis does not suggest a link between variable MGOs and nucleotide/amino acid substitution rates (Supplementary Data S4).
Mitogenome characteristics vary within and between decapod infraorders
Applying the Hyper-Empirical Relative Mitochondrial Evolutionary Speed (HERMES) index developed by Plazzi, et al.41, the amount of mitochondrial evolution was estimated based on several genomic features found to be phylogenetically congruent in their tested datasets. These genomic features include the root-to-tip distance for each species (RtoTdist), ML distance from the E. pacifica outgroup (MLdist), percentage of Unassigned Regions (URs), amount of mitochondrial identical gene arrangements (AMIGA) for PCGs and strand usage skew (SUskew). The HERMES index appears to be informative for our dataset with reasonable goodness-of-fit statistics: Tucker-Lewis Index (TLI)70 = 0.939; root mean square of the residuals (SRMR) = 0.053; root mean squared error of approximation (RMSEA) = 0.058, all close to boundaries suggested by Hu and Bentler71. The highest communality was for AMIGA at 85.5% whereas other variables scored at only 10 to 20%, resulting in a mean of 29.1% (i.e. the HERMES index accounts for 29.1% of the total variability of the source matrix). Since gene order is the key parameter in this system, the HERMES factor analysis in Fig. 3 separates individuals into two distinct groups. The first group exhibits low HERMES index and consists of individuals with MGO that is identical to or highly similar to the pancrustacean ground pattern (Gr) while the second group comprises mostly of individuals that have undergone tdrl events resulting in large rearrangements of mitochondrial PCGs (Fig. 2).

HERMES index across Decapoda. Species are listed horizontally by suborder, infraorder, superfamilies and families to highlight differences among various taxonomic groups.
Several notable observations can be made on the clustering of data points based on various mitogenomic features, as observed in principal component analysis (PCA) plots in Fig. 4. The plot in Fig. 4a is based on the same five variables in the previous HERMES analysis. While most individuals with MGOs identical or similar to the pancrustacean ground pattern tend to form a tight cluster, some of these are distinctly separated from others based on the proportion of their unassigned regions, most of them either containing more than one control region (e.g. Metanephrops, Munida) or long intergenic regions (e.g. Geothelphusa, Longpotamon) (Fig. 4a). Additionally, Fig. 4b–d summarise nucleotide composition, asymmetry (skew) information and amino acid composition respectively. The AT content separates most of the brachyuran crabs from caridean shrimps, whereas GC-skew distinguishes the two Astacidea superfamilies (Astacoidea, Parastacoidea) (Fig. 4b). The two burrowing mud shrimp infraorders, Axiidea and Gebiidea, are often clearly split into different quadrants for plots based on nucleotide features, whereas data points representing anomuran species are generally widely scattered across all PCA plots.

Mitogenomic PCA plots. Principal component analyses using Pearson’s correlation based on various characteristics of the mitogenome. (a) shows the PCA plot based on the same five variables in the HERMES analysis, with the first two principal components accounting for 59.39% of the dataset variability. Additionally, (b–d) summarise nucleotide composition, asymmetry (skew) information and amino acid composition with 91.97%, 56.98% and 56.98% of each dataset variability, respectively, in the first two principal components. Data points are labelled with the first 2 or 3 letters of the genus followed by the first 3 letters of the species name; e.g. ‘ChDes’ for Cherax destructor.
Discussion
This study adds to the growing list that has benefited from the use of museum-preserved specimens for mitogenomic studies37,72,73,74,75,76,77, with mitogenomes for 21 species strategically sampled to fill important taxonomic gaps, particularly in under-represented decapod infraorders. The higher level topologies of all three Maximum likelihood trees reconstructed in this study are congruent at the base and top of the trees (Fig. 1), consistently recovering: [1] The suborder Dendrobranchiata as sister group to the rest of egg-brooding Pleocyemata, [2] Caridea as basal to Pleocyemata, [3] Stenopodidea as sister to all of Reptantia, [4] Axiidea and Gebiidea as separate infraorders, and [5] Anomura and Brachyura placed as sister taxa high in the tree, generally in agreement with previous reports1,11,36,37 though with points of distinction with others2,3,6,78,79. While the higher level topology of the Bayesian-inferred tree generally exhibits similar observations, there are deviations with respect to the relationship between Caridea and Stenopodidea (recovered as sister clades) and the shuffle in positions of Axiidea, Anomura and Brachyura. Generally, incongruent topologies show that inter-relationships among lobster and crayfish infraorders still remain unresolved1,2,6,7,78,80,81, requiring further taxonomic sampling and/or additional nuclear gene information. It is also noteworthy that the taxonomically-impoverished infraorder Procarididea5,82 is absent from our study, but is considered by most to likely be one of the basal lineages within the Decapoda5.
The phylogenetic analysis presented in this study represents one of the most comprehensive samplings of decapod species, based on mitochondrial data. While genome-based phylogenies are still rare for the Decapoda due to the generally large sizes of its genomes (1 to 40 Gbp), the increasingly lower costs of sequencing are enabling studies to target more genomic loci through genome skimming or anchored hybrid enrichment methods83,84,85. However, these still lack the fine-grained resolution obtained in this study. Two noteworthy studies that have been recently published have attempted to elucidate decapod inter-relationships based on genomic (Wolfe, et al.84; 94 decapod species, 410 loci, greater than 86 000 bp) or transcriptomic data (Schwentner, et al.86; 16 decapod species, 81 to 684 orthogroups, 17 690 to 242 530 amino acid positions). Mitogenome-based maximum likelihood phylogenies in this study share similarities in parts of their topologies with that obtained from these studies, specifically in the recovery of shrimp and prawn groups as basal clades and the traditional Meiura (Brachyura + Anomura) as more derived lineages, increasing the confidence of these nodes based on support from different underlying genetic data. Nevertheless, several nodes are still in contention. This study recovers a paraphyletic Caridea and Stenopodidea clade whereas both Wolfe, et al.84 and Schwentner, et al.86 recovered these infraorders as sister clades. The position of Achelata also remains unresolved in most trees as well as the interrelationships among crayfish and lobster groups, as well as the positions of the traditional thalassinids (Axiidea, Gebiidea). Overall, topologies inferred in this study are generally more similar to that obtained by Wolfe, et al.84.
The contrasting MGO patterns across all available decapod mitogenomes have revealed some interesting findings. Most notably, a number of highly-rearranged gene orders are observed, occurring in unequal frequencies across various decapod infraorders or at lower taxonomic levels, with a level of diversity higher than or at least comparable to the levels observed in other metazoan groups42,47,49,63,87,88. We see the prevalence of the pancrustacean ground pattern (Gr)67, and/or other highly-similar MGOs in infraorders at both the basal and more derived positions, indicating that this highly-conserved ground pattern is likely to be ancestral for decapods, with modified MGOs within each infraorder being more derived traits35. Hence, instances of clade-specific MGO patterns or MGO “hot spots” suggest that the utility of MGOs as synapomorphies is amplified for certain decapod groups, acting as unifying evolutionary signatures that can be used to either support or question existing classifications at a range of taxonomic levels31,32,34,35,62,66,67,69. For example, brachyuran species associated with the Varunidae (superfamily Grapsoidea) and Macrophthalmidae (superfamily Ocypodoidea) that form sister clades31,66,89,90 also share the Br2 MGO pattern, which provides complementary support to other phylogenetic studies that call for a re-evaluation of classifications for the two large paraphyletic superfamilies33,89,90,91. Nonetheless, it is equally important that we continue to be cautious when evaluating MGO information within a phylogenetic context42,66,92, keeping in mind the caveats related to potential homoplastic or convergent arrangements42,87,93,94, a saturation of signals from reversible rearrangement events95,96 or possibly inaccurate inferences from unresolved phylogenies66.
In contrast to findings that found a positive correlation between gene rearrangements and elevated nucleotide substitution rates in insect and bivalve mitogenomes41,45, our correlation analysis indicates no such association for the Decapoda (Spearman correlation in Supplementary Data S4). Although the exact drivers of MGO rearrangements remain unclear, other studies have noted broad associations with adaptations to extreme ecological niches and lifestyles34,62,66,68,97,98. Similarly, accelerated rates of MGO rearrangements occur in various decapod lineages such as burrowing crayfish34, freshwater crabs66, and other species adapted to extreme deep-sea environments or temperatures66,98. However, again, our study indicates no such correlation between MGOs and ecologically-similar decapod groups. For example, freshwater crabs and crayfish (southern hemisphere parastacids) display highly variable MGOs, while other freshwater groups such as the shrimps and northern hemisphere crayfish maintain the same gene order among all sampled species (although the members of northern hemisphere crayfish group have a large distinctive rearrangement that acts as a synapomorphy for the superfamily). The same observation applies to other crab and shrimp species inhabiting deep sea vent niches, or burrowing axiid mud shrimps, which exhibit only a few rearranged MGOs.
However, ecological transitions and the evolution of a distinct Bauplan or the adoption of different lifestyles (e.g. parasitism or adaptation to freshwater) may be achieved via different evolutionary pathways99,100,101 and the consequences for energy demands for an organism may be just one of a plethora of factors including thermal tolerance102, aerial exposure103, sensitivity to salinity104, light105 or metal concentrations106. In addition to the regulation of their transcription and translation107,108, mitochondria are very dynamic structures often undergoing continuous fusion, fission and motility, events that can potentially affect their bioenergetic capacities109. However, the extent of these rearrangements and the cues that trigger them are still poorly understood. Thus, we do not rule out an association of heightened MGO rearrangements and profound changes at deep taxonomic levels (e.g. the two groups of crayfish) to clade-specific adaptations and ecological/life history transitions, but these cannot be confidently determined until we achieve a better understanding of the stressors involved at the cellular and physiological level and the associated adaptive responses.
Previous studies have compared decapod MGO patterns at lower taxonomic levels across the Decapoda31,32,34,35,60,66,110,111. However, this study is one of the few to investigate the extent of MGO diversity at the ordinal level and a large number of species (246 species), the other being a phylomitogenomic study on 86 malacostracan mitogenomes38. Other large-scale comparative MGO analyses for various metazoan groups have also reported on the distribution of MGOs, some showing relatively high rates of MGO evolution from their respective ancestral patterns. These examples include not only invertebrate groups such as insects, gastropods, lice, barnacles, bivalves and annelids42,56,62,112,113,114, but also vertebrates such as in fish, frogs, salamanders and amphisbaenian lizards87,115,116,117,118. Conversely, there are other studies that show high levels of conservatism across major taxonomic groups such as beetles, echinoids, batoids and elasmobranchs64,65,119,120. While decapod mitogenomes clearly showcase a high diversity of MGO patterns as a whole (59 MGOs across 246 species), it is premature to claim that this degree of diversity is unusual or comparable to that of other metazoan groups since results are influenced by the number of mitogenomes compared (i.e. scale of comparison), the taxonomic level of interest or as a result of unbalanced or biased sampling. We therefore highly recommend interpreting the findings from any comparative analyses with caution especially those still lacking representation for major groups and again, urge for more consideration to be given to better sampling strategies.
Comprehensive studies on the evolutionary trends of mitogenomes (both small and large scale) are also emerging for various animal groups, following the increased availability of mitogenomic resources41,119,121,122,123,124,125,126. To determine if there are other aspects of the decapod mitogenome that may show clade-specific associations, aside from the arrangement of genes, we compared additional aspects of the molecular architecture and composition of mitogenomes across the major decapod groups. Mitogenomes analysed in this study are generally around 16 kbp in length, though some outliers were identified (17–20 kbp) with lengths inflated by a higher proportion of unassigned regions, i.e. containing more than one control region (e.g. Metanephrops species61) or relatively long stretches of intergenic spaces such as in most Potamoidea species127,128. Though proportions of unassigned regions may potentially be the discerning feature for some taxonomic groups129, this notion should also be treated with caution as the sizes of these regions may be influenced by the assembly or annotation methods used to predict genes, exemplified by the identification of three trnL genes by Segawa and Aotsuka127 as opposed to only two copies reported following re-annotations by MITOS in this study10. But generally, decapod mitogenome sizes are relatively stable compared to those of other metazoan groups such as bivalves, nematodes and sponges that exhibit strong heterogeneity in size88, with some larger molecules spanning lengths of over 20 kbp and even up to 48 kbp130.
Strand and nucleotide composition asymmetries are other mitochondrial characteristics that have been noted to vary across animal groups49,50,131,132,133. In this study, we point out observed trends from PCA plots that suggest molecular signatures for certain decapod groups. Similar to insects, annelids and arachnids, decapod mitogenomes are generally AT-rich, as opposed to lower compositional AT bias often observed in chordates (e.g. fishes, birds, reptiles)134. Within the Decapoda itself, recognizable clusters were observed for Caridea (average: 65%, range: 59% to 70%) and Brachyura (average: 71%, range: 65% to 77%) with minor overlaps. Other noteworthy features include positive GC-skew values that appear to be a unifying characteristic for all northern hemisphere freshwater crayfish (Astacoidea) as well as for the four Coenobita anomuran species included in this study, as opposed to negative GC-skew values for most other decapods. Further, species from infraorders Axiidea and Gebiidea are well separated due to substantial differences in compositional bias and asymmetry, which, in addition to previous reports from phylogenetic and MGO analyses6,35,135,136, is consistent with their status as separate infraorders instead of united in what was once the Thalassinidea1,78,137,138. On the other hand, data points for anomuran species are dispersed across all plots, highlighting their higher plasticity and mitogenomic variability within this infraorder notorious for its morphological and ecological diversity, including the convergent evolution of the “crab” form66,69,101,139,140 and taxonomic controversies141,142,143,144.
For all discussed groups with their own unique signatures, it is of interest to see if these patterns still hold when more mitogenomes are included in future analyses. Though this study is limited in scope to comparisons across whole mitogenomes, we recognise that this only skims the surface of the range of possible compositional comparisons (e.g. at the gene level) and that there are a myriad of other factors that can be included in further detailed investigations, e.g. testing different codon sites43,44 or the effects of these asymmetries on nucleotide versus amino acid compositions50,145.
Methods
Sequencing, mitogenome assembly and annotation
Most samples used for this study were from vouchered specimens obtained from museum collections, including the National Taiwan Ocean University (NTOU), Museum Victoria (NMV), Muséum National d’Histoire Naturelle (MNHN), Australian Museum (AM) and Museum and Art Gallery of the Northern Territory (MAGNT). Using the Sokolov method146, genomic DNA was extracted from the tissue samples of 21 crustacean species (Table 1) representing seven decapod infraorders (Gebiidea: 6, Polychelida: 5, Achelata: 3, Stenopodidea: 3, Astacidea: 2, Axiidea: 1, Caridea: 1), processed using Nextera-based library preparation (Illumina, USA) and sequenced at low coverage on the Illumina MiSeq platform located at the Monash University Malaysia Genomics Facility to generate paired-end short reads (2 × 250 bp), as previously described8. Low quality sequences were trimmed with Trimmomatic v.0.36147 (illuminaclip:2:30:10, avgqual:20, leading:3, trailing:3, minlen:50), followed by contaminant-filtering (i.e. sequences of bacterial origin) with kraken v.0.10.5-beta148 using its precompiled database (minikraken_20141208). Resulting high-quality reads were then assembled de novo with IDBA-UD v.1.1.1149 to recover a complete mitogenome or, if circularity was not obtained, MITObim v.1.89 was used to achieve assemblies using short mtDNA sequences from related species as ‘baits’. Circularised mitogenomes were annotated with MITOS10 to identify gene boundaries for protein-coding genes (PCG), ribosomal RNAs (rRNA) and transfer RNAs (tRNA), which were further adjusted manually based on sequence homology to genes available on NCBI/GenBank150.
Mitogenome-based phylogenetics
Based on combinations of protein-coding genes (PCG) and ribosomal RNA genes (16S, 12S), mitogenome sequences from 246 decapod species, with Euphausia pacifica (Euphausiacea) as the outgroup, maximum-likelihood trees were constructed with the MitoPhAST v3.0 pipeline11, which automates sequence alignments, trimming of ambiguous regions, sequence concatenation, partitioning by gene (and codon position for nucleotides), model testing and tree-building by combining a series of tools151,152,153,154,155,156,157,158. Analyses were carried out on three datasets:
-
I.
13 mitochondrial PCGs (nucleotides).
-
II.
13 mitochondrial PCGs + 12 S + 16 S (nucleotides).
-
III.
13 mitochondrial PCGs (amino acids).
All Maximum-likelihood trees reconstructed from the three mitochondrial-based datasets (Datasets I to III) were rooted with E. pacifica (Euphausiacea), which is often treated as sister clade to the Decapoda159. Support at each node is evaluated with 1000 ultrafast bootstrap replicates (UFBoot)160 and 1000 SH-aLRT replicates (SH)161.
Mitochondrial gene order (MGO) analysis
Prior to gene order analyses, public mitogenome entries downloaded from GenBank were manually inspected for inaccuracies via cross-checks against results from the MITOS webserver10. Any discrepancies found (e.g. incorrect strands specified, missing genes, mislabelling, etc) were corrected and new GenBank files were generated (Supplementary Data S5). Mitogenomes were then processed through the MitoPhAST v3.0 pipeline, which compares and clusters MGOs into groups according to gene order patterns66. Similar to Tan, et al.66, the output Fasta format file containing MGO for every mitogenome was pre-processed to remove missing genes in one or more species and also to retain only a single copy of duplicated genes. Using each of the generated Maximum-likelihood trees (nucleotide- and amino acid-based) as a phylogenetic guide, gene re-arrangement pathways and putative ancestral MGOs were reconstructed with TreeREx v.1.8554. To obtain more accurate inferences, TreeREx results were further used to guide pairwise comparisons using web-based CREx55 to enable the inclusion of fuller sets of genes that would have been excluded in the TreeREx analysis. The interactive Tree of Life (iTOL) online phylogenetic tool162 was used to illustrate the distribution of MGOs across various infraorders in the phylogenetic tree.
Comparison analyses of other mitochondrial features
Excluding individuals with partial mitogenomes, Hyper-Empirical Relative Mitochondrial Evolutionary Speed (HERMES) index is generated for each of 239 individuals using HERMES v.1.041 to estimate the amount of mitochondrial evolution. Given a maximum likelihood tree, the program computes the root-to-tip distance for each species (RtoTdist) and ML distance from the E. pacifica outgroup (MLdist) then merges these with other mitochondrial characteristics including the percentage of Unassigned Regions (URs), amount of mitochondrial identical gene arrangements (AMIGA) for PCGs and strand usage skew (SUskew). Also, codon usage counts in each individual were obtained with EMBOSS v.6.6.0163 with minor adjustments for the Invertebrate Mitochondrial Code, followed by the calculation of relative synonymous codon usage (RSCU) values by taking the ratio of the actual number of times a codon appears to the expected frequency of the codon if all synonymous codons for the same amino acid are used equally164. Various mitogenome characteristics were then summarised by Principal Component Analysis (PCA) using Pearson correlation carried out with XLSTAT v.2018.5.52460165.
Episodic selection, evolutionary rates and spearman correlation test
Without making an a priori assumption on the likelihood of specific lineages undergoing episodic positive selection, the aBSREL method166 implemented in the command-line version of the HyPhy v.2.3.11 package167 was used on the codon alignment of each mitochondrial protein-coding gene to test each branch in the maximum -likelihood phylogeny inferred from Dataset I, to inspect whether a proportion of sites have evolved under positive selection (ω > 1). Further, evolutionary rates were estimated from the same codon alignments using BEAST v.2.5.0168. An uncorrelated, lognormal relaxed clock model was applied to each partition and the Yule Model was used as the tree prior since taxa consist of individuals from different species, with Euphausia pacifica set as outgroup. Two BEAST MCMC runs of 6 × 109 were performed and convergence was checked with Tracer v.1.7.1169, applying a burn-in of 20% and checking for sufficient Effective Sample Size (ESS) (>200). Finally, trees from both MCMC runs were combined and a maximum clade credibility tree was constructed. Rates were visualised with Figtree v.1.4.3170. Correlations among different MGOs, habitat and substitution rates were measured by the Spearman correlation coefficient carried out with XLSTAT v.2018.5.52460165, details in Supplementary Data S4.
Conclusion
Moving forward, further research is needed to contrast the composition and architecture of mitogenomes across other metazoan groups to determine if our observed trends are common or specific to the Decapoda. As more mitogenomic data is made available, coupled with bioinformatics tools such as the MGO analysis feature in the MitoPhAST pipeline, it becomes increasingly feasible to conduct more complete and larger-scale comparisons for any animal group of interest in the future, the bottleneck now being to ensure that annotations in public database entries are accurate, which in this study was still a manual process. We have been fortunate to benefit from mitogenomes generated by various research groups that have contributed to enriching these resources for the order Decapoda but also realise that there is still need for new mitogenomes, carefully sampled to achieve a more balanced representation of infraorders. More importantly, by demonstrating what is possible for large-scale comparative MGO analyses in this study, it is our hope to inspire the undertaking of future research equivalent to or surpassing the extent of this study, contributing as a collective to the eventual reformation of the field of mitochondrial genomics.
Data Availability
Assembled mitogenomes are available on NCBI repository (See Table 1 for GenBank SRA accession numbers). Raw datasets generated during and/or analysed during the current study are available from the corresponding author on reasonable request.
References
Ahyong, S. T. & O’Meally, D. Phylogeny of the Decapoda Reptantia: resolution using three molecular loci and morphology. The Raffles Bulletin of Zoology 52, 673–693 (2004).
Crandal, K. A., Harris, D. J. & Fetzner, J. W. The monophyletic origin of freshwater crayfish estimated from nuclear and mitochondrial DNA sequences. Proceedings of the Royal Society of London. Series B: Biological Sciences 267, 1679–1686, https://doi.org/10.1098/rspb.2000.1195 (2000).
Porter, M. L., Pérez-Losada, M. & Crandall, K. A. Model-based multi-locus estimation of decapod phylogeny and divergence times. Molecular Phylogenetics and Evolution 37, 355–369, https://doi.org/10.1016/j.ympev.2005.06.021 (2005).
Schultz, M. B. et al. Evolution underground: A molecular phylogenetic investigation of Australian burrowing freshwater crayfish (Decapoda: Parastacidae) with particular focus on Engaeus Erichson. Molecular Phylogenetics and Evolution 50, 580–598, https://doi.org/10.1016/j.ympev.2008.11.025 (2009).
Bracken, H. D., De Grave, S., Toon, A., Felder, D. L. & Crandall, K. A. Phylogenetic position, systematic status, and divergence time of the Procarididea (Crustacea: Decapoda). Zoologica Scripta 39, 198–212, https://doi.org/10.1111/j.1463-6409.2009.00410.x (2010).
Bracken, H. D., Toon, A., Felder, D. L. & Martin, J. W. The decapod tree of life: compiling the data and moving toward a consensus of decapod evolution. Arthropod Systematics & Phylogeny 67, 99–116 (2009).
Boisselier-Dubayle, M.-C. et al. The phylogenetic position of the ‘living fossils’ Neoglyphea and Laurentaeglyphea (Decapoda: Glypheidea). Comptes Rendus Biologies 333, 755–759, https://doi.org/10.1016/j.crvi.2010.08.007 (2010).
Gan, H. M., Schultz, M. B. & Austin, C. M. Integrated shotgun sequencing and bioinformatics pipeline allows ultra-fast mitogenome recovery and confirms substantial gene rearrangements in Australian freshwater crayfishes. BMC Evolutionary Biology 14, 19, https://doi.org/10.1186/1471-2148-14-19 (2014).
Hahn, C., Bachmann, L. & Chevreux, B. Reconstructing mitochondrial genomes directly from genomic next-generation sequencing reads—a baiting and iterative mapping approach. Nucleic Acids Research 41, e129–e129, https://doi.org/10.1093/nar/gkt371 (2013).
Bernt, M. et al. MITOS: Improved de novo metazoan mitochondrial genome annotation. Molecular Phylogenetics and Evolution 69, 313–319, https://doi.org/10.1016/j.ympev.2012.08.023 (2013).
Tan, M. H., Gan, H. M., Schultz, M. B. & Austin, C. M. MitoPhAST, a new automated mitogenomic phylogeny tool in the post-genomic era with a case study of 89 decapod mitogenomes including eight new freshwater crayfish mitogenomes. Molecular Phylogenetics and Evolution 85, 180–188, https://doi.org/10.1016/j.ympev.2015.02.009 (2015).
Wyman, S. K., Jansen, R. K. & Boore, J. L. Automatic annotation of organellar genomes with DOGMA. Bioinformatics 20, 3252–3255, https://doi.org/10.1093/bioinformatics/bth352 (2004).
Ratnasingham, S. & Hebert, P. D. N. BOLD: The Barcode of Life Data System (http://www.barcodinglife.org). Molecular Ecology Notes 7, 355–364, https://doi.org/10.1111/j.1471-8286.2007.01678.x (2007).
Smith, D. R. The past, present and future of mitochondrial genomics: have we sequenced enough mtDNAs? Briefings in Functional Genomics 15, 47–54, https://doi.org/10.1093/bfgp/elv027 (2016).
Sanitá Lima, M., Woods, L. C., Cartwright, M. W. & Smith, D. R. The (in)complete organelle genome: exploring the use and nonuse of available technologies for characterizing mitochondrial and plastid chromosomes. Molecular Ecology Resources 16, 1279–1286, https://doi.org/10.1111/1755-0998.12585 (2016).
DeSalle, R. Comments on Smith (2015)—The past, present and future of mitochondrial genomics: have we sequenced enough mtDNAs. Briefings in Functional Genomics 15, 373–373, https://doi.org/10.1093/bfgp/elv052 (2016).
Timm, L. & Bracken-Grissom, H. D. The Forest for the Trees: Evaluating Molecular Phylogenies with an Emphasis on Higher-Level Decapoda. Journal of Crustacean Biology 35, 577–592, https://doi.org/10.1163/1937240X-00002371 (2015).
De Grave, S. et al. A Classification of Living and Fossil Genera of Decapod Crustaceans (2009).
Chávez, E. A. Potential production of the Caribbean spiny lobster (Decapoda, Palinura) fisheries. Crustaceana 82, 1393–1412 (2009).
Chavez, E. A. & Gorostieta, M. Bioeconomic assessment of the red spiny lobster fishery of Baja California, Mexico. California Cooperative Oceanic Fisheries Investigation Report 51, 153–161 (2010).
Dobson, M. Freshwater crabs in Africa. Freshwater Forum21(2010).
Cumberlidge, N. & Clark, P. F. In Studies on Brachyura: a Homage to Danièle Guinot 61–74 (Brill, 2010).
Hickerson, M. J. & Cunningham, C. W. Dramatic Mitochondrial Gene Rearrangements in the Hermit Crab Pagurus longicarpus (Crustacea, Anomura). Molecular Biology and Evolution 17, 639–644, https://doi.org/10.1093/oxfordjournals.molbev.a026342 (2000).
Miller, A. D., Murphy, N. P., Burridge, C. P. & Austin, C. M. Complete Mitochondrial DNA Sequences of the Decapod Crustaceans Pseudocarcinus gigas (Menippidae) and Macrobrachium rosenbergii (Palaemonidae). Marine Biotechnology 7, 339–349, https://doi.org/10.1007/s10126-004-4077-8 (2005).
Miller, A. D., Nguyen, T. T. T., Burridge, C. P. & Austin, C. M. Complete mitochondrial DNA sequence of the Australian freshwater crayfish, Cherax destructor (Crustacea: Decapoda: Parastacidae): a novel gene order revealed. Gene 331, 65–72, https://doi.org/10.1016/j.gene.2004.01.022 (2004).
Wilson, K., Cahill, V., Ballment, E. & Benzie, J. The Complete Sequence of the Mitochondrial Genome of the Crustacean Penaeus monodon: Are Malacostracan Crustaceans More Closely Related to Insects than to Branchiopods? Molecular Biology and Evolution 17, 863–874, https://doi.org/10.1093/oxfordjournals.molbev.a026366 (2000).
Yamauchi, M. M., Miya, M. U. & Nishida, M. Complete mitochondrial DNA sequence of the Japanese spiny lobster, Panulirus japonicus (Crustacea: Decapoda). Gene 295, 89–96, https://doi.org/10.1016/S0378-1119(02)00824-7 (2002).
Yamauchi, M. M., Miya, M. U. & Nishida, M. Complete mitochondrial DNA sequence of the swimming crab, Portunus trituberculatus (Crustacea: Decapoda: Brachyura). Gene 311, 129–135, https://doi.org/10.1016/S0378-1119(03)00582-1 (2003).
Ma, H. et al. The complete mitochondrial genome sequence and gene organization of the mud crab (Scylla paramamosain) with phylogenetic consideration. Gene 519, 120–127, https://doi.org/10.1016/j.gene.2013.01.028 (2013).
Shi, G. et al. The complete mitochondrial genomes of Umalia orientalis and Lyreidus brevifrons: The phylogenetic position of the family Raninidae within Brachyuran crabs. Marine Genomics 21, 53–61, https://doi.org/10.1016/j.margen.2015.02.002 (2015).
Basso, A. et al. The highly rearranged mitochondrial genomes of the crabs Maja crispata and Maja squinado (Majidae) and gene order evolution in Brachyura. Scientific Reports 7, 4096, https://doi.org/10.1038/s41598-017-04168-9 (2017).
Tan, M. H., Gan, H. M., Lee, Y. P., Poore, G. C. B. & Austin, C. M. Digging deeper: new gene order rearrangements and distinct patterns of codons usage in mitochondrial genomes among shrimps from the Axiidea, Gebiidea and Caridea (Crustacea: Decapoda). PeerJ 5, e2982, https://doi.org/10.7717/peerj.2982 (2017).
Ji, Y.-K. et al. Mitochondrial Genomes of two Brachyuran Crabs (Crustacea: Decapoda) and Phylogenetic Analysis. Journal of Crustacean Biology 34, 494–503, https://doi.org/10.1163/1937240X-00002252 (2014).
Gan, H. M. et al. More evolution underground: Accelerated mitochondrial substitution rate in Australian burrowing freshwater crayfishes (Decapoda: Parastacidae). Molecular Phylogenetics and Evolution 118, 88–98, https://doi.org/10.1016/j.ympev.2017.09.022 (2018).
Lin, F.-J. et al. Evolution and phylogeny of the mud shrimps (Crustacea: Decapoda) revealed from complete mitochondrial genomes. BMC Genomics 13, 631, https://doi.org/10.1186/1471-2164-13-631 (2012).
Shen, H., Braband, A. & Scholtz, G. Mitogenomic analysis of decapod crustacean phylogeny corroborates traditional views on their relationships. Molecular Phylogenetics and Evolution 66, 776–789, https://doi.org/10.1016/j.ympev.2012.11.002 (2013).
Tan, M. H. et al. More limbs on the tree: mitogenome characterisation and systematic position of ‘living fossil’ species Neoglyphea inopinata and Laurentaeglyphea neocaledonica (Decapoda: Glypheidea: Glypheidae). Invertebrate Systematics 32, 448–456, https://doi.org/10.1071/IS17050 (2018).
Shen, X., Tian, M., Yan, B. & Chu, K. Phylomitogenomics of Malacostraca (Arthropoda: Crustacea). Acta Oceanologica Sinica 34, 84–92, https://doi.org/10.1007/s13131-015-0583-1 (2015).
Ballard, J. W. O. & Whitlock, M. C. The incomplete natural history of mitochondria. Molecular Ecology 13, 729–744, https://doi.org/10.1046/j.1365-294X.2003.02063.x (2004).
Hurst, G. D. D. & Jiggins, F. M. Problems with mitochondrial DNA as a marker in population, phylogeographic and phylogenetic studies: the effects of inherited symbionts. Proceedings of the Royal Society B: Biological Sciences 272, 1525–1534, https://doi.org/10.1098/rspb.2005.3056 (2005).
Plazzi, F., Puccio, G. & Passamonti, M. Comparative Large-Scale Mitogenomics Evidences Clade-Specific Evolutionary Trends in Mitochondrial DNAs of Bivalvia. Genome Biology and Evolution 8, 2544–2564, https://doi.org/10.1093/gbe/evw187 (2016).
Babbucci, M., Basso, A., Scupola, A., Patarnello, T. & Negrisolo, E. Is It an Ant or a Butterfly? Convergent Evolution in the Mitochondrial Gene Order of Hymenoptera and Lepidoptera. Genome Biology and Evolution 6, 3326–3343, https://doi.org/10.1093/gbe/evu265 (2014).
Perna, N. T. & Kocher, T. D. Patterns of nucleotide composition at fourfold degenerate sites of animal mitochondrial genomes. Journal of Molecular Evolution 41, 353–358, https://doi.org/10.1007/bf00186547 (1995).
Reyes, A., Gissi, C., Pesole, G. & Saccone, C. Asymmetrical directional mutation pressure in the mitochondrial genome of mammals. Molecular Biology and Evolution 15, 957–966, https://doi.org/10.1093/oxfordjournals.molbev.a026011 (1998).
Shao, R., Dowton, M., Murrell, A. & Barker, S. C. Rates of Gene Rearrangement and Nucleotide Substitution Are Correlated in the Mitochondrial Genomes of Insects. Molecular Biology and Evolution 20, 1612–1619, https://doi.org/10.1093/molbev/msg176 (2003).
Chen, H., Sun, S., Norenburg, J. L. & Sundberg, P. Mutation and Selection Cause Codon Usage and Bias in Mitochondrial Genomes of Ribbon Worms (Nemertea). Plos One 9, e85631, https://doi.org/10.1371/journal.pone.0085631 (2014).
Weigert, A. et al. Evolution of mitochondrial gene order in Annelida. Molecular Phylogenetics and Evolution 94, 196–206, https://doi.org/10.1016/j.ympev.2015.08.008 (2016).
Guerra, D., Ghiselli, F. & Passamonti, M. The largest unassigned regions of the male- and female-transmitted mitochondrial DNAs in Musculista senhousia (Bivalvia Mytilidae). Gene 536, 316–325, https://doi.org/10.1016/j.gene.2013.12.005 (2014).
Eberhard, J. R. & Wright, T. F. Rearrangement and evolution of mitochondrial genomes in parrots. Molecular Phylogenetics and Evolution 94, 34–46, https://doi.org/10.1016/j.ympev.2015.08.011 (2016).
Min, X. J. & Hickey, D. A. DNA Asymmetric Strand Bias Affects the Amino Acid Composition of Mitochondrial Proteins. DNA Research 14, 201–206, https://doi.org/10.1093/dnares/dsm019 (2007).
Zhou, T., Shen, X., Irwin, D. M., Shen, Y. & Zhang, Y. Mitogenomic analyses propose positive selection in mitochondrial genes for high-altitude adaptation in galliform birds. Mitochondrion 18, 70–75, https://doi.org/10.1016/j.mito.2014.07.012 (2014).
Luo, Y. et al. Mitochondrial genome analysis of Ochotona curzoniae and implication of cytochrome c oxidase in hypoxic adaptation. Mitochondrion 8, 352–357, https://doi.org/10.1016/j.mito.2008.07.005 (2008).
Eo, S. H. & DeWoody, J. A. Evolutionary rates of mitochondrial genomes correspond to diversification rates and to contemporary species richness in birds and reptiles. Proceedings of the Royal Society B: Biological Sciences 277, 3587–3592, https://doi.org/10.1098/rspb.2010.0965 (2010).
Bernt, M., Merkle, D. & Middendorf, M. An Algorithm for Inferring Mitogenome Rearrangements in a Phylogenetic Tree. 143–157 (2008).
Bernt, M. et al. CREx: inferring genomic rearrangements based on common intervals. Bioinformatics 23, 2957–2958, https://doi.org/10.1093/bioinformatics/btm468 (2007).
Yoshizawa, K. et al. Mitochondrial phylogenomics and genome rearrangements in the barklice (Insecta: Psocodea). Molecular Phylogenetics and Evolution 119, 118–127, https://doi.org/10.1016/j.ympev.2017.10.014 (2018).
Tan, M. H., Gan, H. M., Lee, Y. P. & Austin, C. M. The complete mitogenome of the Morton Bay bug Thenus orientalis (Lund, 1793) (Crustacea: Decapoda: Scyllaridae) from a cooked sample and a new mitogenome order for the Decapoda. Mitochondrial DNA Part A 27, 1277–1278, https://doi.org/10.3109/19401736.2014.945554 (2016).
Gan, H. Y., Gan, H. M., Tan, M. H., Lee, Y. P. & Austin, C. M. The complete mitogenome of the hermit crab Clibanarius infraspinatus (Hilgendorf, 1869), (Crustacea; Decapoda; Diogenidae) – a new gene order for the Decapoda. Mitochondrial DNA Part A 27, 4099–4100, https://doi.org/10.3109/19401736.2014.1003862 (2016).
Bai, J. et al. The complete mitochondrial genome of Huananpotamon lichuanense (Decapoda: Brachyura) with phylogenetic implications for freshwater crabs. Gene 646, 217–226, https://doi.org/10.1016/j.gene.2018.01.015 (2018).
Jia, X.-n et al. The complete mitochondrial genome of Somanniathelphusa boyangensis and phylogenetic analysis of Genus Somanniathelphusa (Crustacea: Decapoda: Parathelphusidae). Plos One 13, e0192601, https://doi.org/10.1371/journal.pone.0192601 (2018).
Ahn, D.-H., Min, G.-S., Park, J.-K. & Kim, S. The complete mitochondrial genome of the red-banded lobster Metanephrops thomsoni (Crustacea, Astacidea, Nephropidae): a novel gene order. Mitochondrial DNA Part A 27, 2663–2664, https://doi.org/10.3109/19401736.2015.1043536 (2016).
Cameron, S. L. Insect mitochondrial genomics: implications for evolution and phylogeny. Annual review of entomology 59, 95–117 (2014).
Zhang, P. et al. Efficient Sequencing of Anuran mtDNAs and a Mitogenomic Exploration of the Phylogeny and Evolution of Frogs. Molecular Biology and Evolution 30, 1899–1915, https://doi.org/10.1093/molbev/mst091 (2013).
Gaitán-Espitia, J. D., Solano-Iguaran, J. J., Tejada-Martinez, D. & Quintero-Galvis, J. F. Mitogenomics of electric rays: evolutionary considerations within Torpediniformes (Batoidea; Chondrichthyes). Zoological Journal of the Linnean Society 178, 257–266, https://doi.org/10.1111/zoj.12417 (2016).
Amaral, C. R. L., Pereira, F., Silva, D. A., Amorim, A. & de Carvalho, E. F. The mitogenomic phylogeny of the Elasmobranchii (Chondrichthyes). Mitochondrial DNA Part A 29, 867–878, https://doi.org/10.1080/24701394.2017.1376052 (2018).
Tan, M. H. et al. ORDER within the chaos: Insights into phylogenetic relationships within the Anomura (Crustacea: Decapoda) from mitochondrial sequences and gene order rearrangements. Molecular Phylogenetics and Evolution 127, 320–331, https://doi.org/10.1016/j.ympev.2018.05.015 (2018).
Boore, J. L., Lavrov, D. V. & Brown, W. M. Gene translocation links insects and crustaceans. Nature 392, 667, https://doi.org/10.1038/33577 (1998).
Dowton, M. & Austin, A. D. Evolutionary dynamics of a mitochondrial rearrangement “hot spot” in the Hymenoptera. Molecular Biology and Evolution 16, 298–309, https://doi.org/10.1093/oxfordjournals.molbev.a026111 (1999).
Morrison, C. L. et al. Mitochondrial gene rearrangements confirm the parallel evolution of the crab-like form. Proceedings of the Royal Society of London. Series B: Biological Sciences 269, 345–350, https://doi.org/10.1098/rspb.2001.1886 (2002).
Tucker, L. R. & Lewis, C. A reliability coefficient for maximum likelihood factor analysis. Psychometrika 38, 1–10, https://doi.org/10.1007/BF02291170 (1973).
Hu, L. T. & Bentler, P. M. Cutoff criteria for fit indexes in covariance structure analysis: Conventional criteria versus new alternatives. Structural Equation Modeling: A Multidisciplinary Journal 6, 1–55, https://doi.org/10.1080/10705519909540118 (1999).
Andersen, J. C. & Mills, N. J. DNA Extraction from Museum Specimens of Parasitic Hymenoptera. Plos One 7, e45549, https://doi.org/10.1371/journal.pone.0045549 (2012).
Tin, M. M.-Y., Economo, E. P. & Mikheyev, A. S. Sequencing Degraded DNA from Non-Destructively Sampled Museum Specimens for RAD-Tagging and Low-Coverage Shotgun Phylogenetics. Plos One 9, e96793, https://doi.org/10.1371/journal.pone.0096793 (2014).
Aznar-Cormano, L. et al. An improved taxonomic sampling is a necessary but not sufficient condition for resolving inter-families relationships in Caridean decapods. Genetica 143, 195–205, https://doi.org/10.1007/s10709-014-9807-0 (2015).
Besnard, G. et al. Valuing museum specimens: high-throughput DNA sequencing on historical collections of New Guinea crowned pigeons (Goura). Biological Journal of the Linnean Society 117, 71–82, https://doi.org/10.1111/bij.12494 (2016).
McCormack, J. E., Tsai, W. L. E. & Faircloth, B. C. Sequence capture of ultraconserved elements from bird museum specimens. Molecular Ecology Resources 16, 1189–1203, https://doi.org/10.1111/1755-0998.12466 (2016).
Timmermans, M. J. T. N., Viberg, C., Martin, G., Hopkins, K. & Vogler, A. P. Rapid assembly of taxonomically validated mitochondrial genomes from historical insect collections. Biological Journal of the Linnean Society 117, 83–95, https://doi.org/10.1111/bij.12552 (2016).
Dixon, C., Ahyong, S. & Schram, F. A new hypothesis of decapod phylogeny. Crustaceana 76, 935–975, https://doi.org/10.1163/156854003771997846 (2003).
Tsang, L. M., Ma, K. Y., Ahyong, S. T., Chan, T. Y. & Chu, K. H. Phylogeny of Decapoda using two nuclear protein-coding genes: Origin and evolution of the Reptantia. Molecular Phylogenetics and Evolution 48, 359–368, https://doi.org/10.1016/j.ympev.2008.04.009 (2008).
Schram, F. & Ahyong, S. The higher affinities of Neoglyphea inopinata in particular and the Glypheoidea (Decapoda, Reptantia) in general. Crustaceana 75, 629–635, https://doi.org/10.1163/156854002760095651 (2002).
Bracken-Grissom, H. D. et al. The Emergence of Lobsters: Phylogenetic Relationships, Morphological Evolution and Divergence Time Comparisons of an Ancient Group (Decapoda: Achelata, Astacidea, Glypheidea, Polychelida). Systematic Biology 63, 457–479, https://doi.org/10.1093/sysbio/syu008 (2014).
De Grave, S. & Fransen, C. Carideorum catalogus: the recent species of the dendrobranchiate, stenopodidean, procarididean and caridean shrimps (Crustacea: Decapoda). Zoologische Mededelingen (2011).
Lemmon, A. R., Emme, S. A. & Lemmon, E. M. Anchored Hybrid Enrichment for Massively High-Throughput Phylogenomics. Systematic Biology 61, 727–744, https://doi.org/10.1093/sysbio/sys049 (2012).
Wolfe, J. M. et al. A phylogenomic framework, evolutionary timeline and genomic resources for comparative studies of decapod crustaceans. Proceedings of the Royal Society B: Biological Sciences 286, 20190079, https://doi.org/10.1098/rspb.2019.0079 (2019).
Grandjean, F. et al. Rapid recovery of nuclear and mitochondrial genes by genome skimming from Northern Hemisphere freshwater crayfish. Zoologica Scripta 46, 718–728, https://doi.org/10.1111/zsc.12247 (2017).
Schwentner, M., Richter, S., Rogers, D. C. & Giribet, G. Tetraconatan phylogeny with special focus on Malacostraca and Branchiopoda: highlighting the strength of taxon-specific matrices in phylogenomics. Proceedings of the Royal Society B: Biological Sciences 285, 20181524, https://doi.org/10.1098/rspb.2018.1524 (2018).
Macey, J. R., Papenfuss, T. J., Kuehl, J. V., Fourcade, H. M. & Boore, J. L. Phylogenetic relationships among amphisbaenian reptiles based on complete mitochondrial genomic sequences. Molecular Phylogenetics and Evolution 33, 22–31, https://doi.org/10.1016/j.ympev.2004.05.003 (2004).
Gissi, C., Iannelli, F. & Pesole, G. Evolution of the mitochondrial genome of Metazoa as exemplified by comparison of congeneric species. Heredity 101, 301, https://doi.org/10.1038/hdy.2008.62, https://www.nature.com/articles/hdy200862#supplementary-information (2008).
Schubart, C. D., Cannicci, S., Vannini, M. & Fratini, S. Molecular phylogeny of grapsoid crabs (Decapoda, Brachyura) and allies based on two mitochondrial genes and a proposal for refraining from current superfamily classification. Journal of Zoological Systematics and Evolutionary Research 44, 193–199, https://doi.org/10.1111/j.1439-0469.2006.00354.x (2006).
Kitaura, J., Wada, K. & Nishida, M. Molecular Phylogeny of Grapsoid and Ocypodoid Crabs with Special Reference to the Genera Metaplax and Macrophthalmus. Journal of Crustacean Biology 22, 682–693, https://doi.org/10.1163/20021975-99990281 (2002).
Tsang, L. M. et al. Evolutionary History of True Crabs (Crustacea: Decapoda: Brachyura) and the Origin of Freshwater Crabs. Molecular Biology and Evolution 31, 1173–1187, https://doi.org/10.1093/molbev/msu068 (2014).
Boore, J. L. & Brown, W. M. Big trees from little genomes: mitochondrial gene order as a phylogenetic tool. Current Opinion in Genetics &. Development 8, 668–674, https://doi.org/10.1016/S0959-437X(98)80035-X (1998).
Mindell, D. P., Sorenson, M. D. & Dimcheff, D. E. Multiple independent origins of mitochondrial gene order in birds. Proceedings of the National Academy of Sciences 95, 10693–10697 (1998).
Stöger, I. et al. Monoplacophoran mitochondrial genomes: convergent gene arrangements and little phylogenetic signal. BMC Evolutionary Biology 16, 274, https://doi.org/10.1186/s12862-016-0829-3 (2016).
Perseke, M. et al. Evolution of mitochondrial gene orders in echinoderms. Molecular Phylogenetics and Evolution 47, 855–864, https://doi.org/10.1016/j.ympev.2007.11.034 (2008).
Moret, B. M. E., Wang, L.-S., Warnow, T. & Wyman, S. K. New approaches for reconstructing phylogenies from gene order data. Bioinformatics 17, S165–S173, https://doi.org/10.1093/bioinformatics/17.suppl_1.S165 (2001).
Nakajima, Y. et al. The mitochondrial genome sequence of a deep-sea, hydrothermal vent limpet, Lepetodrilus nux, presents a novel vetigastropod gene arrangement. Marine Genomics 28, 121–126, https://doi.org/10.1016/j.margen.2016.04.005 (2016).
Zhuang, X. & Cheng, C. H. C. ND6 Gene “Lost” and Found: Evolution of Mitochondrial Gene Rearrangement in Antarctic Notothenioids. Molecular Biology and Evolution 27, 1391–1403, https://doi.org/10.1093/molbev/msq026 (2010).
Tshudy, D. & Sorhannus, U. L. F. Pectinate claws in decapod crustaceans: convergence in four lineages. Journal of Paleontology 74, 474–486, https://doi.org/10.1666/0022-3360(2000)074<0474:PCIDCC>2.0.CO;2 (2000).
Murphy, N. P. & Austin, C. M. Phylogenetic relationships of the globally distributed freshwater prawn genus Macrobrachium (Crustacea: Decapoda: Palaemonidae): biogeography, taxonomy and the convergent evolution of abbreviated larval development. Zoologica Scripta 34, 187–197, https://doi.org/10.1111/j.1463-6409.2005.00185.x (2005).
Tsang, L. M., Chan, T.-Y., Ahyong, S. T. & Chu, K. H. Hermit to King, or Hermit to All: Multiple Transitions to Crab-like Forms from Hermit Crab Ancestors. Systematic Biology 60, 616–629, https://doi.org/10.1093/sysbio/syr063 (2011).
Kankondi, S. L., McQuaid, C. D. & Tagliarolo, M. Influence of respiratory mode on the thermal tolerance of intertidal limpets. Plos One 13, e0203555, https://doi.org/10.1371/journal.pone.0203555 (2018).
Greenaway, P. Terrestrial adaptations in the anomura (Crustacea: Decapoda). Memoirs of Museum Victoria 60, 13–26 (2003).
Vereshchagina, K. P. et al. Salinity modulates thermotolerance, energy metabolism and stress response in amphipods Gammarus lacustris. PeerJ 4, e2657, https://doi.org/10.7717/peerj.2657 (2016).
Frank, T. M. & Widder, E. A. Comparative study of behavioral-sensitivity thresholds to near-UV and blue-green light in deep-sea crustaceans. Marine Biology 121, 229–235, https://doi.org/10.1007/bf00346730 (1994).
Hand, S. C. & Somero, G. N. Energy metabolism pathways of hydrothermal vent animals: adaptations to a food-rich and sulfide-rich deep-sea environment. The Biological Bulletin 165, 167–181, https://doi.org/10.2307/1541362 (1983).
Joseph, A.-M., Pilegaard, H., Litvintsev, A., Leick, L. & Hood, D. A. Control of gene expression and mitochondrial biogenesis in the muscular adaptation to endurance exercise. Essays In Biochemistry 42, 13, https://doi.org/10.1042/bse0420013 (2006).
D’Souza, A. R. & Minczuk, M. Mitochondrial transcription and translation: overview. Essays In Biochemistry 62, 309, https://doi.org/10.1042/EBC20170102 (2018).
Porat-Shliom, N., Harding, O. J., Malec, L., Narayan, K. & Weigert, R. Mitochondrial Populations Exhibit Differential Dynamic Responses to Increased Energy Demand during Exocytosis. In Vivo. iScience 11, 440–449, https://doi.org/10.1016/j.isci.2018.12.036 (2019).
Chen, J. et al. Characterization of four new mitogenomes from Ocypodoidea & Grapsoidea, and phylomitogenomic insights into thoracotreme evolution. Gene 675, 27–35, https://doi.org/10.1016/j.gene.2018.06.088 (2018).
Sun, Se, Hui, M., Wang, M. & Sha, Z. The complete mitochondrial genome of the alvinocaridid shrimp Shinkaicaris leurokolos (Decapoda, Caridea): Insight into the mitochondrial genetic basis of deep-sea hydrothermal vent adaptation in the shrimp. Comparative Biochemistry and Physiology Part D: Genomics and Proteomics 25, 42–52, https://doi.org/10.1016/j.cbd.2017.11.002 (2018).
Grande, C., Templado, J. & Zardoya, R. Evolution of gastropod mitochondrial genome arrangements. BMC Evolutionary Biology 8, 61, https://doi.org/10.1186/1471-2148-8-61 (2008).
Tsang, L. M., Shen, X., Cheang, C. C., Chu, K. H. & Chan, B. K. K. Gene rearrangement and sequence analysis of mitogenomes suggest polyphyly of Archaeobalanid and Balanid barnacles (Cirripedia: Balanomorpha). Zoologica Scripta 46, 729–739, https://doi.org/10.1111/zsc.12246 (2017).
Oceguera-Figueroa, A. et al. Comparative Mitogenomics of Leeches (Annelida: Clitellata): Genome Conservation and Placobdella-Specific trnD Gene Duplication. Plos One 11, e0155441, https://doi.org/10.1371/journal.pone.0155441 (2016).
Miya, M. et al. Major patterns of higher teleostean phylogenies: a new perspective based on 100 complete mitochondrial DNA sequences. Molecular Phylogenetics and Evolution 26, 121–138, https://doi.org/10.1016/S1055-7903(02)00332-9 (2003).
Mueller, R. L. & Boore, J. L. Molecular Mechanisms of Extensive Mitochondrial Gene Rearrangement in Plethodontid Salamanders. Molecular Biology and Evolution 22, 2104–2112, https://doi.org/10.1093/molbev/msi204 (2005).
Xia, Y. et al. The evolution of mitochondrial genomes in modern frogs (Neobatrachia): nonadaptive evolution of mitochondrial genome reorganization. BMC Genomics 15, 691, https://doi.org/10.1186/1471-2164-15-691 (2014).
Satoh, T. P., Miya, M., Mabuchi, K. & Nishida, M. Structure and variation of the mitochondrial genome of fishes. BMC Genomics 17, 719, https://doi.org/10.1186/s12864-016-3054-y (2016).
Sheffield, N. C., Song, H., Cameron, S. L. & Whiting, M. F. A Comparative Analysis of Mitochondrial Genomes in Coleoptera (Arthropoda: Insecta) and Genome Descriptions of Six New Beetles. Molecular Biology and Evolution 25, 2499–2509, https://doi.org/10.1093/molbev/msn198 (2008).
Bronstein, O. & Kroh, A. The first mitochondrial genome of the model echinoid Lytechinus variegatus and insights into Odontophoran phylogenetics. Genomics, https://doi.org/10.1016/j.ygeno.2018.04.008 (2018).
Lagisz, M., Poulin, R. & Nakagawa, S. You are where you live: parasitic nematode mitochondrial genome size is associated with the thermal environment generated by hosts. Journal of Evolutionary Biology 26, 683–690, https://doi.org/10.1111/jeb.12068 (2013).
Lavrov, D. V. & Pett, W. Animal Mitochondrial DNA as We Do Not Know It: mt-Genome Organization and Evolution in Nonbilaterian Lineages. Genome Biology and Evolution 8, 2896–2913, https://doi.org/10.1093/gbe/evw195 (2016).
Bernt, M. et al. A comprehensive analysis of bilaterian mitochondrial genomes and phylogeny. Molecular Phylogenetics and Evolution 69, 352–364, https://doi.org/10.1016/j.ympev.2013.05.002 (2013).
Plazzi, F., Puccio, G. & Passamonti, M. Burrowers from the Past: Mitochondrial Signatures of Ordovician Bivalve Infaunalization. Genome Biology and Evolution 9, 956–967, https://doi.org/10.1093/gbe/evx051 (2017).
Song, S.-N., Tang, P., Wei, S.-J. & Chen, X.-X. Comparative and phylogenetic analysis of the mitochondrial genomes in basal hymenopterans. Scientific Reports 6, 20972, https://doi.org/10.1038/srep20972 (2016).
Tang, B.-P. et al. Characterisation of the complete mitochondrial genome of Helice wuana (Grapsoidea: Varunidae) and comparison with other Brachyuran crabs. Genomics 110, 221–230, https://doi.org/10.1016/j.ygeno.2017.10.001 (2018).
Segawa, R. D. & Aotsuka, T. The mitochondrial genome of the Japanese freshwater crab, Geothelphusa dehaani (Crustacea: Brachyura): Evidence for its evolution via gene duplication. Gene 355, 28–39, https://doi.org/10.1016/j.gene.2005.05.020 (2005).
Wang, Y. et al. The complete mitochondrial genome of freshwater crab Sinopotamon xiushuiense (Decapoda: brachyura: Potamoidea). Mitochondrial DNA Part B 1, 750–752, https://doi.org/10.1080/23802359.2016.1209094 (2016).
Ghikas, D. V., Kouvelis, V. N. & Typas, M. A. The complete mitochondrial genome of the entomopathogenic fungus Metarhizium anisopliae var. anisopliae: gene order and trn gene clusters reveal a common evolutionary course for all Sordariomycetes, while intergenic regions show variation. Archives of Microbiology 185, 393, https://doi.org/10.1007/s00203-006-0104-x (2006).
Hou, Y. et al. Complete mitochondrial genome of Ark shell Scapharca subcrenata. Mitochondrial DNA Part A 27, 939–940, https://doi.org/10.3109/19401736.2014.926495 (2016).
Clare, E. L. et al. Diversity: Applications of a Sentinel Gene Approach. Journal of Molecular Evolution 66, 362–367, https://doi.org/10.1007/s00239-008-9088-2 (2008).
Foster, P. G., Jermiin, L. S. & Hickey, D. A. Nucleotide Composition Bias Affects Amino Acid Content in Proteins Coded by Animal Mitochondria. Journal of Molecular Evolution 44, 282–288, https://doi.org/10.1007/pl00006145 (1997).
Sun, Se, Li, Q., Kong, L. & Yu, H. Multiple reversals of strand asymmetry in molluscs mitochondrial genomes, and consequences for phylogenetic inferences. Molecular Phylogenetics and Evolution 118, 222–231, https://doi.org/10.1016/j.ympev.2017.10.009 (2018).
Castellana, S., Vicario, S. & Saccone, C. Evolutionary Patterns of the Mitochondrial Genome in Metazoa: Exploring the Role of Mutation and Selection in Mitochondrial Protein–Coding Genes. Genome Biology and Evolution 3, 1067–1079, https://doi.org/10.1093/gbe/evr040 (2011).
Chu, K., Tsang, L., Ma, K., Chan, T. & Ng, P. In Decapod crustacean phylogenetics 101–112 (CRC Press, 2016).
Robles, R., Tudge, C. C., Dworschak, P. C., Poore, G. & Felder, D. Molecular phylogeny of the Thalassinidea based on nuclear and mitochondrial genes. Decapod crustacean phylogenetics 18, 309 citation_lastpage= 326 (2009).
Poore, G. C. A phylogeny of the families of Thalassinidea (Crustacea: Decapoda) with keys to families and genera. Memoirs of the Museum of Victoria 54, 79–120 (1994).
Scholtz, G. & Richter, S. Phylogenetic systematics of the reptantian Decapoda (Crustacea, Malacostraca). Zoological Journal of the Linnean Society 113, 289–328, https://doi.org/10.1111/j.1096-3642.1995.tb00936.x (1995).
Cunningham, C. W., Blackstone, N. W. & Buss, L. W. Evolution of king crabs from hermit crab ancestors. Nature 355, 539, https://doi.org/10.1038/355539a0 (1992).
Noever, C. & Glenner, H. The origin of king crabs: hermit crab ancestry under the magnifying glass. Zoological Journal of the Linnean Society 182, 300–318, https://doi.org/10.1093/zoolinnean/zlx033 (2018).
Ahyong, S. T., Schnabel, K. E. & Maas, E. W. Anomuran phylogeny: new insights from molecular data. Decapod crustacean phylogenetics 18, 16 (2009).
Schnabel, K. E., Ahyong, S. T. & Maas, E. W. Galatheoidea are not monophyletic – Molecular and morphological phylogeny of the squat lobsters (Decapoda: Anomura) with recognition of a new superfamily. Molecular Phylogenetics and Evolution 58, 157–168, https://doi.org/10.1016/j.ympev.2010.11.011 (2011).
Ahyong, S. T., Baba, K., Macpherson, E. & Poore, G. C. A new classification of the Galatheoidea (Crustacea: Decapoda: Anomura). Zootaxa 2676, 57–68 (2010).
McLaughlin, P. A., Lemaitre, R. & Sorhannus, U. Hermit Crab Phylogeny: A Reappraisal and Its “Fall-Out”. Journal of Crustacean Biology 27, 97–115, https://doi.org/10.1651/S-2675.1 (2007).
Masta, S. E., Longhorn, S. J. & Boore, J. L. Arachnid relationships based on mitochondrial genomes: Asymmetric nucleotide and amino acid bias affects phylogenetic analyses. Molecular Phylogenetics and Evolution 50, 117–128, https://doi.org/10.1016/j.ympev.2008.10.010 (2009).
Sokolov, E. P. An improved method for DNA isolation from mucopolysaccharide-rich molluscan tissues. Journal of Molluscan Studies 66, 573–575, https://doi.org/10.1093/mollus/66.4.573 (2000).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120, https://doi.org/10.1093/bioinformatics/btu170 (2014).
Wood, D. E. & Salzberg, S. L. Kraken: ultrafast metagenomic sequence classification using exact alignments. Genome Biology 15, R46, https://doi.org/10.1186/gb-2014-15-3-r46 (2014).
Peng, Y., Leung, H. C. M., Yiu, S. M. & Chin, F. Y. L. IDBA-UD: a de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinformatics 28, 1420–1428, https://doi.org/10.1093/bioinformatics/bts174 (2012).
Clark, K., Karsch-Mizrachi, I., Lipman, D. J., Ostell, J. & Sayers, E. W. GenBank. Nucleic Acids Research 44, D67–D72, https://doi.org/10.1093/nar/gkv1276 (2016).
Abascal, F., Zardoya, R. & Telford, M. J. TranslatorX: multiple alignment of nucleotide sequences guided by amino acid translations. Nucleic Acids Research 38, W7–W13, https://doi.org/10.1093/nar/gkq291 (2010).
Nguyen, L.-T., Schmidt, H. A., von Haeseler, A. & Minh, B. Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Molecular Biology and Evolution 32, 268–274, https://doi.org/10.1093/molbev/msu300 (2015).
Kück, P. & Longo, G. C. FASconCAT-G: extensive functions for multiple sequence alignment preparations concerning phylogenetic studies. Frontiers in Zoology 11, 81, https://doi.org/10.1186/s12983-014-0081-x (2014).
Katoh, K. & Standley, D. M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Molecular Biology and Evolution 30, 772–780, https://doi.org/10.1093/molbev/mst010 (2013).
Castresana, J. Selection of Conserved Blocks from Multiple Alignments for Their Use in Phylogenetic Analysis. Molecular Biology and Evolution 17, 540–552, https://doi.org/10.1093/oxfordjournals.molbev.a026334 (2000).
Scordis, P., Flower, D. R. & Attwood, T. K. FingerPRINTScan: intelligent searching of the PRINTS motif database. Bioinformatics 15, 799–806, https://doi.org/10.1093/bioinformatics/15.10.799 (1999).
Eddy, S. R. Accelerated Profile HMM Searches. Plos Computational Biology 7, e1002195, https://doi.org/10.1371/journal.pcbi.1002195 (2011).
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. Journal of Molecular Biology 215, 403–410, https://doi.org/10.1016/S0022-2836(05)80360-2 (1990).
Vereshchaka, A. L., Kulagin, D. N. & Lunina, A. A. A phylogenetic study of krill (Crustacea: Euphausiacea) reveals new taxa and co-evolution of morphological characters. Cladistics 0, https://doi.org/10.1111/cla.12239 (2018).
Minh, B. Q., Nguyen, M. A. T. & von Haeseler, A. Ultrafast approximation for phylogenetic bootstrap. Molecular biology and evolution 30, 1188–1195, https://doi.org/10.1093/molbev/mst024 (2013).
Guindon, S. et al. New Algorithms and Methods to Estimate Maximum-Likelihood Phylogenies: Assessing the Performance of PhyML 3.0. Systematic Biology 59, 307–321, https://doi.org/10.1093/sysbio/syq010 (2010).
Letunic, I. & Bork, P. Interactive tree of life (iTOL) v3: an online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Research 44, W242–W245, https://doi.org/10.1093/nar/gkw290 (2016).
Rice, P., Longden, I. & Bleasby, A. EMBOSS: the European molecular biology open software suite. Trends in genetics 16, 276–277 (2000).
Sharp, P. M. & Li, W.-H. The codon adaptation index-a measure of directional synonymous codon usage bias, and its potential applications. Nucleic Acids Research 15, 1281–1295, https://doi.org/10.1093/nar/15.3.1281 (1987).
Addinsoft, S. XLSTAT V. 2015.1. 01: Data Analysis and Statistics Software for Microsoft Excel. Addinsoft: Paris, France (2015).
Smith, M. D. et al. Less Is More: An Adaptive Branch-Site Random Effects Model for Efficient Detection of Episodic Diversifying Selection. Molecular Biology and Evolution 32, 1342–1353, https://doi.org/10.1093/molbev/msv022 (2015).
Pond, S. L. K. & Muse, S. V. in Statistical methods in molecular evolution 125–181 (Springer, 2005).
Bouckaert, R. et al. BEAST 2: A Software Platform for Bayesian Evolutionary Analysis. PLOS Computational Biology 10, e1003537, https://doi.org/10.1371/journal.pcbi.1003537 (2014).
Rambaut, A., Drummond, A. J., Xie, D., Baele, G. & Suchard, M. A. Posterior Summarization in Bayesian Phylogenetics Using Tracer 1.7. Systematic Biology 67, 901–904, https://doi.org/10.1093/sysbio/syy032 (2018).
Rambaut, A. FigTree v1. 4. (2012).
Acknowledgements
This work was supported by the Monash University Malaysia Tropical Medicine and Biology Platform, Deakin Genomics Centre and Ministry of Science and Technology, Taiwan. This is contribution # 146 from the Center for Coastal Oceans Research in the Institute of Water and Environment at Florida International University. The authors would like to thank Joanne Taylor and Genefor Walker-Smith (Museum Victoria), Laure Corbari and Paula Martin-Lefevre (Muséum national d’Histoire naturelle), as well as Shane Ahyong (Australian Museum) for the provision of museum tissue samples used in this study. The authors would also like to thank Frederic Grandjean, Alastair Richardson and Michael Hammer for their contribution of other samples. We also thank Kristina von Rintelen for her advice relating to freshwater shrimps (Atyidae).
Author information
Authors and Affiliations
Contributions
C.A. is the principal investigator and contributed to the concept and design of the study. C.A., H.B. and T.Y. provided samples for the experiment while M.T., H.G. and Y.L. performed the laboratory work. M.T. analysed the data, prepared figures and tables, and wrote the paper. All authors contributed to the critical review and revision of the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Tan, M.H., Gan, H.M., Lee, Y.P. et al. Comparative mitogenomics of the Decapoda reveals evolutionary heterogeneity in architecture and composition. Sci Rep 9, 10756 (2019). https://doi.org/10.1038/s41598-019-47145-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-47145-0
This article is cited by
-
The mitochondrial genome of Bottapotamon fukienense (Brachiura: Potamidae) is fragmented into two chromosomes
BMC Genomics (2024)
-
Drivers of interlineage variability in mitogenomic evolutionary rates in Platyhelminthes
Heredity (2024)
-
Morphological and histological description of the midgut caeca in true crabs (Malacostraca: Decapoda: Brachyura): origin, development and potential role
BMC Zoology (2022)
-
Exploring mitogenome evolution in Branchiopoda (Crustacea) lineages reveals gene order rearrangements in Cladocera
Scientific Reports (2022)
-
Comparative mitochondrial genome analysis of Varunidae and its phylogenetic implications
Acta Oceanologica Sinica (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
