
Abstract
Amyotrophic lateral sclerosis (ALS) is an incurable and fatal neurodegenerative disease. Increasing the chances of success for future clinical strategies requires more in-depth knowledge of the molecular basis underlying disease heterogeneity. We recently laid the foundation for a molecular taxonomy of ALS by whole-genome expression profiling of motor cortex from sporadic ALS (SALS) patients. Here, we analyzed copy number variants (CNVs) occurring in the same patients, by using a customized exon-centered comparative genomic hybridization array (aCGH) covering a large panel of ALS-related genes. A large number of novel and known disease-associated CNVs were detected in SALS samples, including several subgroup-specific loci, suggestive of a great divergence of two subgroups at the molecular level. Integrative analysis of copy number profiles with their associated transcriptomic data revealed subtype-specific genomic perturbations and candidate driver genes positively correlated with transcriptional signatures, suggesting a strong interaction between genomic and transcriptomic events in ALS pathogenesis. The functional analysis confirmed our previous pathway-based characterization of SALS subtypes and identified 24 potential candidates for genomic-based patient stratification. To our knowledge, this is the first comprehensive “omics” analysis of molecular events characterizing SALS pathology, providing a road map to facilitate genome-guided personalized diagnosis and treatments for this devastating disease.
Similar content being viewed by others

Introduction
Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disorder, characterized by progressive loss and degeneration of motor neurons in both the motor cortex, brainstem and spinal cord, and is usually fatal due to respiratory failure within 3–5 years of onset1. The disease has an incidence of 2.6 per 100,000 individuals-years and prevalence rates of around 6–7/100,000 in Europe, making it the most common adult-onset motor neuron disease2. About 5–10% of ALS cases show a family history (FALS), while the remainder of cases are classified as sporadic (SALS), and are probably associated to a polygenic and multifactorial etiology3,4,5.
The remarkable advances in genome technologies over the last years have led to a huge progress in deciphering the genes and pathways involved in ALS pathogenesis. From the discovery of the first ALS-associated gene SOD1, several candidate-gene or genome-wide association studies (GWAS) have identified multiple single-nucleotide polymorphisms (SNPs) affecting potentially ALS-associated genes, including C9orf72, TDP43, FUS, MATR3, UBQLN2, VCP and OPTN6,7,8,9. In this context, a recent large-scale genome-wide association study identified a common missense variant and several rare loss-of-function (LOF) mutations within the microtubule motor protein-encoding gene, KIF5A, as candidate ALS risk factors, further supporting perturbations in cytoskeletal function play an important role in ALS and offering a potential target for drug development10,11.
In addition to the contribution of SNPs, which account for only a limited number of familial and sporadic ALS cases, evidence suggests that other genomic variants, such as copy-number variations (CNVs), that change gene dose rather than gene function, may exert a more pronounced effect on the onset and rate of disease progression5,12,13. In particular, the involvement of CNVs in ALS susceptibility has been clearly highlighted in two ALS genome-wide association studies, where multiple rare CNVs were shown to represent a more important risk factor for SALS than common CNVs14,15.
The complexity of its molecular architecture has completely transformed the way we think about ALS, leading us to reconsider the traditional classification and therapeutic systems. In fact, despite intensive research efforts, the precise causes of ALS remain unknown and there is no cure for this devastating disease. The absence of effective treatments can be due in part by the complex and heterogeneous clinical, biochemical and molecular features of ALS, which is also supported by clinical studies on Edaravone (MCI-186), a free radical scavenger recently approved by FDA for ALS treatment showing effectiveness only in specific sub-cohorts of patients16. Developing a robust molecular disease portrait that can explain the heterogeneity of ALS is thus fundamental to improve our understanding of the precise molecular mechanisms underlying disease pathogenesis and develop effective treatments for patients.
Our research group has recently characterized the transcriptional profiles of motor cortex samples from control and SALS patients, grouping these on the basis of their similarities measured over the most “hypervariable genes” (9.646 genes with a standard deviation >1.5). Unsupervised hierarchical clustering analysis allowed to discriminate controls from SALS patients and clearly distinguished two greatly divergent SALS subtypes, each associated with differentially expressed genes (DEGs) and biological pathways5,17,18. In particular, the most representative functional processes deregulated in SALS1 were annotated as involved in the regulation of chemotaxis, immunity, and cell adhesion and communication. Deregulated genes in SALS2, in turn, were selectively associated with cytoskeleton organization, regulation of transport and mitochondrial oxidative phosphorylation5,17. While these findings are consistent with previous evidence about the crucial role of these pathogenetic mechanisms in ALS17,19,20,21,22,23,24, they suggest for the first time the differential involvement of these mechanisms in specific subsets of ALS patients, offering a useful starting point for the further development of personalized diagnostics and targeted therapies.
While our work lays the foundation for a molecular taxonomy of ALS, very little information is so far available from the single-omic analysis, which makes difficult to discriminate genes critical to ALS pathogenesis (driver genes) from non-relevant genes (passenger genes). An integrated and comprehensive view of multiple genomic data types (such as genome and transcriptome) may provide a powerful potential for defining disease subgroups and their molecular drivers, allowing for an overall understanding of the complex molecular networks that drive ALS pathogenesis at yet another level of systemic complexity.
In this study, we applied the customized exon-centric comparative genomic hybridization array (aCGH) NeuroArray platform, designed to target genes associated with ALS as well as genes associated with other neurological disorders25, to analyze copy number variants (CNVs) in 40 motor cortex samples of control (10) and SALS (30) patients, clinically and transcriptomically characterized in our previous work17,25. Next, we provided the first comprehensive integrative analysis of genomic aberrations with expression data derived from the same patients to identify specific chromosomal regions and genes with concordant alterations in DNA and RNA profiles that may represent promising key molecular candidates for SALS. Finally, functional pathway and network analyses were carried out to gain further insights into the molecular complexity of ALS and reveal novel and yet unrecognized biomarkers and therapeutic targets, potentially useful for the development of personalized medicine in ALS.
Results
Transcriptomically distinct SALS patient subgroups show specific copy number alterations
The customized exon-centric NeuroArray aCGH platform was used to identify DNA copy number alterations in 30 SALS patients and 10 controls. A total of 1472 CNVs were detected in SALS, including 780 losses and 692 gains (Fig. 1a). The chromosomal distribution of all CNVs across the 30 SALS genomes tested is plotted in Fig. 1b. To reduce individual heterogeneities and identify ALS-related significant CNVs, we focused on those that occurred in at least 10% SALS samples. Accordingly, a total of 488 significant CNVs ranging in size from 7 bp to 5.9 Mb were identified in SALS patients, including 271 losses and 217 gains (Fig. 1c, Supplementary Table 1). Recurrent CNVs were dispersed in the chromosome 1 to 22, with the most frequent amplifications (76.7%) found in chromosome 14, followed by amplifications in chromosome 17 (70%) (Supplementary Table 1). The most common linkage rate in SALS mapped to chromosome 20 with a frequency of 80%, followed by chromosome 1 with a frequency of 76.6% (Supplementary Table 1).

Significant copy number regions in SALS patient subgroups. (a) Graphical overview of CNV regions detected in SALS patients by NeuroArray platform. The bars represents the number of regions that may be involved in CNV detected in SALS patients (ADM-2 aberration filter: threshold = 6; Log2 ratio ≥ 0.5 and 3 consecutive interval probes), both with and without the assignment into the hierarchically determined two sets (SALS1 and SALS2). The blue bar represents the number of deleted regions and the red bar represents the number of amplified regions. (b) Chromosome distribution of CNVs detected with high-resolution custom exon-centered NeuroArray aCGH from SALS patients. The horizontal axis represents different chromosomes and the vertical axis represents the number of regions of each chromosome that may be involved in CNV. (c) Graphical overview of recurrent gains or losses (occurred in at least 10% of the SALS patients), both with and without the assignment into the hierarchically determined two sets (SALS1 and SALS2). The blue bar represents the number of deleted regions and the red bar represents the number of amplified regions that occurred at a high frequency (≥10%) in our cohort of SALS patients.
To identify subgroup-specific genomic signatures, we analyzed CNV events taking into account the previously characterized transcriptome-based stratification of SALS patients in the two subgroups, SALS1 and SALS2. Overall, 813 aberrant regions were associated with SALS1 and 659 with SALS2 patients (Fig. 1a). Among these, 335 CNVs (218 losses and 117 gains) were detected as frequently altered in SALS1 patients, while 308 (135 losses and 173 gains) were frequently associated with the SALS2 subgroup (Fig. 1c, Table 1 and Supplementary Tables 2–3). Interestingly, a large number of these recurrent amplifications and deletions were detected exclusively in SALS patients (absent in the control samples) (Table 2).
To investigate the reliability of our results and further confirm the potential functional implications of the detected CNVs in ALS pathogenesis, we interrogated our data for overlap with genomic aberrations previously associated to ALS cases available in publicly available databases (i.e., CNVD) and published PubMed literature. Highly similar genomic altered patterns were observed, supporting the functional importance of these regions in disease etiopathogenesis (Table 3).
Identifying CNV signature genes in SALS
To identify ALS driver genes from aberrant regions, the recurrent CNVs in SALS were annotated and filtered out for genes previously linked to ALS and other neurological diseases as causative and/or susceptibility factors and included in the NeuroArray design. A total of 406 significant CNV genes were obtained, including 251 duplications and 161 deletions (Fig. 2a, Supplementary Table 4). Among these, 36 were previously identified as ALS-linked genes (Supplementary Table 4). The same analysis was also performed on the most frequent subgroup-specific CNV regions, revealing 310 genes as the most significantly altered CNV-genes in SALS1 (137 duplicated and 174 deleted) and 454 genes in SALS2 patients (320 duplicated and 140 deleted). Among these, 28/310 SALS1 and 39/454 SALS2 CNV genes were already associated to ALS (Fig. 2a, Supplementary Tables 5 and 6).

Integrative analysis of DNA copy number and expression variation in SALS patients reveals a good number of overlapping ALS candidate genes. (a) Graphical overview of the most frequent (≥10%) CNV-embedded genes detected in SALS patients, both with and without the assignment into the hierarchically determined two sets (SALS1 and SALS2). (b,c) Venn diagrams compare the number of protein-coding genes obtained from CNV analyses with the genes found to be differentially expressed in SALS1 (b) and SALS2 (c) patients.
Integrated analysis of CNVs and gene expression profiling identify candidate ALS-driver genes
To determine whether genomic aberrations contribute to global gene expression patterns in SALS, the identified CNV genes were checked for overlap with the DEGs previously detected in the same patient cohort17. We identified 70 overlapping CNV genes (29 duplications and 42 deletions) that were also differentially expressed in SALS1 patients and 246 CNV-driven DEGs in SALS2 patients (173 duplicated and 76 deleted) (Fig. 2b,c, Supplementary Tables 7 and 8). Among these, 35 CNV-driven genes (50%) in SALS1 and 112 CNV-driven genes (45%) in SALS2 showed a positive association between gene expression and DNA copy number changes, including 77 up-regulated genes (24 in SALS1 and 53 in SALS2) and 70 down-regulated genes (11 in SALS1 and 59 in SALS2) (Fig. 2b,c, Supplementary Tables 7 and 8). Interestingly, several CNV-driven genes were SALS-patient specific (not detected in 10 controls) and most of them were previously linked to ALS.
To demonstrate that the correlation found in our work is meaningful, we also performed a “control experiment” in which we evaluated the overlap between the genomic and transcriptomic data between two random groups within the total SALS samples (different from SALS1 and SALS2). We observed a very low overlap between CNV genes and differentially expressed genes in these randomized disease-related subgroups, confirming the appropriateness/accuracy of our analysis (Supplementary Fig. 2 and Supplementary Table 14).
Computational systems biology analysis identified distinct drivers and pathways in SALS molecular subtypes
To gain further insights into the biological role of identified CNV-driven DEGs in SALS, functional annotation and pathway enrichment analyses were performed by using specialized bioinformatics tools and databases (i.e., Enrichr, IPA, Metacore).
According to GO analysis, the CNV-driven DEGs in SALS1 were significantly enriched in biological processes such as regulation of cellular component organization (GO:0051129, P value = 0.0002), DNA conformation change (GO:0071103, P value = 0.0004) and regulation of neuron death (GO:1901214, P value = 0.0012), whereas regulation of synaptic transmission (GO:0050804, P value = 3.45E−12) and learning and memory (GO:0007611, P value = 5.26E−12) were the most enriched in SALS2 (Fig. 3a, Supplementary Table 9). On the basis of molecular function, the CNV-driven DEGs in SALS1 prominently accumulated in small conjugating protein ligase binding (GO:0044389, P value = 0.0004) and ubiquitin protein ligase binding (GO:0031625, P value = 0.0004), while the CNV-driven DEGs in SALS2 were enriched in amino acid binding (GO:0016597, P value = 7.00126E−06) and transcription factor binding (GO:0008134, P value = 6.6232E-05) (Fig. 3a, Supplementary Table 9).

Functional enrichment analysis for GO and pathway map ontologies revealed significant biological processes associated with the candidate CNV-driven genes in SALS. (a) Pie charts represent the top 10 enriched (P < 0.05) GO terms for the 70 CNV-encompassed DEGs in SALS1 and SALS2 patients. The GO terms were subdivided into three GO categories: biological processes, molecular functions and cellular components. Enrichment analyses were performed using the Enrichment Analysis tool in Enrichr. For each category, GO terms or biological features represented in CNV-driven differently expressed genes are indicated. (b) Representation of the top 20 most significantly enriched (P value < 0.05) canonical pathway maps associated with the candidate CNV-driven genes in SALS1 and SALS2 patients. A histogram of statistical significance (−log P value) is shown: the list is arranged in descending order with the most significant pathways at the top. The analysis was performed using the MetaCore™ pathway analysis suite.
Further pathways enrichment analysis identified an immune/inflammatory response, cytoskeleton remodeling and apoptotic processes as the major deregulated processes in SALS (Fig. 3b, Supplementary Table 10). In particular, Huntington’s disease signaling, T cell activation, Apoptosis and survival signaling were the most significantly enriched pathways in both SALS patient subgroups, with AKT1, NFKB1 and SOS as the major key involved genes. In addition, ubiquitin-proteasome pathway, immune-cell mediated inflammatory response and apoptosis were significantly up-regulated in SALS1, while axonal guidance, oxidative stress and inflammatory intracellular signaling cascades were mainly up-regulated in SALS2 (Fig. 4a). Notably, the majority of the enriched pathway for CNV-driven DEGs in SALS patients represented processes already associated with ALS pathogenesis, such as immune response, cell adhesion and cell communication (Fig. 4a).

Definition of SALS subtype-specific genomic signature using pathway and network analyses. (a) A representative illustration showing the functional correlation between SALS associated CNV-driven genes and their biological processes. Interaction map represents the most promising candidate genes showing a positive correlation between gene expression and underlying genomic changes, grouped on the basis of the main biological processes associated with them. The map was created using the MetaCore Pathway Map Creator tool (GeneGo). Gene expression and CNV values are presented on the map as ‘thermometer-like’ figures with SALS1 patients data represented as thermometer #1 and SALS2 patients as #2. Genes associated with overexpression and CNV gain regions are labeled with red dots while genes associated with downregulated expression and homozygous or heterozygous deleted CNVs are labeled with blue dots. A detailed legend for the network objects is shown in the Supplementary Fig. 1. (b) Functional network of known and predicted interactions of the most promising candidate CNV-driven genes. The network was produced by the Search Tool for the Retrieval of Interacting Genes/Proteins (STRING) v10 (http://string-db.org/) using default settings. Proteins are represented by spheres. Lines linking proteins indicate evidence for interactions: a red line indicates the presence of gene fusion (genes that are sometimes fused into single open reading frames); a green line – gene neighborhood (genes that reside within 300 bp on the same strand in the genome); a blue line – co-occurrence (gene families whose occurrence patterns across genomes show similarities); a purple line - experimental evidence (interaction extracted from protein-protein interaction databases); a yellow line – text mining (interaction extracted from scientific literature); a light blue line - database (interaction extracted from curated databases); a black line – co-expression (proteins whose genes are co-expressed in the same or in other species).
To better understand the interactions of the CNV-driven genes and identify the best candidate genes in SALS, a protein-protein interaction (PPI) network analysis of their encoding products was performed, revealing a highly interconnected functional network, also including a greater number of ALS-associated genes (Fig. 4b). The PPI network consisted of 147 nodes and 2787 edges, including 46 CNV-driven DEGs in SALS. Node degree ≥10 was selected as the threshold. UBA52, RPS27A and HIST2H3A were selected as the hub genes.
To assess the value of CNV-driven genes as potential biomarkers for patient-specific diagnosis and prognosis, we reviewed and analyzed the literature on the CNV-driven genes exhibiting the same expression tendencies. A total of 24 candidate gene markers were selected, including 6 up-regulated CNV-driven DEGs in SALS1 and 18 deregulated (10 up-regulated and 8 down-regulated) CNV-driven genes in SALS2. Interestingly, some of these candidate genes (TIMP2, AKT1, MMP9, CST3, SMN1 and SMN2) were previously associated with susceptibility to ALS while the remaining 18 genes (GAA, KIF1A, MC1R, MECP2, ALPL, HSPG2, L1CAM, PLEC, STK11, CSF1, F2, GSTM3, TRAF2, HSPA5, HTT, IL6, LETMD1, SOAT1) represent novel candidate mediators for disease progression. The set of CNV-driver genes also included many patient-specific ‘druggable’ genes that may represent good candidates for the development of personalized, molecularly targeted therapies for SALS patients (Table 4).
Discussion
In this work, we reported the first fully integrated analysis of CNVs and gene expression profiling derived from the same SALS patients to provide a more comprehensive genomic framework for dissecting molecular heterogeneity of ALS and identify the DEGs with alterations in genomic segments that may represent novel potential markers and/or therapeutic targets.
Taking advantage of the custom-made NeuroArray platform, designed to uncover CNVs in clinically relevant genes for ALS and other neurological diseases25, we performed an exon-focused evaluation of structural imbalances occurring in motor cortex samples from 30 SALS and 10 control patients trascriptomically characterized in our previous work17. A large number of aberrations were detected in over 10% of SALS patients, with the highest number of gains documented at chromosomes 14 and 17, and the majority of losses found on the p arm of chromosome 1 and at cytoband 20q11.22-q11.23 (Fig. 1c and Supplementary Table 1). Notably, at 17p region the SREBF1, a gene encoding a lipogenic transcription factor whose expression levels were increased in the spinal cords of FALS and SALS patients as well as in ALS animal models, and whose direct causative role in excitotoxicity-induced neuronal cell death, has been extensively established26,27,28.
Our analysis also revealed distinct genomic signatures associated with two previously characterized transcriptome-based SALS subgroups. In particular, the loss of chromosome 1p was the most common chromosomal aberration in SALS1 (~90%), while 60–70% of the SALS2 patients showed simultaneous deletion of precise regions of chromosomes 3q and 18p (Fig. 1c, Table 1, Supplementary Table 1)29. It is interesting to note that genes in these CNV regions showed the same subgroup-specific expressional tendencies. In fact, the 1p33 deletion includes STIL, a gene involved in neural protection and survival and whose expression was down-regulated in SALS1, suggesting that a “loss-of-function” of this gene may contribute to render motor neurons vulnerable to excitotoxic insults in these patients30. Likewise, SALS2 patients carried the deletion of 18p11.22 that encompassed NDUFV2, one of the many components of the mitochondrial oxidative phosphorylation pathway, whose expression levels were decreased in the same patient subgroup. Defects in this subunit have been associated with altered energy production, mitochondrial dysfunction and oxidative stress, representing a risk factor for several neuronal diseases, including ALS31.
Compared with previous CNV reports32, some genomic aberrations identified in our study partially or totally overlap with those previously associated with ALS, further supporting them as disease susceptibility variants (Table 3). Among these, of particular interest is the deletion of chromosomal region 15q22.2-q22.31 that was detected exclusively in SALS patients and not altered in any controls in our study nor listed in the Database of Genomic Variants – DGV.
Besides strengthening previously reported results, we identified new potential susceptibility loci that were overrepresented in SALS patients and absent in controls (Table 2). Among these, we distinguished the deletion at the chromosome 1q32.2, which encompassed CR1 (also known as CD35), a member of the human regulator of complement activation gene cluster33,34. This gene encodes one of the major immune adherence receptors and plays an important role in immune complex processing and clearance via reducing activation of classical and alternative complement cascade activity. Deletion of CR1 is consistent with the lower mRNA expression level of this receptor in SALS patients, providing support for aberrant complement regulation as a part of ALS process and highlighting the potential use of complement molecules as disease biomarkers35,36. Another interesting association is represented by a 162.362-kb duplicated region in 1p36.12 spanning several neuronal genes, including the FBXO42 encoding an important member of the F-box protein family involved in the ubiquitin-proteasome system and already known to be associated with ALS (Table 2)37. Of particular interest, three CNV regions (17q12, 22q11.21; 6q13) were selectively detected in specific SALS patient subgroups and absent in all control individuals, providing the basis for a CNV-based molecular classification of the disease (Table 2). In particular, the deletion at the 6q13 region harbors the SLC17A5 gene encoding sialin, a vesicular excitatory amino acid transporter, whose loss-of-function leads to defect in myelin structure and function, contributing to the disruption of axonal integrity and the motor phenotype38,39. The increase in gene copy numbers for the 17q12 region encompassing several chemokines (i.e., CCL5 and CCL3) is consistent with the observation that high mRNA expression levels of these chemokines may increase activation of the inflammatory system and changes in blood-brain barrier permeability, two key mechanisms implicated in, and possibly aggravating, motor neuron damage40,41.
By integrating the analysis of CNVs and gene expression profiling in the same patients and tissue samples, we found that 71.2% of CNV genes were differentially expressed in SALS patients in comparison to controls, the majority of them were cluster specific, further suggestive of the great divergence of two SALS subgroups at the molecular level (Fig. 2b,c, Supplementary Tables 7 and 8). Among these genes, 49% showed a positive association between CNV and mRNA expression, suggesting them as potential driver genes in ALS. These included some genes (SYNE1, SDK1, EEF1D, GSDMD, TIMP2, CST3, ALAD, AKT1, EPHA4, SPAST, SMN1, SETX, ATXN2, TBK1, SPG11, TAF15, MMP9) previously reported as potential risk factors in ALS, as well as novel candidates whose association with ALS was previously unappreciated. These genes may represent overlapping genetic signatures among different neurological condition, providing additional features for exploring ALS pathogenesis. To name a few of those detected exclusively in SALS patients (i.e., absent from controls) are ALDH4A1, a component of the mitochondrial matrix contributing to protect cells from oxidative stress42; BIN1, the most significant late-onset susceptibility locus for Alzheimer’s disease whose alterations in expression levels and splicing seem to induce muscle weakness and T tubule alterations43, and RABGAP1L, a GTPase-activating protein whose loss-of-function exacerbates neuronal loss44.
On the other hand, our analysis identified a number of CNV genes, including known ALS-related genes (i.e., NIPA1, NAIP, VPS54 and GRIN3B), which do not appear to exert any apparent influence on expression levels, suggesting that the expression of these genes may be not gene-dose dependent and that they are likely to represent secondary ‘passenger’ events in ALS pathogenesis. Genes differently regulated at the transcriptomic and genomic level (i.e., showing no or negative correlation), in turn, could result from other, non-CNV-related regulatory mechanisms, such as those associated with gene mutation, promoter methylation, and non-coding RNA regulation (Fig. 2b,c, Supplementary Tables 7 and 8). An example is KIF5A, a gene encoding a neuronal kinesin heavy chain that acts as a molecular motor and whose genetic alteration was recently associated with ALS susceptibility10. SALS2 patients showed a selective downregulation of KIF5A not overlapping with any CNV, suggesting reduced mRNA expression of this gene is probably due to defective splicing events and/or loss-of-function variants rather than to deletion within its genomic region. It is interesting to note that in the same subgroup we also observed defects in axonal transport as well as a general downregulation of genes involved in mitochondrial oxidative phosphorylation machinery, suggesting a link between loss-of-function events in KIF5A and impaired transport and dysfunction of mitochondria in ALS17,45.
Integrated analysis of CNVs and corresponding expression data represents an effective approach to elucidate mediators and mechanisms involved in ALS. However, it has become clear that the process of motor neuronal degeneration is complex and requires many genomic alterations acting in concert. This also emerged by our analysis of PPI network constructed for CNV-driven DEGs that revealed a great number of close interconnections and identified three hub genes (UBA52, RPS27A and HIST2H3A) involved in inflammatory and immune responses (Fig. 4b)46. We also put forward a ‘systems biology’ analysis to identify biological processes and signaling pathways that were overly represented by CNV-driven genes in both SALS patient subgroups. Functional enrichment analysis of these candidate genes showed that they were mainly involved in immune/inflammatory signaling, neuronal migration, differentiation and survival, and neurite outgrowth, supporting the concept that these pathways may crosstalk with each other to drive the disease pathogenesis (Fig. 4a). In particular, Huntington’s disease (HD) signaling and Protein folding and processing were among the top canonical pathways for both SALS patient subgroups, supporting the possibility of a causal link between protein aggregation, neurotoxicity and disease severity in ALS and other neurological diseases, like HD47,48 (Fig. 4a). Among the most important candidate genes involved in these processes is AKT1, a gene encoding a cAMP-dependent serine-threonine kinase that was amplified in both SALS subgroup and overexpressed in SALS2. Our results are supported by previous evidence that abnormal AKT activation is implicated in several cellular mechanisms involved in ALS, such as the altered elimination of toxic protein aggregates, increasing oxidative stress and rendering cells susceptible to ROS-triggered cell death49. Our PPI network analysis identified AKT as a highly interconnected node, suggesting that alterations in this protein are not mere passenger events, but may have a great impact on one or more signaling pathways that are recurrently involved in ALS. Pharmacological inhibition of AKT and its downstream pathways has already demonstrated neuroprotective effects by modulating the activation state of microglial cells during neuroinflammation, and promoting cellular clearance in neurodegenerative storage diseases, suggesting a potential role of AKT inhibitors in ALS treatment50,51.
It is interesting to note that although partially complementary and convergent, the two SALS patient clusters showed different significantly deregulated processes and mediators. In particular, antigen processing and presentation, and extracellular matrix organization were the most significantly enriched pathways for the CNV-driven genes in SALS1, while the pathways of highest significance in SALS2 were associated with axonal guidance, oxidative stress and inflammatory intracellular signaling cascades (Fig. 4a). Therefore, a careful monitoring of these signaling cascades may help to better diagnose the specific subtype of ALS and optimize treatment strategies. To this regard, the convergent functional analysis of CNV-driven genes also pinpointed known and novel candidate therapeutic targets and biomarkers for early diagnosis, molecular subtyping and targeted therapy in SALS (Table 4). In particular, among CNV-driven genes in SALS1 showing the same expressional tendencies between DNA copy number and mRNA expression, we distinguished some genes (KIF1A, MC1R and MECP2) that were not previously implicated in ALS, representing new candidates for molecularly guided diagnosis and treatments. Within the cluster of CNV-driven genes in SALS2, we found a large number of new candidate ALS genes mainly implicated in oxidative and inflammatory signaling cascades52,53,54,55,56,57. Among these, identification of gain and overexpression of TRAF2 was in accordance with previous evidence that correlated elevated expression levels of this gene with inflammatory processes in PD and other neurological disorders58,59. On the contrary, copy number loss and reduced expression levels HSPA5 in SALS2 may reflect the suppression of neuroprotective role of this molecule against ER stress-associated cell death, leading to oxidative stress and alterations in calcium homeostasis, and rendering neurons vulnerable to degeneration60,61,62,63,64. Concordantly, pharmacological activation of HSPA5 and its co-chaperones has demonstrated to exert neuroprotective effects on motor neurons of ALS by reducing ER stress-mediated cell death, supporting a translational potential for HSPA5 induction as a therapy against ALS and other neurologic disorders65.
Overall, our study provided the first comprehensive and integrated map of genomic and transcriptional events characterizing different SALS subtypes, revealing key drivers and etiopathogenic mechanisms that may have been masked by considering SALS pathology as a single entity. Despite the most obvious limitation in this research was that of a small and unbalanced number of samples, our brain tissue samples represent a vanishingly rare resource for investigating molecular mechanisms underlying neurological disorders. The importance of an integrative analysis such as the one presented here, emerges from recent published data that highlight the presence of somatic changes in brain tissue of patients affected by neurological diseases66,67. Moreover, the custom-designed platform used in this study has some disadvantages, including limited use for discovering novel genes or gene features and inability to detect nucleotide repeat expansions or balanced structural chromosomal abnormalities. However, the possibility to simultaneously detect multiple genes involved in neurological disorders may allow for differential diagnosis between common neurological disorders, refine the genotype-phenotype correlations and explore the potential genetic overlapping signatures among different neurological conditions.
In conclusion, the present study proposes the use of a multi-omics analysis as a promising approach for the identification of somatic alteration and candidate drivers in ALS for defining disease subtype and directing molecular targeted clinical trials that more accurately reflect inter-individual differences among patients. Future more in-depth functional and integrative omics studies will be necessary to verify our findings and explore the impact of candidate genes on the outcome of the disease.
Material and Methods
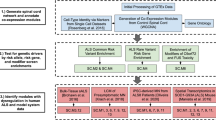
The analysis workflow is shown in Fig. 5 and described below.

Experimental workflow of multi-omics analysis for characterization of CNV-driven differentially expressed genes in SALS patients. The workflow depicts the steps performed in this study, from data acquisition to the visualization, validation and export of results in various output formats. See Materials and Methods section for details.
Subject cohorts and sample preparation
All samples were provided by the Department of Pathology of the Academic Medical Center (University of Amsterdam). This cohort included motor cortex samples from 30 patients with clear SALS diagnosis and 10 control individuals collected as previously described17, and whose clinico-pathological parameters are detailed in the Supplementary Table 12. Informed consent was obtained for the use of brain tissues and for access to medical records for research purposes, and approval was obtained from the relevant local ethical committees for medical research. All experiments were performed in accordance with relevant guidelines and regulations of both institutions. Genomic DNA was extracted from 10μm-thick sections using the QIAamp Fast DNA Tissue Kit according to the manufacturer’s instructions (QIAGEN, Hilden, Germany). The extracted genomic DNA was quantified by using the NanoDrop ND-1000 spectrophotometer (Thermo Fisher Scientific, MA, USA), and assessed for quality by microcapillary electrophoresis on 2100 Bioanalyzer (Agilent Technologies, Palo Alto, CA).
NeuroArray aCGH processing and data analysis
High-resolution exon-centered analysis of CNVs was done using an 8 × 60 K custom exon-centric NeuroArray platform v.2.0 (Agilent Technologies, Santa Clara, CA), tailored to detect single/multi-exon deletions and duplications in a large panel of ALS-related genes (n = 154) and to others additional neurological disorders (Supplementary Table 13)25. Details concerning the NeuroArray aCGH platform can be found in Supplementary Materials. DNA labeling and hybridization on NeuroArray were performed according to the manufacturer’s protocol (Agilent Technologies, Santa Clara, CA). Briefly, DNA test and a reference of the same sex (Euro Reference, Agilent Technologies, Santa Clara, CA), both at the concentration of 500 ng, were double digested with RsaI and AluI for 2 hours at 37 °C. After heat inactivation of the enzymes at 65 °C for 20 min, each digested sample was labeled by random priming by using the genomic DNA Enzymatic Labelling Kit (Agilent Technologies, Santa Clara, CA) for 2 hours using Cy5-dUTP for patient DNAs and Cy3-dUTP for reference DNAs. Labeled products were column purified by using the SureTag DNA Labeling Kit Purification Columns (Agilent Technologies, Santa Clara, CA). After probe denaturation and pre-annealing with Cot-1 DNA, hybridization was performed at 65 °C with rotation for 24 hr. After two washing steps, arrays were scanned at 3 µm resolution using an Agilent G4900DA SureScan Microarray Scanner System and aCGH image data were processed using Agilent’s Feature Extraction software to assess the array spot quality as well as check signal and background intensity statistics in the default setting. Feature extracted raw data was normalized, analyzed and visualized using Agilent CytoGenomics v. 4.0.3.12 and Genomic Workbench v. 7.0.4.0 software (Agilent Technologies, Santa Clara, CA, USA). Briefly, after filtering for saturated and non-uniform probes, data were normalized by GC correction with a window size of 2 kb and Diploid Peak Centralization. The Centralization Normalization Algorithm with a threshold of 6.0 and a bin size of 10 was also used for detecting aberrant regions or regions of constant CNVs. Aberrations were detected by the Aberration Detection Method II algorithm (ADM-2), with a sensitivity threshold of 6.0 and moving average window of 2 Mb, which permits to identify all aberrant intervals in a given sample with consistently high or low log ratios based on the statistical score. An aberration filter was applied for identifying copy number alterations; changes were considered as true positive events with a minimum log2 ratio test/control of ±0.5 and a minimum of 3 consecutive probes. Positive statistical score meant amplification, while a negative score indicated deletion. Human reference sequence hg19 assembly was used to define the genomic coordinates of detected CNVs. Raw data of the microarrays are available at NCBI’s Gene Expression Omnibus (GEO) with the accession number GSE107375.
Identification of significantly altered genomic regions and CNV-encompassed genes
For statistical analysis, the ALS samples were divided into two groups (SALS1 and SALS2) based on their previously characterized gene expression profiles17. The chromosomal distribution and the frequency of the copy number gains and losses in both SALS subgroups were also investigated. Using ADM-2 generated interval based amplification and deletion data, penetrance analysis was performed to find the percentage of samples that share aberrations in a particular genomic region among multiple samples. A recurrent CNV was called when the gains or losses occurred in at least 10% of the SALS patients, both with and without the assignment into the hierarchically determined two sets. Multiple amplifications and deletions were counted as separate events. Aberrant intervals were also filtered taking into account those occurring in at least 10% of the cases and absent in individual controls. In addition, to assess the effective relations between the detected CNVs and ALS pathogenesis, we compared these aberrant regions with those previously associated with ALS via screening of publicly available databases (i.e., CNVD) and published literature.
Frequent amplifications and deletions observed in both SALS patient subgroups were reviewed and annotated to the human hg19 reference genome and then were screened out for only genes included in the NeuroArray aCGH design. In addition, significant probe signals were clustered for pathologies according to their location on causative or susceptibility genes through a homemade script on R-platform, in order to search for CNVs in candidate genes of ALS disease68.
Source of gene expression data
Gene expression data set E-MTAB-2325 was downloaded from EBI ArrayExpress database, which was annotated using the platform of GPL6480 (Agilent-014850 Whole Human Genome Microarray 4 × 44 K G4112F). A total of 40 samples were selected out, including 10 normal and 30 SALS motor cortex samples. Raw signal values were thresholded to 1, log2 transformed, normalized to the 50th percentile, and baselined to the median of all samples using GeneSpringGX v.14.5 (Agilent Technologies, Italy). Fold change (FC) values were calculated between SALS patients and individual controls. Positive FC meant over-expression, whereas negative FC indicated under-expression. Probes not corresponding to an ENTREZ ID were removed. In cases where several probes corresponded to one ENTREZ ID, the probe showing the highest variance over all samples was chosen for further analysis. Genes that showed a significant P value < 0.05 (one-way ANOVA followed by the Benjamini and Hochberg False Discovery Rate and the Tukey’s Post Hoc test) and FC ≥ ± 1.5 were considered differentially expressed and were taken for further analysis.
Integration of the aCGH and gene expression data
To assess the contribution of genomic aberrations to global gene expression pattern changes in SALS and identify CNV-associated DEGs, we performed an integrated analysis of differential expression values and the corresponding DNA copy number changes through a gene-by-gene approach. In particular, each gene expression measurement was assigned to the corresponding copy number probe interrogating the same named gene. A CNV-driven gene was defined when the gene expression trend was consistent with the copy number change (i.e., up-regulated gene transcript with a chromosomal amplification and down-regulated gene transcript with a chromosomal deletion).
Functional enrichment and biological network analysis
The function of CNV-associated DEGs in SALS patients was annotated and analyzed according to the three organizing principles of Gene Ontology (BP: Biological Process, MF: Molecular Function, CC: Cellular Component) by using the enrichment analysis tool Enrichr 69. To interpret the biological significance of CNV-driven genes in the context of known biological pathways, we used QIAGEN’s Ingenuity Pathway Analysis (IPA®; http://www.ingenuity.com/) and MetaCore repository (Clarivate Analytics, Philadelphia, United States) 70. Both these programs identify significantly enriched biological pathways and signaling cascades that are associated with a given list of genes by calculating the hypergeometric distribution. P-value < 0.05 was set as the threshold to filter out significant terms. In addition, to increase the statistical power of our analysis, we compared our results with three other pathway enrichment analysis tools and databases (KEGG; Reactome; Panther) and selected signaling pathways that were identified as significantly deregulated by two or more platforms.
To better understand the interactions of the CNV-driven genes and identify the best candidate genes in SALS, an extended protein-protein interaction (PPI) network of their encoding products was predicted by using the STRING database 71 and visualized with the Cytoscape v.3.5.0 software 72. The extended network was constructed by using the CNV-driven genes as seed molecules and setting a high level of confidence between molecular interactions (high confidence score of at least 0.8) and a maximum number of interactions to 100. In order to identify the “Hub” nodes, a network topology analysis was performed by using the Cytoscape plug-in NetworkAnalyzer based on topological parameters 73. The relative importance of the genes in each network, meaning their ability to hold together the communicating nodes in a biological network, was determined based on the node centrality measure setting the topological parameter “node degree” ≥10. Nodes with high degree (hub genes) represented the genes having important biological functions: the higher the value, the higher the relevance of the gene in connecting regulatory molecules. Likewise, values of edge betweenness were mapped with the edge size: high values of this parameter correspond to a large edge size. The final PPI network was visualized based on node degree and edge betweenness parameters.
Finally, target and biomarker assessment tools in both IPA and Metacore were used to screen candidate CNV-driven genes in order to identify potential candidate biomarkers and therapeutic targets for ALS.
Real-Time quantitative polymerase chain reaction (RT-qPCR) validation
To confirm the reliability of our data, we validated the NeuroArray CGH results performing ad hoc real-time quantitative polymerase chain reaction (RT-qPCR) assays. Briefly, we used DNA extracted from the motor cortex samples of 15 donors assayed by NeuroArray, including 5 controls, 4 SALS1 and 6 SALS2 (Supplementary Table 11). From the list of CNV-driven DEGs genes, we selected 5 candidates (GAA, KIF1A, CSF1, TRAF2, HSPA5) on the basis of their potential clinical relevance as patient-specific biomarkers and therapeutic targets (Supplementary Table 11). The primer sets flanking the putative exonic imbalances were designed using the PrimerBlast tool (http://www.ncbi.nlm.nih.gov/tools/primer-blast/) as described 74 and were available in Supplementary Table 11. RT-qPCR was performed in triplicate using the LightCycler 1.5 (Roche Diagnostics, Germany). Cycling conditions were 95 °C for 15 s followed by 40 cycles of 95 °C (5 s), 60 °C (15 s) and one cycle of 95 °C (15 s), 60 °C (60 s), 95 °C (15 s). The data were analyzed by the 2 − ΔΔCt method that requires a healthy control sample (diploid) as a calibrator in all amplifications 75. 2 − ΔΔCt ≥ 1.4 or ≤0.6 was defined as copy number gain or loss, respectively, whereas 2 − ΔΔCt values included from 0.8 to 1.2 were considered as normal diploid. As calibrator control, we used the same DNA reference hybridized in NeuroArray experiments. The specific PCR products were confirmed by the results of melting curve analysis and agarose gel electrophoresis.
Ethics approval and consent to participate
Experiments involving human participants have been approved by an ethical committee (Ethics Committee of the Amsterdam Academic Medical Center, approved protocol: W11_073) for medical research and have been performed in accordance with ethical standards. Informed consent Informed consent was obtained from all individual participants included in the study.
Data Availability
All data generated during this study are included in this published article and the additional files. Transcriptional data are available at EBI ArrayExpress database with the accession number E-MTAB-2325. Raw data from NeuroArray aCGH analysis are available at NCBI’s Gene Expression Omnibus (GEO) with the accession number GSE107375.
References
Wijesekera, L. C. & Leigh, P. N. Amyotrophic lateral sclerosis. Orphanet journal of rare diseases 4, 3, https://doi.org/10.1186/1750-1172-4-3 (2009).
Costa, J. & de Carvalho, M. Emerging molecular biomarker targets for amyotrophic lateral sclerosis. Clinica chimica acta; international journal of clinical chemistry 455, 7–14, https://doi.org/10.1016/j.cca.2016.01.011 (2016).
Simpson, C. L. & Al-Chalabi, A. Amyotrophic lateral sclerosis as a complex genetic disease. Biochimica et biophysica acta 1762, 973–985, https://doi.org/10.1016/j.bbadis.2006.08.001 (2006).
Bruijn, L. I., Miller, T. M. & Cleveland, D. W. Unraveling the mechanisms involved in motor neuron degeneration in ALS. Annual review of neuroscience 27, 723–749, https://doi.org/10.1146/annurev.neuro.27.070203.144244 (2004).
Morello, G., Spampinato, A. G. & Cavallaro, S. Molecular Taxonomy of Sporadic Amyotrophic Lateral Sclerosis Using Disease-Associated. Genes. Frontiers in neurology 8, 152, https://doi.org/10.3389/fneur.2017.00152 (2017).
Volk, A. E., Weishaupt, J. H., Andersen, P. M., Ludolph, A. C. & Kubisch, C. Current knowledge and recent insights into the genetic basis of amyotrophic lateral sclerosis. Medizinische Genetik: Mitteilungsblatt des Berufsverbandes Medizinische Genetik e.V 30, 252–258, https://doi.org/10.1007/s11825-018-0185-3 (2018).
Al-Chalabi, A., van den Berg, L. H. & Veldink, J. Gene discovery in amyotrophic lateral sclerosis: implications for clinical management. Nature reviews. Neurology 13, 96–104, https://doi.org/10.1038/nrneurol.2016.182 (2017).
Pampalakis, G. et al. New molecular diagnostic trends and biomarkers for amyotrophic lateral sclerosis. Human mutation, https://doi.org/10.1002/humu.23697 (2018).
Caroline, E. The genetic basis of amyotrophic lateral sclerosis: recent breakthroughs. Advances in Genomics and Genetics. 5, 327–345, https://doi.org/10.2147/AGG.S57397 (2015).
Nicolas, A. et al. Genome-wide Analyses Identify KIF5A as a Novel ALS Gene. Neuron 97, 1268–1283 e1266, https://doi.org/10.1016/j.neuron.2018.02.027 (2018).
Brenner, D. et al. Hot-spot KIF5A mutations cause familial ALS. Brain: a journal of neurology 141, 688–697, https://doi.org/10.1093/brain/awx370 (2018).
Redon, R. et al. Global variation in copy number in the human genome. Nature 444, 444–454, https://doi.org/10.1038/nature05329 (2006).
Lupski, J. R. Genomic rearrangements and sporadic disease. Nature genetics 39, S43–47, https://doi.org/10.1038/ng2084 (2007).
Blauw, H. M. et al. Copy-number variation in sporadic amyotrophic lateral sclerosis: a genome-wide screen. The Lancet. Neurology 7, 319–326, https://doi.org/10.1016/S1474-4422(08)70048-6 (2008).
Wain, L. V. et al. The role of copy number variation in susceptibility to amyotrophic lateral sclerosis: genome-wide association study and comparison with published loci. PloS one 4, e8175, https://doi.org/10.1371/journal.pone.0008175 (2009).
Katyal, N. & Govindarajan, R. Shortcomings in the Current Amyotrophic Lateral Sclerosis Trials and Potential Solutions for Improvement. Frontiers in Neurology 8, https://doi.org/10.3389/fneur.2017.00521 (2017).
Aronica, E. et al. Molecular classification of amyotrophic lateral sclerosis by unsupervised clustering of gene expression in motor cortex. Neurobiology of disease 74, 359–376, https://doi.org/10.1016/j.nbd.2014.12.002 (2015).
Morello, G. & Cavallaro, S. Transcriptional analysis reveals distinct subtypes in amyotrophic lateral sclerosis: implications for personalized therapy. Future medicinal chemistry 7, 1335–1359, https://doi.org/10.4155/fmc.15.60 (2015).
Lederer, C. W., Torrisi, A., Pantelidou, M., Santama, N. & Cavallaro, S. Pathways and genes differentially expressed in the motor cortex of patients with sporadic amyotrophic lateral sclerosis. BMC genomics 8, 26, https://doi.org/10.1186/1471-2164-8-26 (2007).
Carter, B. J., Anklesaria, P., Choi, S. & Engelhardt, J. F. Redox modifier genes and pathways in amyotrophic lateral sclerosis. Antioxidants & redox signaling 11, 1569–1586, https://doi.org/10.1089/ARS.2008.2414 (2009).
Billaud, M. et al. Regulation of cellular communication by signaling microdomains in the blood vessel wall. Pharmacological reviews 66, 513–569, https://doi.org/10.1124/pr.112.007351 (2014).
Zhao, W., Beers, D. R. & Appel, S. H. Immune-mediated mechanisms in the pathoprogression of amyotrophic lateral sclerosis. Journal of neuroimmune pharmacology: the official journal of the Society on NeuroImmune Pharmacology 8, 888–899, https://doi.org/10.1007/s11481-013-9489-x (2013).
Shao, H., He, Y., Li, K. C. & Zhou, X. A system mathematical model of a cell-cell communication network in amyotrophic lateral sclerosis. Molecular bioSystems 9, 398–406, https://doi.org/10.1039/c2mb25370d (2013).
Bendotti, C. et al. Inter- and intracellular signaling in amyotrophic lateral sclerosis: role of p38 mitogen-activated protein kinase. Neuro-degenerative diseases 2, 128–134, https://doi.org/10.1159/000089617 (2005).
La Cognata, V. et al. A customized high-resolution array-comparative genomic hybridization to explore copy number variations in Parkinson’s disease. Neurogenetics 17, 233–244, https://doi.org/10.1007/s10048-016-0494-0 (2016).
Taghibiglou, C., Lu, J., Mackenzie, I. R., Wang, Y. T. & Cashman, N. R. Sterol regulatory element binding protein-1 (SREBP1) activation in motor neurons in excitotoxicity and amyotrophic lateral sclerosis (ALS): Indip, a potential therapeutic peptide. Biochemical and biophysical research communications 413, 159–163, https://doi.org/10.1016/j.bbrc.2011.08.011 (2011).
Porstmann, T. et al. SREBP activity is regulated by mTORC1 and contributes to Akt-dependent cell growth. Cell metabolism 8, 224–236, https://doi.org/10.1016/j.cmet.2008.07.007 (2008).
Saxena, S. et al. Neuroprotection through excitability and mTOR required in ALS motoneurons to delay disease and extend survival. Neuron 80, 80–96, https://doi.org/10.1016/j.neuron.2013.07.027 (2013).
Kaminsky, E. B. et al. An evidence-based approach to establish the functional and clinical significance of copy number variants in intellectual and developmental disabilities. Genetics in medicine: official journal of the American College of Medical Genetics 13, 777–784, https://doi.org/10.1097/GIM.0b013e31822c79f9 (2011).
Collaboration, A. et al. Jet energy resolution in proton-proton collisions at [Formula: see text] recorded in 2010 with the ATLAS detector. The European physical journal. C, Particles and fields 73, 2306, https://doi.org/10.1140/epjc/s10052-013-2306-0 (2013).
Nishioka, K. et al. Genetic variation of the mitochondrial complex I subunit NDUFV2 and Parkinson’s disease. Parkinsonism & related disorders 16, 686–687, https://doi.org/10.1016/j.parkreldis.2010.09.007 (2010).
Morello, G. et al. Copy Number Variations in Amyotrophic Lateral Sclerosis: Piecing the Mosaic Tiles Together through a Systems Biology Approach. Molecular neurobiology, https://doi.org/10.1007/s12035-017-0393-x (2017).
Renton, A. E., Chio, A. & Traynor, B. J. State of play in amyotrophic lateral sclerosis genetics. Nature neuroscience 17, 17–23, https://doi.org/10.1038/nn.3584 (2014).
Brouwers, N. et al. Alzheimer risk associated with a copy number variation in the complement receptor 1 increasing C3b/C4b binding sites. Molecular psychiatry 17, 223–233, https://doi.org/10.1038/mp.2011.24 (2012).
Bahia El Idrissi, N. et al. Complement activation at the motor end-plates in amyotrophic lateral sclerosis. Journal of neuroinflammation 13, 72, https://doi.org/10.1186/s12974-016-0538-2 (2016).
Goldknopf, I. L. et al. Complement C3c and related protein biomarkers in amyotrophic lateral sclerosis and Parkinson’s disease. Biochemical and biophysical research communications 342, 1034–1039, https://doi.org/10.1016/j.bbrc.2006.02.051 (2006).
Leger, B. et al. Human skeletal muscle atrophy in amyotrophic lateral sclerosis reveals a reduction in Akt and an increase in atrogin-1. FASEB journal: official publication of the Federation of American Societies for Experimental Biology 20, 583–585, https://doi.org/10.1096/fj.05-5249fje (2006).
Prolo, L. M., Vogel, H. & Reimer, R. J. The lysosomal sialic acid transporter sialin is required for normal CNS myelination. The Journal of neuroscience: the official journal of the Society for Neuroscience 29, 15355–15365, https://doi.org/10.1523/JNEUROSCI.3005-09.2009 (2009).
Scekic-Zahirovic, J. et al. Motor neuron intrinsic and extrinsic mechanisms contribute to the pathogenesis of FUS-associated amyotrophic lateral sclerosis. Acta neuropathologica 133, 887–906, https://doi.org/10.1007/s00401-017-1687-9 (2017).
Pharoah, D. S. et al. Expression of the inflammatory chemokines CCL5, CCL3 and CXCL10 in juvenile idiopathic arthritis, and demonstration of CCL5 production by an atypical subset of CD8+ T cells. Arthritis research & therapy 8, R50, https://doi.org/10.1186/ar1913 (2006).
Garbuzova-Davis, S. & Sanberg, P. R. Blood-CNS Barrier Impairment in ALS patients versus an animal model. Frontiers in cellular neuroscience 8, 21, https://doi.org/10.3389/fncel.2014.00021 (2014).
Marchitti, S. A., Brocker, C., Stagos, D. & Vasiliou, V. Non-P450 aldehyde oxidizing enzymes: the aldehyde dehydrogenase superfamily. Expert opinion on drug metabolism & toxicology 4, 697–720, https://doi.org/10.1517/17425255.4.6.697 (2008).
Chapuis, J. et al. Increased expression of BIN1 mediates Alzheimer genetic risk by modulating tau pathology. Molecular psychiatry 18, 1225–1234, https://doi.org/10.1038/mp.2013.1 (2013).
Yao, Y. et al. A striatal-enriched intronic GPCR modulates huntingtin levels and toxicity. eLife 4, https://doi.org/10.7554/eLife.05449 (2015).
Campbell, P. D. et al. Unique function of Kinesin Kif5A in localization of mitochondria in axons. The Journal of neuroscience: the official journal of the Society for Neuroscience 34, 14717–14732, https://doi.org/10.1523/JNEUROSCI.2770-14.2014 (2014).
Hegde, A. N. & Upadhya, S. C. Role of ubiquitin-proteasome-mediated proteolysis in nervous system disease. Biochimica et biophysica acta 1809, 128–140, https://doi.org/10.1016/j.bbagrm.2010.07.006 (2011).
Chhetri, S. K., Dayanandan, R., Bindman, D., Craufurd, D. & Majeed, T. Amyotrophic lateral sclerosis and Huntington’s disease: neurodegenerative link or coincidence? Amyotrophic lateral sclerosis & frontotemporal degeneration 15, 145–147, https://doi.org/10.3109/21678421.2013.817586 (2014).
Buck, E. et al. Comparison of Sirtuin 3 Levels in ALS and Huntington’s Disease-Differential Effects in Human Tissue Samples vs. Transgenic Mouse Models. Frontiers in molecular neuroscience 10, 156, https://doi.org/10.3389/fnmol.2017.00156 (2017).
Los, M., Maddika, S., Erb, B. & Schulze-Osthoff, K. Switching Akt: from survival signaling to deadly response. BioEssays: news and reviews in molecular, cellular and developmental biology 31, 492–495, https://doi.org/10.1002/bies.200900005 (2009).
de Oliveira, A. C. et al. Pharmacological inhibition of Akt and downstream pathways modulates the expression of COX-2 and mPGES-1 in activated microglia. Journal of neuroinflammation 9, 2, https://doi.org/10.1186/1742-2094-9-2 (2012).
Palmieri, M. et al. mTORC1-independent TFEB activation via Akt inhibition promotes cellular clearance in neurodegenerative storage diseases. Nature communications 8, 14338, https://doi.org/10.1038/ncomms14338 (2017).
Satoh, J., Asahina, N., Kitano, S. & Kino, Y. A Comprehensive Profile of ChIP-Seq-Based Olig2 Target Genes in Motor Neuron Progenitor Cells Suggests the Possible Involvement of Olig2 in the Pathogenesis of Amyotrophic Lateral Sclerosis. Journal of central nervous system disease 7, 1–14, https://doi.org/10.4137/JCNSD.S23210 (2015).
Nishimura, A. L. et al. A novel locus for late onset amyotrophic lateral sclerosis/motor neurone disease variant at 20q13. Journal of medical genetics 41, 315–320 (2004).
Martinez-Muriana, A. et al. CSF1R blockade slows the progression of amyotrophic lateral sclerosis by reducing microgliosis and invasion of macrophages into peripheral nerves. Scientific reports 6, 25663, https://doi.org/10.1038/srep25663 (2016).
de Oliveira, G. P., Alves, C. J. & Chadi, G. Early gene expression changes in spinal cord from SOD1(G93A) Amyotrophic Lateral Sclerosis animal model. Frontiers in cellular neuroscience 7, 216, https://doi.org/10.3389/fncel.2013.00216 (2013).
Lee, J. K. et al. MST1 functions as a key modulator of neurodegeneration in a mouse model of ALS. Proceedings of the National Academy of Sciences of the United States of America 110, 12066–12071, https://doi.org/10.1073/pnas.1300894110 (2013).
Ferraiuolo, L. et al. Transcriptional response of the neuromuscular system to exercise training and potential implications for ALS. Journal of neurochemistry 109, 1714–1724, https://doi.org/10.1111/j.1471-4159.2009.06080.x (2009).
Chung, J. Y. et al. Elevated TRAF2/6 expression in Parkinson’s disease is caused by the loss of Parkin E3 ligase activity. Laboratory investigation; a journal of technical methods and pathology 93, 663–676, https://doi.org/10.1038/labinvest.2013.60 (2013).
Alves, C. J., Maximino, J. R. & Chadi, G. Dysregulated expression of death, stress and mitochondrion related genes in the sciatic nerve of presymptomatic SOD1(G93A) mouse model of Amyotrophic Lateral Sclerosis. Frontiers in cellular neuroscience 9, 332, https://doi.org/10.3389/fncel.2015.00332 (2015).
Kanekura, K., Suzuki, H., Aiso, S. & Matsuoka, M. ER stress and unfolded protein response in amyotrophic lateral sclerosis. Molecular neurobiology 39, 81–89, https://doi.org/10.1007/s12035-009-8054-3 (2009).
Cai, Y. et al. Interplay of endoplasmic reticulum stress and autophagy in neurodegenerative disorders. Autophagy 12, 225–244, https://doi.org/10.1080/15548627.2015.1121360 (2016).
Shiihashi, G. et al. Mislocated FUS is sufficient for gain-of-toxic-function amyotrophic lateral sclerosis phenotypes in mice. Brain: a journal of neurology 139, 2380–2394, https://doi.org/10.1093/brain/aww161 (2016).
Booth, L. et al. GRP78/BiP/HSPA5/Dna K is a universal therapeutic target for human disease. Journal of cellular physiology 230, 1661–1676, https://doi.org/10.1002/jcp.24919 (2015).
Casas, C. GRP78 at the Centre of the Stage in Cancer and Neuroprotection. Frontiers in neuroscience 11, 177, https://doi.org/10.3389/fnins.2017.00177 (2017).
Wang, M., Wey, S., Zhang, Y., Ye, R. & Lee, A. S. Role of the unfolded protein response regulator GRP78/BiP in development, cancer, and neurological disorders. Antioxidants & redox signaling 11, 2307–2316, https://doi.org/10.1089/ARS.2009.2485 (2009).
Nishioka, M., Bundo, M., Iwamoto, K. & Kato, T. Somatic mutations in the human brain: implications for psychiatric research. Molecular psychiatry, https://doi.org/10.1038/s41380-018-0129-y (2018).
Fullard, J. F. et al. Assessment of somatic single-nucleotide variation in brain tissue of cases with schizophrenia. Translational psychiatry 9, 21, https://doi.org/10.1038/s41398-018-0342-0 (2019).
Team, R. C. (ISBN 3-900051-07-0, 2014).
Kuleshov, M. V. et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic acids research 44, W90–97, https://doi.org/10.1093/nar/gkw377 (2016).
Nikolsky, Y., Ekins, S., Nikolskaya, T. & Bugrim, A. A novel method for generation of signature networks as biomarkers from complex high throughput data. Toxicol.Lett. 158, 20–29 (2005).
Szklarczyk, D. et al. STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic acids research 43, D447–452, https://doi.org/10.1093/nar/gku1003 (2015).
Shannon, P. et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome research 13, 2498–2504, https://doi.org/10.1101/gr.1239303 (2003).
Assenov, Y., Ramirez, F., Schelhorn, S. E., Lengauer, T. & Albrecht, M. Computing topological parameters of biological networks. Bioinformatics 24, 282–284, https://doi.org/10.1093/bioinformatics/btm554 (2008).
D’haene, B., Vandesompele, J. & Hellemans, J. Accurate and objective copy number profiling using real-time quantitative PCR. Methods 50, 262–270, https://doi.org/10.1016/j.ymeth.2009.12.007 (2010).
Patitucci, A. et al. Comparison of different techniques for detecting 17p12 duplication in CMT1A. Neuromuscular Disorders 15, 488–492, https://doi.org/10.1016/j.nmd.2005.04.006 (2005).
Acknowledgements
Authors gratefully acknowledge Valentina La Cognata for her significant contribution during the array design, and Cristina Calì, Alfia Corsino, Maria Patrizia D’Angelo, Francesco Marino, Francesco Rabboni for their administrative and technical support. This work was supported by the Italian Ministry of Education, Universities and Research through grant CTN01_00177_817708 and the international PhD programs in Neuroscience and Computer Science of the University of Catania.
Author information
Authors and Affiliations
Contributions
G.M. and M.G. performed experiments, analyzed and interpreted data. G.M. wrote the manuscript. A.G.S. performed bioinformatics analyses. E.A. conducted tissue sample preparation. S.S., V.D. and F.L.C. participated in revising the manuscripts. S.C. conceived, directed, and supervised the project. All authors have read and approved the final version of this manuscript and agreed to be accountable for all aspects of the work. Correspondence and requests for materials should be addressed to the author at sebastiano.cavallaro@cnr.it.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Morello, G., Guarnaccia, M., Spampinato, A. et al. Integrative multi-omic analysis identifies new drivers and pathways in molecularly distinct subtypes of ALS. Sci Rep 9, 9968 (2019). https://doi.org/10.1038/s41598-019-46355-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-46355-w
This article is cited by
-
Increased copy-number variant load of associated risk genes in sporadic cases of amyotrophic lateral sclerosis
Cellular and Molecular Life Sciences (2024)
-
Multiomic ALS signatures highlight subclusters and sex differences suggesting the MAPK pathway as therapeutic target
Nature Communications (2024)
-
Unsupervised machine learning identifies distinct ALS molecular subtypes in post-mortem motor cortex and blood expression data
Acta Neuropathologica Communications (2023)
-
Molecular subtypes of ALS are associated with differences in patient prognosis
Nature Communications (2023)
-
DNA sequence features underlying large-scale duplications and deletions in human
Journal of Applied Genetics (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
