
Abstract
Multidrug-resistant (MDR) Salmonella enterica has been deemed a high-priority pathogen by the World Health Organization. Two hundred and sixty-four Salmonella enterica isolates recovered over a 16-year period (2000 to 2016) from the poultry and swine production chains, in Brazil, were investigated by whole-genome sequencing (WGS). Most international lineages belonging to 28 serovars, including, S. enterica serovars S. Schwarzengrund ST96, S. Typhimurium ST19, S. Minnesota ST548, S. Infantis ST32, S. Heidelberg ST15, S. Newport ST45, S. Brandenburg ST65 and S. Kentucky ST198 displayed MDR and virulent genetic backgrounds. In this regard, resistome analysis revealed presence of qnrE1 (identified for the first time in S. Typhimurium from food chain), qnrB19, qnrS1, blaCTX-M-8, blaCTX-M-2 and blaCMY-2 genes, as well as gyrA mutations; whereas ColpVC, IncHI2A, IncHI2, IncFIA, Incl1, IncA/C2, IncR, IncX1 and po111 plasmids were detected. In addition, phylogenetic analysis revealed multiple independent lineages such as S. enterica serovars S. Infantis, S. Schwarzengrund, S. Minnesota, S. Kentucky and S. Brandenburg. In brief, ocurrence and persistence of international lineages of S. enterica serovars in food production chain is supported by conserved genomes and wide virulome and resistome.
Similar content being viewed by others

Introduction
Salmonella enterica displaying resistance to fluoroquinolone and third-generation cephalosporin remains one of the most pressing global concerns, being deemed a high-priority pathogen by the World Health Organization (WHO)1. In this regard, quinolone efflux pumps (oqxA/oqxB) and plasmid-mediated quinolone resistance (PMQR) genes, such as qnrB and qnrS variants have contributed to the increase in fluoroquinolone resistance2,3,4. On the other hand, the coexistence of PMQR and extended-spectrum beta-lactamases (ESBLs), most CTX-M-type, in S. enterica isolates is being increasingly reported at the human-animal interface worldwide4,5,6.
Since, there is a growing understanding that the food production chain plays an important role both in the transmission of antibiotic-resistant pathogens and in their evolution and dissemination4. Genomic investigation of high-priority S. enterica serovars is a fundamental component of epidemiological surveillance work, particularly in major food-producing countries. Therefore, we have conducted a genomic investigation of S. enterica isolates recovered over a 16-year period from the poultry and swine meat production chains, in Brazil, which currently is one of the largest exporters of chicken and swine meat. In this regard, our results highlight persistence and dissemination of international lineages of multidrug-resistant S. enterica serovars exhibiting a highly virulent genetic background, in the food production chain.
Materials and Methods
Bacterial isolates
During a national surveillance study, 264 nontyphoidal Salmonella enterica (NTS) isolates recovered over a 16-year period (2000 to 2016), from the poultry and swine production chains, in Brazil, have been subjected to antimicrobial resistance screening. Specifically, in this study, we have focused on high-priority S. enterica isolates (n = 43) harboring fluoroquinolones, extended-spectrum β-lactamase (ESBL), and/or plasmid-mediated AmpC (pAmpC) resistance genes. Non-clinical samples were obtained from different points of poultry and swine production chain (Supplementary-Table 1) representing four different geographic regions in Brazil: South (Parana and Santa Catarina), Southeast (Sao Paulo and Minas Gerais), Midwest (Distrito Federal), and Northeast (Bahia) (Supplementary-Table 1). Bacterial isolation and serotyping were performed as previously reported7,8,9. All isolates (n = 43) underwent phenotypic and molecular characterization by Kirby-Bauer disc diffusion (Cefar), as well as broth microdilution using Sensititre® Gram Negative Plates (Trek Diagnostic Systems, OH) and whole-genome sequencing, respectively. Mininmum inhibitory concentration (MIC) values were interpreted according to Clinical and Laboratory Standards Institute (CLSI)10 and the National Antimicrobial Resistance Monitoring System (NARMS)11.
Whole-Genome sequencing
DNA extraction was performed using a commercial kit (QiAmp tissue, Qiagen, Germany) according to manufacturer’s guidelines. Genomic DNA of S. enterica isolates (n = 43) harboring fluoroquinolones, ESBL and/or pAmpC resistance genes were sequenced at a 300-bp paired-end-read using the Nextera XT library preparation kit at the MiSeq platform (Illumina, San Diego, CA).
Genomic data analysis
FastQ data were transferred into CLC genomic workbench 10.1.1 (Qiagen) and reads underwent strict quality control as previously described12. Subsequently, the software has ensured the appropriate size of the read length (300-bp), as well GC% content around 50%. Sequences obtained were de novo assembled using default settings in CLC workbench 10.1.1 (Qiagen). Resulting contigs were used to determine resistome (ResFinder 3.1), plasmidome (PlasmidFinder 2.0), multilocus sequence typing (MLST 2.0), plasmid MLST (pMLST 2.0), Salmonella pathogenicity island (SPIfinder 1.0), using default settings for all databases such as select threshold for %ID (≥90%) available in Center for Genomic Epidemiology (www.genomicepidemiology.org).
Assemblies were annotated with PROKKA version 1.14-dev13. The core genome was defined and aligned using Roary software version 3.11.2, with the BlastP threshold set to 95%14. The Roary software performs an all-against-all Blast of sequences produced by an initial CD-Hit clustering. The BlastP threshold was selected to include the mode of hits over a range of thresholds from 60 to 100%, while simultaneously providing a strict criterion to determine a set of vertically inherited core genes that can confidently be used to infer a phylogeny. A pan-genome gene presence-absence matrix was visualized with Phandango15 using the gene presence/absence output from Roary. Single nucleotide polymorphisms were extracted from the alignment using SNP-sites version 2.3.316.
Phylogeny was reconstructed using RAxML version 8.2.12 with a General Time Reversible Model with Gamma distribution for rate heterogeneity17. The resulting phylogeny was tested against 600 bootstrap replications and the number of replications necessary was determined by implementing the majority rule, autoMR convergence criteria in the RAxML software18. The resulting phylogeny was visualized and annotated using iTol version 319.
Sequence data accession number
Sequence data were deposited as part of the GenomeTrakr Project: Thakur Molecular Epidemiology Laboratory, NC State University. We deposited all sequences (n = 43) in GenBank and their accession numbers are listed in Table 1.
Results
Salmonella isolates and distribution of serotypes
Amongst 264 S. enterica isolates, we identified 28 serotypes distributed in the six regions studied (Supplementary-Table 1). Most isolates included S. Heidelberg (n = 81), S. Typhimurium (n = 43), S. Infantis (n = 35), S. Schwarzengrund (n = 21), and S. Enteritidis (n = 20). The remaining sixty-four isolates were classified in 23 different serotypes, including rarely reported S. Abony (n = 6), S. Isangi (n = 4), S. Rochdale (n = 3), S. Saphra (n = 2), S. Orion (n = 2) S. Ouakam (n = 1), S. Grumpensis (n = 1), S. Carrau (n = 1), S. Abaetetuba (n = 1), and S. Idikan (n = 1) (Supplementary-Table 1).
In this study, 264 isolates were screened for resistance to fluoroquinolones and/or broad-spectrum cephalosporins, of which 40 isolates were considered as high priority Salmonella strains due to broad-spectrum cephalosporin- or fluoroquinolone-resistance profiles1. Then, genomic investigation was performed on 43 isolates, including high priority Salmonella strains displaying an MDR (n = 36), defined as resistant to three or more classes of antimicrobial compounds20; or a quinolone-resistant profile (n = 4). Additionally, 3 representative pan-susceptible Salmonella serovars S. Infantis (SI690), S. Minnesota (SMi294) and S. Kentucky (SK497) were also investigated by WGS, for comparative analysis (Table 1).
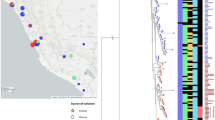
Moreover, of the six Brazilian states studied, Sao Paulo (SP) harbored nineteen S. enterica isolates beloging to different serovars such as S. Typhimurium (n = 7), S. Schwarzengrund (n = 6), S. Infantis (n = 2), S. Minnesota (n = 2), S. Brandenburg (n = 1) and S. Heidelberg (n = 1). Salmonella Infantis (n = 1) and S. Brandenburg (n = 1) were isolated from the Santa Catarina (SC) state while the Distrito Federal (DF) harbored S. Schwarzengrund (n = 13), S. Newport (n = 2) and S. Minnesota (n = 1). Finally, the state of Minas Gerais (MG) harbored serotypes S. Schwarzengrund (n = 1), S. Minnesota (n = 1), and S. Heidelberg (n = 1), followed by Parana (PR) and Bahia (BA) with occurrence of S. Typhimurium (n = 2) and S. Kentucky (n = 1), respectively as summarized in Table 1 and Fig. 1.

Distribution of S. enterica isolates (n = 43) harboring fluoroquinolones, extended-spectrum β-lactamase (ESBL), and/or plasmid-mediated AmpC (pAmpC) resistance genes and antimicrobial resistance genes (ARGs) over a 16-year period in Brazil. The map showing the distribution of S. enterica (n = 43) was created using an online service (https://mapchart.net/). Footnotes: *DF, Distrito Federal; MG, Minas Gerais; SP, São Paulo; PR, Paraná; SC, Santa Catarina; BA, Bahia.
Resistome
While these 43 Salmonella isolates reimaned susceptible to colistin and carbapenems, additional genes encoding resistance to aminoglycoside [aadA1, aadA2, aac(6′), aac(3)-Iva, aph(4)-Ia, aac(3)-IIa], sulfonamide [sul1, sul2], tetracycline [tet(A), tet(B)], trimethoprim [drfA1], phenicol [floR], streptomycin [strA, strB], fosfomycin [fosA7], macrolide [inu(F)], and quaternary ammonium [qacEdelta1] were confirmed by WGS as shown in Table 1. All of these genes have been previously reported in Salmonella isolates from variety sources including food and human5,21.
Among 43 selected isolates, which presented genes conferring resistance to fluoroquinolones, ESBLs and/or pAmpC, 36 carried both PMQR and β-lactams genes, whereas 7 isolates presented PMQR but not β-lactams encoding genes. These isolates belonged to eight serovars including S. Schwarzengrund (n = 20), S. Typhimurium (n = 9), S. Minnesota (n = 4), S. Infantis (n = 3), S. Heidelberg (n = 2), S. Newport (n = 2), S. Brandenburg (n = 2) and S. Kentucky (n = 1).
The PMQR qnrB19 gene, (n = 32) [8 isolates were only positive for qnrB19, and 24 co-produced CTX-M-2, CTX-M-8 or CMY-2 genes] was the most commom quinolone resistance gene observed followed by qnrE1 (n = 8) [1 was positive for TEM-1A and 7 co-produced TEM-1B], qnrS1 (n = 2) [both co-produced TEM-1B], and oqxA/oqxB (n = 1) [co-produced TEM-1A].
The highest PMQR gene diversity was observed in strains isolated from samples collected in Sao Paulo which harbored qnrE1 (n = 6), qnrB19 (n = 9), qnrS1 (n = 2) and oqxA/B (n = 1). Subsequently, Distrito Federal [n = 16; (qnrB19)], Minas Gerais [n = 3; (qnrB19)], Santa Catarina [n = 2; (qnrB19)], Parana [n = 2; (qnrE1) and Bahia [n = 1; (qnrB19)] harbored different genes. The ESBL/pAampC distribution is shown in Table 1. Among these 43 isolates the most frequent source of S. enterica was poultry (34/43; 79%), followed by swine (5/43; 11.6%) and different sources (4/43; 9.3%), including chicken cage after cleaning (n = 3) and swab (n = 1) (Table 1).
In addition to overall resistance, the MIC results reveals 13 resistance patterns among these S. enterica isolates (n = 43) (Table 1). The MIC values ranged from 0.5 µg/mL to 512 µg/mL among 14 antibiotics tested. The majority of quinolone-resistant phenotypes (QRP) isolates presented high-level resistance (MIC ≥32 µg/mL) to nalidixic acid and range between 0.5 to 8 µg/mL for ciprofloxacin. Additionally, most QRP isolates were resistant to levofloxacin (third-generation quinolone), moxifloxacin (fourth generation quinolone) and enrofloxacin (veterinary use only) by using Kirby-Bauer disc diffusion method. Regarding broad-spectrum cephalosporin-resistant, all isolates harbouring blaCTX-M-type presented high-level resistance against ceftriaxone (MIC >64 µg/ml) and ceftiofur (>8 µg/ml). Besides ceftriaxone and ceftiofur (veterinary use only), the isolates that carried blaCMY-2 had high-level resistance against cefoxitin (>32 µg/ml). Further, fourty isolates were resistant to ciprofloxacin and interestingly, one isolate (S. Infantis) was ciprofloxacin-resistant but nalidixic acid susceptible. Lastly, a total of 33 genes in this collection encoded resistance to β-lactams, aminoglicosydes, sulphonamides, tetracycline, phenicols, trimethoprim, microlides, fosfomycin and amonium quaternary (Fig. 2).

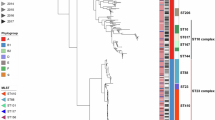
(A) Reconstructed phylogeny based on the core genome (3533 genes) of the 42 Salmonella strains. Percent of bootstrap samples in which nodes appeared are shown. The serotype of each isolate is labeled on its respective branch. Color strips depict the year, source, and geographic location of isolation, respectively. Poultry sources include broiler chicken, chicken wing, chicken wing paddle, chicken thigh, chicken feet, chicken breast, chicken cage after cleaning, chicken carcass, and Mechanically recovered chicken meat. Swine sources include: swine stomach, liver, muscle, and pork. Environmental sources include slaughterhouse and swab. (B) Presence and absence of selected antimicrobial resistance genes are shown, black indicating presence. (C) The gene presence/absence matrix depicts pan-genome variation.
Quinolone resistance-determining region (QRDR) among Salmonella enterica isolates
Thirteen isolates (30.2%) exhibited a single mutation in gyrA at codons Ser83 and Asp87, which has been most frequently reported in S. enterica22. Of these, eight S. Typhimurium presented mutation at codon Ser83Tyr, one S. Typhimurium isolate at codon Asp87Asn, two S. Brandenburg isolates at codon Asp87Gly, and two S. Heidelberg isolates at codon Ser83Phe. While gyrB, parC and parE were not identified, this single mutation in gyrA was sufficient to promote resistance at >32 µg/mL and >4 µg/mL for nalidixic acid and ciprofloxacin, respectively; it was particularly observed among S. Typhimurium isolates carrying qnrE1 and gyrA mutation, as shown in Table 2. Notably, the isolates, which harbored the combination between gyrA mutations and qnrE1, played a markedly greater role than gyrA and qnrB19 in mediating quinolone resistance (Table 2).
Identification of international lineages among fluoroquinolone- and cephalosporin-resistant Salmonella enterica serovars
We obtained among 43 S. enterica isolates a total of 9 different sequence types (STs) including the most frequently observed ST96 [S. Schwarzengrund (n = 20)], ST19 [S. Typhimurium (n = 8)] and ST548 [S. Minnesota (n = 4)]. As described above for S. Schwarzengrund, S. Typhimurium, and S. Minnesota, all STs were consistently associated with their respective serotypes: S. Infantis [ST32 (n = 3; 100%)], S. Heidelberg [ST15 (n = 2; 100%)], S. Newport [ST45 (n = 2; 100%)], S. Brandenburg [ST65 (n = 2; 100%)], and also the clinically important ST198 [S. Kentucky (n = 1; 100%)]. Only the S. Typhimurium contained two sequence types, being ST19 (n = 8) and ST3438 (n = 1), highlighting their genetic diversity.
Mobilome
The most common plasmid incompatibility group in our collection was ColpVC (n = 27; 62%). Furthermore, a range of plasmids previously associated with multidrug-resistant foodborne bacteria which have been associated with clinical settings harbored: IncHI2A (n = 24; 55.8%), IncHI2 (n = 24; 55.8%), IncFIA (n = 8; 18.6%), Incl1 (n = 6; 14%), IncA/C2 (n = 5; 11.6%), IncR (n = 3; 7%), IncX1 (n = 2; 4.6%), po111 (n = 1; 2.3%) (Table 1).
The eight S. Typhimurium qnrE1-positive isolates exhibited identical genetic content, regardless of the source (swine or chicken) or year of isolation (2000, 2013, 2015) (Fig. 3). These plasmids were composed by IRL (inverted repeat left)-ISEcp1-IRR (inverted repeat right)-qnrE1-araJ-ahp in a total of 4,659-bp and were different from previous reports23,24 (Fig. 3).

(A–E) Comparison of the genetic environments of qnrE1 gene. (F) Genetic environment of qnrS1. (G) Representative qnrB19 plasmid for 29 S. enterica isolates. Genes, different plasmids and shotgun sequences were extracted from GenBank database. Arrows indicate the positions and directions of the genes; Δ indicates the truncated gene. Regions with >99% identity are indicated in blue shadow.
Twenty-nine S. enterica containing qnrB19 genes had the plasmid sequences closed at 2,989-bp and each plasmid shared the same incompatibility group, ColpVC (Fig. 3). These small plasmids were identical to qnrB19 that were identified in E. coli in Brazil in 2016 (KX452393.1). They also shared 99% of identity with qnrB19-plasmids from Salmonella Muenchen in the United States in 2017 (KY991368.1) and from S. enterica serovars in Canada in 2018 (CP030230.1). The remaining three isolates had large plasmids ranging from 3,159-bp (S. Schwarzengrund) to 37,696-bp (S. Heidelberg). Notably, the qnrB19-backbone (37,696-bp) of S. Heidelberg (SH159) isolated from a chicken cage after cleaning in this study, showed 100% identity with S. Heidelberg previously reported from human, animal and food sources in Canada in 2016 (CP016580.1) denoting intercontinental spread of bacteria harboring these genes.
The genetic context of qnrS1 on S. Infantis had approximately 4,755-bp and this gene was surrounded upstream by truncated tnpR and downstream by truncated ISKpn19 that were carried on a core quinolone resistance genetic enviroment (Fig. 3). Additionally, the genetic context of blaCTX-M-8 showed that this gene was flanked by two copies of IS26; one which was located 1,922-bp upstream followed by the transposase (tnpA) and IS26 located 869-bp downstream for a total of 3,668-bp (IS26-tnpA-blaCTX-M-8-IS26). These data indicated that this genetic platform has the same mobilization apparatus of CTX-M-8-producing E. coli, which has been primarily isolated from retail chicken meat imported from Brazil25. Lastly, IncA/C2 was associated with sul2/tetA in S. Heidelberg, while IncHI2 carried blaCTX-M-2 and Incl1 was responsible by dissemination of blaCMY-2.
Virulome and Salmonella pathogenicity Island
All 43 Salmonella genomes were analyzed for virulence factors using Prokka anotation and SPIfinder 1.0 (https://cge.cbs.dtu.dk/services/SPIFinder/). Several virulence factors were shared between S. enterica serotypes. All isolates were positive for invA and slyA genes, which are responsible for host invasion and cytolysin production, respectively. Most S. enterica displayed important virulence factors involved in pathogenicity process. For example, fimH (n = 43) is an adhesin responsible for host cell specific recognition26. Further, were detected in all isolates components of Salmonella Pathogenicity Island (SPI) composed of virulence genes such as phoP, phoQ, pagP, sipA, sipB, sipC, and mgtA. The genes phoP and phoQ are responsible for the control of HilE expression which in turn regulates the expression of SPI-127. Additionally, the others virulence genes (pagP, sipABC, mgtA) are involved in modification of lipid A (intracelular survival and ions transport), mechanisms not only involved in pathogenicity, but also in AMR28.
Salmonella Pathogenicity Islands harbor a variety of virulence genes, of which most are chromosomally located. These genes are required for interaction among Salmonella spp. and hosts29. Most of them are integrated with SPIs and the majority of the Salmonella isolates possess SPI-129. However, the absence or partial deletions of these genetic components in certain circumstances does not interfere in the ability to cause disease, remaining potentially pathogenic presenting infection process such as invasion, intracellular survival and replication29. In fact, our findings revealed that not all serovars harbor SPI-1 as shown in Table 3. In this regard, a limitation of this study was the lack of an in vivo analysis for confirmation of the virulence behavior. However, our results confirm previous reports of association between SPI and serotype30 and highlight risk factors associated with Salmonella host infection21.
Interestingly, regarding virulome, we identified IS200 only among S. Brandenburg isolates. This isertion sequence was recently described in S. Typhimurium to be involved in host gene expression31,32.
Phylogenetic and evolutionary dinamics of S. enterica isolates
The core genome used for phylogeny reconstruction represented a sizeable portion of the pan-genome, 3533 out of 8286 total genes in the pan-genome (Fig. 2). Each serotype was represented by a monophyletic clade on the reconstructed phylogeny, and bootstrap values of these clades were greater than 99, representing a high confidence in the phylogenetic topology. Genomic variation was observed between serovars, and specifically, antimicrobial resistance genes varied by serovar. The quinolone resistance gene allele was largely similar within serovar and varied between serovars (Fig. 2). Isolates did not cluster by year, source, or geographic location across the phylogeny.
The investigation of genomic diversity between Salmonella isolates is useful from an epidemiological perspective. We observed that specifically in S. enterica serotype and sequence type are the main drivers for cluster analysis, as most of the time isolates were clustered together by serotype and not by resistance profile, year, source or geographic location. Additionally, our results reveal high similarity among serotype regardless of the year of isolation suggesting the widespread distribution and persistence of Salmonella strains in Brazil.
Although the evolutionary relatedness in S. enterica has been improved in the last decade due broad molecular approach studies33, most often, remains difficult to determine when contamination begins34 or which isolate is considered a common evolutionary ancestor. In this concern, SNP trees were reconstructed using Salmonella isolates (n = 508) retrieved from GenomeTrakr. Ten different clades were identified as shown in Supplementary Fig. S1 and Supplementary Fig. S2. Of these, five were monophyletic [clades-A, B, E, H, J], and five appear to be from novel clades, since our analysis revealed multiple independent lineages of S. enterica serovars S. Infantis [clade-C], S. Schwarzengrund [clade-D], S. Minnesota [clade-F], S. Kentucky [clade-G] and S. Brandenburg [clade-I] (Supplementary Fig. S1).
Clade A was composed of 10 MDR S. Typhimurium isolates recovered from Brazil. Besides our 7 isolates, 1 isolate was recovered in 2009 from an industrialized product (CFSAN033917), as well as 2 isolates were recovered from swine collected in 2012 (CFSAN068037) and 2015 (UFRGS-SA052). Non-association with international strains was observed. In this regard, most likely this clade is endemic in the Brazilian food sector. Most isolates (n = 13) clustered in clade B were clinical or host-associated recovered in USA (PRJNA230403). These isolates grouped together with S. Infantis isolates (SI017 and SI018) from this study. Interestingly, S. Infantis SI690 clustered with 2 pan-susceptible S. Infantis identified in 2016 by this study, constituting a new clade, named clade C.
All isolates from clade D were MDR S. Schwarzengrund recovered from different sources in Brazil. These 21 isolates did not cluster with isolates from other countries. Clade E was mostly constituted by S. Minnesota isolated in the United Kingdom (Supplementary Fig. S1). S. Minnesota isolates SMi124, SMi160 and SSc153 were clustered with 3 S. Minnesota isolated from Gallus gallus in Brazil and 38 S. Minnesota isolates recovered in UK, all being predominantly found in humans and food (Supplementary Fig. S1). Among S. Minnesota, most showed the same resistance profile and carried qnrB19 and blaCMY-2 supporting clonal spread of this lineage. On the other hand, S. Minnesota SMi294 did not cluster together with the previous clade being nested with another S. Minnesota isolate within clade F (Supplementary Fig. S1). Although this isolate carried qnrB19, the resistance profile was pan-susceptible. Therefore, the resistance genotype appear to be a determining factor to clustering this isolate outside of the clade E.
Regarding clade G, two pan-susceptible S. Kentucky ST198 were nested between each other, which appear not to be related to the highly-drug resistant clade disseminated in Africa35.
Clade H, besides our 2 S. Newport isolates recovered from chicken carcass, included an isolate from pilgrims (SAMEA2673767) carrying blaCTX-M-2.
A novel clade (designated I), clustered only two S. Brandenburg isolates (Supplementary Fig. S1), both from poultry samples collected from two different geographic locations in Brazil (Table 1). These isolates were multidrug-resistant harboring important resistance genes, including qnrB19, blaCMY-2, fosA7, aac(6′)-Iaa, sul2 and tetA (Table 1). Lastly, clade J was composed of 403 S. Heidelberg isolates including strains from Brazil, UK and Germany. Of these isolates, 77 were from our Salmonella collection (Supplementary Fig. S2). S. Heidelberg, currently circulating in Europe, are most likely part of a Brazilian clade that has been the most frequent in Brazil. In this serovar, the presence of the plasmid-mediated blaCMY-2, readily mobilized, seems to be a major public health issue.
Discussion
S. enterica harboring qnrB19 has become the most common PMQR gene observed in Brazil and has been increasing in the US36. In addition to this study describing qnrB19 identified among 8 serotypes, there are only three additional reports worldwide on qnrB19-producing S. Infantis37 S. Heidelberg38 and S. Newport39 isolated from Colombia, Venezuela and Poland, respectively. In this context, this is the first know report of qnrB19 in Salmonella serovars S. Schwarzengrund (poultry and environmental), S. Infantis (environmental), S. Minnesota (poultry), S. Brandenburg (poultry), and S. Kentucky (poultry), given that qnrB19 have been reported in Brazil only in S. Corvallis40, in E. coli41,42 or K. pneumoniae43,44.
Isolates harboring qnrE1 had not previously been reported for Salmonella enterica isolated from poultry and swine. Interestingly, qnrE1 was reported in K. pneumoniae from human in Argentina23 and in K. pneumoniae isolated from a parrot in Brazil24. In this regard, to our knowledge, this is the first report of qnrE1 (poultry and swine) in S. enterica serovar Typhimurium worldwide. These results emphasize the plasticity of this new plasmid and the presence of ISEcp1 might be the key vector to silent spread of this resistance gene. These results will aid in the development of mitigation strategies to limit the global distribution of bacteria harboring these genes, since genomic surveillance study allow us to predict, prevent and manage antibiotic resistance and virulence markers in One Health interface, providing substantial evidences that can be implemented in Brazilian food sector.
Two fluoroquinolone-resistant S. Infantis isolated from swine were found to be carrying qnrS1. Conversely, qnrS1 in Brazil was associated only with E. coli41, K. pneumoniae43,45 or Pseudomonas aeruginosa46. In this regard, the presence of oqxA/oqxB in swine highlights their importance as emergence of qnrS1 and oqxA/oqxB quinolone resistance genes being a public health concern in swine production chain.
Lastly, the high prevalence of PMQR shown in this study is consistent with those other studies that found qnrB19 in 201140, 201241, 201343, 201544, 201742, qnrS1 in 201241, 201343, 201445, 201646 and qnrE1 in 201724, in Brazil highlighting an urgent need to strengthen surveillance. This finding emphasizes the importance of persistence of quinolone resistance genes emerging in the poultry and swine production chain and calls for action to arrest further transmission and dissemination.
Although ESBL resistance genes are well described within the environment-food-human interface6,47, the wide distribution of CTX-M-8, CTX-M-2 and CMY-2 ESBL/pAmpC genes are regarded as a threat to public health, as they are easily transferred horizontally to other foodborne pathogens and commensal bacteria in gut environment48. In fact, the high prevalence of ESBLs/pAmpC strains shown in this study is consistent and highlights the endemic occurrence of broad-spectrum cephalosporin strains in South America49. In addition, this is the first report in Brazil, identifying S. Heidelberg harboring a new gene, fosA7, which encodes resistance to fosfomycin [as previously described in S. Heidelberg, also isolated from broiler chicken in Canada]50.
Out of the nine sequence types found, ST19 was associated with strains isolated from swine and poultry. This ST19 appears to be closely related to the sub-Saharan Africa ST313 clade, which is gastroenteritis-associated and globally distributed51,52. However, based on previous investigations53, as well as showed in clade A (Supplementary Fig. S1), we suggest that S. Typhimurium ST19 strains from Brazil are genetically distinct from those ST19 strains associated with gastroenteritis worldwide, given they did not cluster with international isolates.
Sequence type 32 was associated with strains isolated from swine and chicken cage after cleaning. This ST is highly conserved in S. Infantis and has been associated with a clonal dissemination in food sources and human21,54,55. Conversely, isolates from poultry and environmental sources were associated with ST96 and ST15. Interestingly, S. Schwarzengrund (ST96) was previously associated with ESBL from poultry in Brazil [2013]56, carbapenemase resistant KPC-2-producing S. Schwarzengrund from human in Argentina [2014]57, and most recently with mcr-1-producing S. Schwarzengrund isolated from poultry in Brazil [2018]58. In addition, ST15 which had not been previously reported in Brazil has become the most prevalent and relevant serotype in Brazil (manuscript in preparation). Also of interest, STs 548, 198, 45 and 65 were associated with strains isolated from one source (poultry).
In order to support the current knowledge regard the epidemiological distribution of MDR strains between the food-animal-environmental interface, our results provide valuable information related to distribution of multidrug-resistant S. enterica serovars in food-producing animal settings. In addition to the range of mobile genetic elements identified in our isolate collection, these data provide additional insights into global mobility and genomic plasticity, which contribute to persistence of strains along food chain.
The widespread of multi-drug resistant S. enterica in poultry and swine production chain is concerning due the potential transmission to human in the end of food chain. Given these isolates are resistant to fluoroquinolones and third-generation cephalosporin raises a particular concern, since these antibiotics are the first choice for the treatment of salmonellosis. While, our results provide additional evidences of the global mobilization of international clones of S. enterica, over a 16 year-period, continuous surveillance and additional studies in MDR S. enterica isolated from human, needs to be established as mitigation strategies to limit their global spread.
References
Tacconelli, E. et al. Discovery, research, and development of new antibiotics: the WHO priority list of antibiotic-resistant bacteria and tuberculosis. Lancet Infect Dis 18, 318–327, https://doi.org/10.1016/S1473-3099(17)30753-3 (2018).
Jacoby, G. A., Strahilevitz, J. & Hooper, D. C. Plasmid-mediated quinolone resistance. Microbiol Spectr 2, PLAS-0006-2013, https://doi.org/10.1128/microbiolspec.PLAS-0006-2013 (2014).
Li, L. et al. Spread of oqxAB in Salmonella enterica serotype Typhimurium predominantly by IncHI2 plasmids. J Antimicrob Chemother 68, 2263–8, https://doi.org/10.1093/jac/dkt209 (2013).
Redgrave, L. S., Sutton, S. B., Webber, M. A. & Piddock, L. J. Fluoroquinolone resistance: mechanisms, impact on bacteria, and role in evolutionary success. Trends Microbiol 22, 438–45, https://doi.org/10.1016/j.tim.2014.04.007 (2014).
Li, X. P. et al. Clonal spread of mcr-1 in PMQR-carrying ST34 Salmonella isolates from animals in China. Sci Rep 6, 38511, https://doi.org/10.1038/srep38511 (2016).
Bevan, E. R., Jones, A. M. & Hawkey, P. M. Global epidemiology of CTX-M β-lactamases: temporal and geographical shifts in genotype. J Antimicrob Chemother 72, 2145–2155, https://doi.org/10.1093/jac/dkx146 (2017).
Food and Drug Administration. Bacteriological analytical manual. 8th ed. Gaithersburg, Md.: AOAC International (1998).
Grimont PAD, Weil FX. Antigenic formulae of the Salmonella serovars. Institut Pasteur, Paris, France, Centre Collaborateur OMS de Référence et de Recherche pour les Salmonella 9th ed.; [166 pp.] (2007).
Guibourdenche, M. et al. Supplement 2003–2007 (No. 47) to the White-Kauffmann-Le Minor scheme. Res Microbiol 161, 26–9, https://doi.org/10.1016/j.resmic.2009.10.002 (2010).
Clinical and Laboratory Standards Institute (CLSI). Performance standards for antimicrobial susceptibility testing. 27th ed. CLSI supplement M100. CLSI, Wayne, PA (2017).
U S Food and Drug Administration (FDA). The National Antimicrobial Resistance Monitoring System Manual of Laboratory Methods. Retrieved from, https://www.fda.gov/downloads/AnimalVeterinary/SafetyHealth/AntimicrobialResistance/NationalAntimicrobialResistanceMonitoringSystem/UCM453381.pdf (2015).
Monte, D. F. et al. Genome Sequencing of an Escherichia coli Sequence Type 617 Strain Isolated from Beach Ghost Shrimp (Callichirus major) from a Heavily Polluted Ecosystem Reveals a Wider Resistome against Heavy Metals and Antibiotics. Microbiol Resour Announc 8, e01471–18, https://doi.org/10.1128/MRA.01471-18 (2019).
Seemann, T. Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–9, https://doi.org/10.1093/bioinformatics/btu153 (2014).
Page, A. J. et al. Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics 31, 3691–3, https://doi.org/10.1093/bioinformatics/btv421 (2015).
Hadfield, J. et al. Phandango: an interactive viewer for bacterial population genomics. Bioinformatics 34, 292–293, https://doi.org/10.1093/bioinformatics/btx610 (2017).
Page, A. J. et al. SNP-sites: rapid efficient extraction of SNPs from multi-FASTA alignments. Microb Genom 2, e000056, https://doi.org/10.1099/mgen.0.000056 (2016).
Stamatakis, A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–3, https://doi.org/10.1093/bioinformatics/btu033 (2014).
Pattengale, N. D., Alipour, M., Bininda-Emonds, O. R., Moret, B. M. & Stamatakis, A. How many bootstrap replicates are necessary? J Comput Biol 17, 337–54, https://doi.org/10.1089/cmb.2009.0179 (2010).
Letunic, I. & Bork, P. Interactive tree of life (iTOL) v3: an online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res 44, W242–5, https://doi.org/10.1093/nar/gkw290 (2016).
Magiorakos, A. P. et al. Multidrug-resistant, extensively drug-resistant and pandrug-resistant bacteria: an international expert proposal for interim standard definitions for acquired resistance. Clin Microbiol Infect 18, 268–81, https://doi.org/10.1111/j.1469-0691.2011.03570.x (2012).
Moura, Q. et al. Virulent nontyphoidal Salmonella producing CTX-M and CMY-2 β-lactamases from livestock, food and human infection, Brazil. Virulence 9, 281–286, https://doi.org/10.1080/21505594.2017.1279779 (2018).
Hopkins, K. L., Davies, R. H. & Threlfall, E. J. Mechanisms of quinolone resistance in Escherichia coli and Salmonella: recent developments. Int J Antimicrob Agents 25, 358–73, https://doi.org/10.1016/j.ijantimicag.2005.02.006 (2005).
Albornoz, E. et al. qnrE1, a Member of a New Family of Plasmid-Located Quinolone Resistance Genes, Originated from the Chromosome of Enterobacter Species. Antimicrob. Agents Chemother 61, e02555–16, https://doi.org/10.1128/10.1128/AAC.02555-16 (2017).
Cunha, M. P. V. et al. Complete DNA Sequence of an IncM1 Plasmid Bearing the Novel qnrE1 Plasmid-Mediated Quinolone Resistance Variant and bla(CTX-M-8) from Klebsiella pneumoniae Sequence Type 147. Antimicrob Agents Chemother 61, e00592–17, https://doi.org/10.1128/AAC.00592-17 (2017).
Norizuki, C. et al. Specific bla(CTX-M-8)/IncI1 Plasmid Transfer among Genetically Diverse Escherichia coli Isolates between Humans and Chickens. Antimicrob Agents Chemother 61, e00663–17, https://doi.org/10.1128/AAC.00663-17 (2017).
Liu, F. et al. Novel virulence gene and clustered regularly interspaced short palindromic repeat (CRISPR) multilocus sequence typing scheme for subtyping of the major serovars of Salmonella enterica subsp. enterica. Appl Environ Microbiol 77, 1946–56, https://doi.org/10.1128/AEM.02625-10 (2011).
Tang, T., Cheng, A., Wang, M. & Li, X. Reviews in Salmonella Typhimurium PhoP/PhoQ two-component regulatory system. Rev Med Microbiol 24, 18–21, https://doi.org/10.1097/MRM.0b013e32835a9490 (2013).
Marcus, S. L., Brumell, J. H., Pfeifer, C. G. & Finlay, B. B. Salmonella pathogenicity islands: big virulence in small packages. Microbes Infect 2, 145–56, https://doi.org/10.1016/S1286-4579(00)00273-2 (2000).
Abd El Ghany, M. et al. Genomic and Phenotypic Analyses Reveal the Emergence of an Atypical Salmonella enterica Serovar Senftenberg Variant in China. J Clin Microbiol 54, 2014–22, https://doi.org/10.1128/JCM.00052-16. (2016).
Roer, L. et al. Is the Evolution of Salmonella enterica subsp. Enterica Linked to Restriction-Modification Systems? mSystems 1, e00009–16, https://doi.org/10.1128/mSystems.00009-16 (2016).
Ellis, M. J., Trussler, R. S., Charles, O. & Haniford, D. B. A transposon-derived small RNA regulates gene expression in Salmonella Typhimurium. Nucleic Acids Res 45, 5470–5486, https://doi.org/10.1093/nar/gkx094 (2017).
Ellis, M. J. et al. Silent but deadly: IS200 promotes pathogenicity in Salmonella Typhimurium. RNA Biol 15, 176–181, https://doi.org/10.1080/15476286.2017.1403001 (2018).
Timme, R. E. et al. Phylogenetic diversity of the enteric pathogen Salmonella enterica subsp. enterica inferred from genome-wide reference-free SNP characters. Genome Biol Evol 5, 2109–23, https://doi.org/10.1093/gbe/evt159 (2013).
Monte, D. F., Lincopan, N., Fedorka-Cray, P. & Landgraf, M. Current Insights on High Priority Antibiotic-Resistant Salmonella enterica in Food and Foodstuffs: A review. Curr Opin Food Sci 26, 35–46, https://doi.org/10.1016/j.cofs.2019.03.004 (2019).
Le Hello, S. et al. Highly drug-resistant Salmonella enterica serotype Kentucky ST198-X1: a microbiological study. Lancet Infect Dis 13, 672–679, https://doi.org/10.1016/s1473-3099(13)70124-5 (2013).
Tyson, G. H. et al. Identification of Plasmid-Mediated Quinolone Resistance in Salmonella Isolated from Swine Ceca and Retail Pork Chops in the United States. Antimicrob Agents Chemother 61, e01318–17, https://doi.org/10.1128/AAC.01318-17 (2017).
Karczmarczyk, M. et al. Fanning S. Characterization of antimicrobial resistance in Salmonella enterica food and animal isolates from Colombia: identification of a qnrB19-mediated quinolone resistance marker in two novel serovars. FEMS Microbiol Lett 313, 10–9, https://doi.org/10.1111/j.1574-6968.2010.02119.x (2010).
González, F. & Araque, M. Association of Transferable Quinolone Resistance Determinant qnrB19 with Extended-Spectrum β -Lactamases in Salmonella Give and Salmonella Heidelberg in Venezuela. Int J Microbiol 2013, 1–6, https://doi.org/10.1155/2013/628185 (2013).
Wasyl, D., Hoszowski, A. & Zając, M. Prevalence and characterisation of quinolone resistance mechanisms in Salmonella spp. Vet Microbiol 171, 307–14, https://doi.org/10.1016/j.vetmic.2014.01.040 (2014).
Ferrari, R. et al. Plasmid-mediated quinolone resistance by genes qnrA1 and qnrB19 in Salmonella strains isolated in Brazil. J Infect Dev Ctries 5, 496–8, https://doi.org/10.3855/jidc.1735 (2011).
Paiva, M. C., Nascimento, A. M., Camargo, I. L., Lima-Bittencourt, C. I. & Nardi, R. M. The first report of the qnrB19, qnrS1 and aac(6′)-Ib-cr genes in urinary isolates of ciprofloxacin-resistant Escherichia coli in Brazil. Mem Inst Oswaldo Cruz 107, 687–9, https://doi.org/10.1590/S0074-02762012000500018 (2012).
Cunha, M. P. et al. Coexistence of CTX-M-2, CTX-M-55, CMY-2, FosA3, and QnrB19 in Extraintestinal Pathogenic Escherichia coli from Poultry in Brazil. Antimicrob Agents Chemother 61, e02474–16, https://doi.org/10.1128/AAC.02474-16 (2017).
Viana, A. L. et al. Extended-spectrum β-lactamases in Enterobacteriaceae isolated in Brazil carry distinct types of plasmid-mediated quinolone resistance genes. J Med Microbiol 62, 1326–31, https://doi.org/10.1099/jmm.0.055970-0 (2013).
Martins, W. M. et al. Coproduction of KPC-2 and QnrB19 in Klebsiella pneumoniae ST340 isolate in Brazil. Diagn Microbiol Infect Dis 83, 375–6, https://doi.org/10.1016/j.diagmicrobio.2015.09.003 (2015).
Andrade, L. N. et al. Expansion and evolution of a virulent, extensively drug-resistant (polymyxin B-resistant), QnrS1-, CTX-M-2-, and KPC-2-producing Klebsiella pneumoniae ST11 international high-risk clone. J Clin Microbiol 52, 2530–5, https://doi.org/10.1128/JCM.00088-14 (2014).
Araujo, B. F. et al. Clinical and Molecular Epidemiology of Multidrug-Resistant P. aeruginosa Carrying aac(6′)-Ib-cr, qnrS1 and bla SPM Genes in Brazil. PLoS One 11, e0155914, https://doi.org/10.1371/journal.pone.0155914 (2016).
Sjölund-Karlsson, M. et al. CTX-M-producing non-Typhi Salmonella spp. isolated from humans, United States. Emerg Infect Dis 17, 97–9, https://doi.org/10.3201/eid1701.100511 (2011).
Card, R. M. et al. An In Vitro Chicken Gut Model Demonstrates Transfer of a Multidrug Resistance Plasmid from Salmonella to Commensal Escherichia coli. MBio 8, e00777–17, https://doi.org/10.1128/mBio.00777-17 (2017).
Brown, A. C. et al. CTX-M-65 Extended-Spectrum β-Lactamase-Producing Salmonella enterica Serotype Infantis, United States. Emerg Infect Dis 24, 2284–2291, https://doi.org/10.3201/eid2412.180500 (2018).
Rehman, M. A., Yin, X., Persaud-Lachhman, M. G. & Diarra, M. S. First Detection of a Fosfomycin Resistance Gene, fosA7, in Salmonella enterica Serovar Heidelberg Isolated from Broiler Chickens. Antimicrob Agents Chemother 61, e00410, https://doi.org/10.1128/AAC.00410-17 (2017).
Ramachandran, G. et al. Virulence of invasive Salmonella Typhimurium ST313 in animal models of infection. PLoS Negl Trop Dis 11, e0005697, https://doi.org/10.1371/journal.pntd.0005697 (2017).
Hammarlöf, D. L. et al. Role of a single noncoding nucleotide in the evolution of an epidemic African clade of Salmonella. Proc Natl Acad Sci USA 115, E2614–E2623, https://doi.org/10.1073/pnas.1714718115 (2018).
Panzenhagen, P. H. N. et al. Genetically distinct lineages of Salmonella Typhimurium ST313 and ST19 are present in Brazil. Int J Med Microbiol 308, 306–316, https://doi.org/10.1016/j.ijmm.2018.01.005 (2018).
Hauser, E. et al. Clonal dissemination of Salmonella enterica serovar Infantis in Germany. Foodborne Pathog Dis 9, 352–60, https://doi.org/10.1089/fpd.2011.1038 (2012).
Almeida, F., Pitondo-Silva, A., Oliveira, M. A. & Falcão, J. P. Molecular epidemiology and virulence markers of Salmonella Infantis isolated over 25 years in São Paulo State, Brazil. Infect Genet Evol 19, 145–51, https://doi.org/10.1016/j.meegid.2013.07.004 (2013).
Silva, K. C. et al. Emergence of extended-spectrum–lactamase CTX-M-2-producing Salmonella enterica serovars Schwarzengrund and Agona in poultry farms. Antimicrob Agents Chemother 57, 3458–3459, https://doi.org/10.1128/AAC.05992-11.19 (2013).
Jure, M. A. et al. Emergence of KPC-2-Producing Salmonella enterica Serotype Schwarzengrund in Argentina. Antimicrob Agents Chemother 58, 6335–6, https://doi.org/10.1128/AAC.03322-14 (2014).
Moreno, L. Z. et al. First report of mcr-1-harboring Salmonella enterica serovar Schwarzengrund isolated from poultry meat in Brazil. Diagn Microbiol Infect Dis 0, S0732-8893(18)30561–3, https://doi.org/10.1016/j.diagmicrobio.2018.10.016 (2018).
Acknowledgements
This study was granted by Fundação de Amparo à Pesquisa do Estado de São Paulo-FAPESP, [Food Research Center (FoRC-2013/07914-8) and 2016/03044-7]. The project has been partially developed during D.F.M time as visiting scholars at the North Carolina State University under fellowship grant from FAPESP (2017/15967-5). We are in debt with Dr. Joy Horovitz (NCSU/USA) for the valuable support in the whole-genome sequencing.
Author information
Authors and Affiliations
Contributions
D.F.M., M.L., N.L. and P.J.F.C. performed the conception and design of the study. D.F.M. carried out all in vitro assays and analysed the data. Also, interpreted the results, performed literature review, prepared tables, figures and wrote the manuscript. M.L. acquired the Salmonella enterica isolates, while P.J.F.C., S.T. and S.K. provided the whole-genome sequencing. D.F.M., H.B. and L.C. built the phylogeny data and genetic environments. All authors had the opportunity to review the manuscript.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Monte, D.F., Lincopan, N., Berman, H. et al. Genomic Features of High-Priority Salmonella enterica Serovars Circulating in the Food Production Chain, Brazil, 2000–2016. Sci Rep 9, 11058 (2019). https://doi.org/10.1038/s41598-019-45838-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-45838-0
This article is cited by
-
CRISPR and CRISPR–MVLST reveal conserved spacer distribution and high similarity among Salmonella enterica serovar Infantis genomes from Brazil and other countries
Molecular Genetics and Genomics (2024)
-
Multiobjective response and chemometric approaches to enhance the phytochemicals and biological activities of beetroot leaves: an unexploited organic waste
Biomass Conversion and Biorefinery (2023)
-
Genomic analyses of drug-resistant Salmonella enterica serovar Heidelberg strains isolated from meat and related sources between 2013 and 2017 in the south region of Brazil
Current Genetics (2023)
-
Genomic characteristics and comparative genomics of Salmonella enterica subsp. enterica serovar Schwarzengrund strain S16 isolated from chicken feces
Gut Pathogens (2022)
-
Genome characteristics of clinical Salmonella enterica population from a state public health laboratory, New Hampshire, USA, 2017–2020
BMC Genomics (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
