
Abstract
Genetic isolation of populations over evolutionary time leads to the formation of independent species. We examined a pair of shorebirds – the Kentish Plover Charadrius alexandrinus and the enigmatic White-faced Plover C. dealbatus – which display strong plumage differentiation, yet show minimal genetic divergence based on previous mitochondrial and microsatellite work. Two scenarios may lead to this situation: (1) they represent clinal or poorly diverged populations with limited genomic differentiation despite substantial plumage variation, or (2) they are diverging taxa at the cusp of speciation, with ongoing limited gene flow obliterating signals of differentiation in traditional genetic markers. We compared the genotypes of 98 plovers (59 Kentish Plovers, 35 White-faced Plovers and 4 genomic hybrids) sampled in eastern Asia and Europe using ddRADSeq to harvest over 8000 genome-wide SNPs. In contrast to previous studies, our analyses revealed two well defined genomic clusters, with limited hybridization and a narrow contact zone. We also uncovered significant differences in bill length and further sex-specific differences in size, which may signal differences in mate choice between Kentish and White-faced Plovers. Our results support the hypothesis that this shorebird duo is on the verge of speciation.
Similar content being viewed by others


Introduction
“Firstly, why, if species have descended from other species by insensibly fine gradations, do we not everywhere see innumerable transitional forms? Why is not all nature in confusion, instead of the species being, as we see them, well defined?” – Darwin (1859)1
Speciation is the evolutionary process by which populations differentiate to become distinct species. The formation of barriers to gene flow plays an important role in the speciation process2, with allopatric barriers being its most well recognized driver3. However, there is now growing recognition that variation in intensity of sexual selection or preferences for different traits may also lead to species divergence4,5. Speciation can occur extremely rapidly over the course of a few generations2,6 or over long time scales, and may or may not be accompanied by gene flow from overlapping congenerics7. Consequently, young species can often be difficult to distinguish from divergent populations8,9,10. Biologists often conservatively use the term ‘taxon’ to refer to difficult cases in which there is no consensus on whether a lineage qualifies as a full species or is merely a divergent population.
Shorebirds (order Charadriiformes) are an old avian lineage with high behavioural and morphological diversity11,12,13,14. Charadrius plovers, one of the core clades and the name-sake group, have been an important study subject in evolutionary and ecological research given their unusual diversity in mating and parental care systems, even within a single species15,16. This variation in breeding behaviour has also been linked to population differentiation and diversification, whereby monogamous breeding systems in plovers are thought to be key promoters for population divergence whereas polygamous behaviour may instead slow down divergence5. The advent of Next-Generation Sequencing (NGS) technologies provides us with powerful tools to investigate differentiation across plovers from a genomic perspective17.
Despite the status of Charadrius as an extremely well-studied bird genus, the last few years have seen the discovery of novel populations and taxa18. Among these, Kennerley et al.19 documented the discovery of a distinctly-plumaged East Asian population of the Kentish Plover Charadrius alexandrinus complex that was shown to refer to the generally synonymized taxon name dealbatus. Several aspects regarding the breeding plumage of this rediscovered taxon, such as the extent of white on the face, and minor differences in morphology, render it as distinct as many universally recognized plover species19,20, which has led to its occasional recognition as an independent species named White-faced Plover C. dealbatus (e.g.,21). However, a first molecular inquiry using mitochondrial DNA and microsatellites on museum specimens found no evidence of genetic differentiation between White-faced and Kentish Plovers20. While the two taxa overlap widely in their Southeast Asian winter distribution (Fig. 1), the breeding grounds of the enigmatic White-faced Plover have never been characterised in detail, although they have been found to breed along the southern coastline of China and northernmost Vietnam22. It is therefore unclear to what extent the breeding range of White-faced Plovers overlaps with the breeding range of Kentish Plovers.

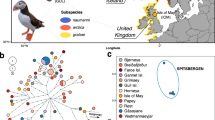
Map of sampling locations and STRUCTURE results at K = 3 for eastern Kentish and White-faced Plover populations. The inset shows global distribution of Kentish and White-faced Plovers22: green areas demarcate year-round resident populations of Kentish Plovers, purple shows breeding ranges of migratory populations and blue areas show wintering regions; the thin dotted line (• • •) outlines the presumed breeding range of White-faced Plovers and the stippled line (−) demarcates their known wintering range. Cross and open diamond refer to sampling localities in Spain and far western China respectively. Samples collected during the breeding season (May–July) are outlined in red whilst those collected during the non-breeding season are outlined in blue. Sampling dates for Taiwan are unknown and thus lack outlines. Sampling locality acronyms are as follows: QH – Qinghai, QD – Qinhuangdao, JP – Japan, SH – Shanghai, YJ – Yuanjiang, WZ – Wenzhou, WL – Wenling, FZ – Fuzhou, PT – Putian, XM – Xiamen, TW – Taiwan, B-Z – Beihai-Zhanjiang. Map modified from https://maps-for-free.com/ (© OpenStreetMap contributors) using Adobe Illustrator CC v.23.0.1 (https://www.adobe.com/products/illustrator.html).
The differentiation in breeding plumage between White-faced and Kentish Plovers in the absence of mitochondrial and microsatellite differentiation has posed a conundrum that is difficult to reconcile. On the one hand, these may represent two populations with poor genomic divergence but substantial plumage differentiation. Under this scenario, we would expect a broad genetic cline across geographic populations (Fig. 2A). On the other hand, they may represent two deeply differentiated subspecies or even young incipient species whose divergence in certain traits does not track their level of genetic diversification, possibly because of continuing low levels of gene flow and introgression. In this case, instead of observing genomic clinality and gradual changes between the two breeding populations, we would find a narrow contact zone of overlap and gene flow (Fig. 2B). Under either scenario, the most important arena in which to study gene flow would be the central to southern coastline of China, an area suspected to be the overlapping breeding zone for both Kentish Plovers and White-faced Plovers.

Two hypotheses for the interaction between Kentish and White-faced Plovers: (A) shallow divergence with widespread gene flow - a genomic cline from northern to southern China, with the greatest differentiation seen at either end of the spectrum of distribution and a broad hybrid zone, and (B) deeper divergence with limited gene flow – the two taxa meet at a narrow contact zone and show limited hybridization.
Here we investigate the two possible scenarios by examining the patterns of genomic differentiation between plovers along the east coast of China. We used a genotyping-by-sequencing approach23 to harvest over 8000 genome-wide SNPs across 98 individuals (~60% Kentish Plovers, ~40% White-faced Plovers). Our taxon sampling covers a broad geographic range to investigate the magnitude of differentiation and dynamics of the contact zone, and includes a densely sampled coastal transect in China, a population from the western distribution end of Kentish Plovers in Spain, and a population from another East Asian subspecies nihonensis that inhabits island archipelagos at the northeastern end of the Kentish Plover breeding distribution. Our sampling incorporates a comprehensive dataset of body measurements to explore whether biometric changes correlate with the genomic clines. Our objective is to establish the nature of genomic connectivity between these two taxa to ascertain how and if two such divergent plumage phenotypes can be maintained in the presence of gene flow.
Results
Population genetic structure and hybridization
We recovered a total of 1,729,307,240 paired-end 150 bp reads from Illumina sequencing of 122 plover samples. Preliminary filtering of the sequencing data included manually removing seven individuals with poor sequencing coverage (less than one million reads). Each of the remaining 115 samples had a mean sequencing depth of 139x. A further nine individuals were removed in PLINK 2.0 as they exhibited more than 30% missing stack data, whilst eight additional individuals that showed evidence for first or second-order kinship with another individual were also pruned. This yielded a final dataset of 98 individuals and 8088 SNPs. The total genotyping rate was 0.990466. Of these 98 individuals, 59 were Kentish Plovers, 35 were White-faced Plovers and four consistently displayed genomic signatures intermediate between the two taxa in both STRUCTURE and PCA (see below).
Our population genetic analysis using STRUCTURE showed a primary division between Kentish Plovers and White-faced Plovers, and subsequent division between allopatric subspecies alexandrinus (the Chinese mainland and Taiwanese samples) and nihonensis (Japanese samples, Figs 1, 3 and S2). Our Netview results mirrored this pattern, with White-faced Plovers forming a tight cluster and Kentish Plovers forming a looser cluster, and with Japanese and Spanish individuals outside the main Kentish Plover cloud (Fig. S1). Our result for the Japanese samples is consistent with the traditional recognition of the Japanese Kentish Plover population as an independent subspecies Charadrius alexandrinus nihonensis, characterized by a longer bill and slightly duller breeding plumage24.
For the remaining samples, individual cluster assignments showed a sharp population genetic break from north to south along the eastern Chinese coastline, with a narrow zone of overlap around the border between the provinces of Fujian and Zhejiang. The majority of birds sampled in Fujian were White-faced Plovers (32 out of 52 individuals, or 62%) and the minority were Kentish Plovers (19 out of 52 individuals, or 37%). More importantly, when we considered only records from the breeding season (April–July, the percentage of White-faced Plovers in northern Fujian increased to 83% (29 out of 35 individuals). Conversely, in southern Zhejiang, 78% of breeders were Kentish and only 22% were White-faced Plovers (7 and 2 individuals respectively; Fig. 1). A single individual in northern Fujian emerged as a genomic hybrid between the two clusters (Fig. 3).

Principal component analysis of the complete final dataset of 98 individuals using 8088 SNPs, explaining 5.51% of the total variance observed. Colours and shapes of locality symbols match those shown in Fig. 1.
In Taiwan, a large island east of Fujian, five of the eight (62.5%) plovers sampled were Kentish Plovers (Fig. 1). The remaining three individuals had genotypes consistent with hybrids between Kentish and White-faced Plovers (Fig. 3).
The results of the Principal Component Analysis (PCA) were consistent with the STRUCTURE results. White-faced and Kentish Plovers separated along PC1, with White-faced Plovers forming a tight cloud, whereas Kentish Plovers were more broadly dispersed along both axes (Fig. 3). The distribution of Kentish Plovers along PC2 from top to bottom roughly matched a continental East-West cline, with samples from Japan on one end and samples from Spain on the other. Samples from western China (Qinghai and Xinjiang) emerged as intermediate between the coastal eastern Chinese and Spanish samples, further reinforcing the East-West clinal distribution.
Our summary statistics also echoed the STRUCTURE results, with White-faced Plovers displaying high genetic divergence and population differentiation from all Kentish Plover populations. Within the Kentish Plover populations, the Japanese population (nihonensis) consistently appeared to be the most distinct, whilst divergences of Spanish and Western Chinese populations mirrored their geographic distance from other populations.
Morphological differentiation
The results of all Shapiro-Wilk tests for normality of the morphological traits were non-significant (P > 0.05 for all tests). Two way ANOVAs showed that White-faced Plovers had longer bills than Kentish Plovers (White-faced Plovers: 17.97 ± 0.64 mm [mean ± SD], Kentish Plovers: 16.94 ± 1.10 mm) (\({F}_{(\mathrm{1,1})}\)= 13.28, P = 0.0008, Table S3), with 27% of the variance in bill length explained by ‘taxon’. Additionally, we found significant species-sex interactions for three other size measurements (Tarsus (\({F}_{(\mathrm{1,1})}\)= 10.17, P = 0.0029); tail (\({F}_{(\mathrm{1,1})}\,\)= 4.714, P = 0.0366) and full body length (\({F}_{(\mathrm{1,1})}\)= 5.95, P = 0.0199): White-faced Plover males were significantly longer than Kentish Plover males in full body length (and had non-significantly longer tarsi and tails), but there was no significant difference between White-faced and Kentish Plover females for all three measurements (Table S3; Fig. 4). The sole hybrid individual for which biometric data were available was female, and exhibited measurements more closely resembling White-faced Plovers than Kentish Plovers (Fig. 4).

Box plots of morphological traits with significant sex-specific interactions for Kentish (left) and White-faced Plovers (right). From top to bottom: Tarsus length, tail length and full length measurements. Males are depicted in blue and females in red. Significance was calculated using student’s t-test.
Discussion
The evolutionary history of the White-faced Plover has long been debated by ornithologists and taxonomists alike. On the one hand, this taxon is uniquely distinct in its breeding plumage, with coloration differences that surpass those of many other Charadrius plover species19,20. On the other hand, evidence from mitochondrial DNA and a set of microsatellites have revealed limited differentiation between White-faced and Kentish Plovers20,25. While they overlap widely in their wintering range across Southeast Asia and southern China, their breeding range dynamics are little understood, and it is not known whether their breeding distributions meet in a clinal fashion or if there is a discrete contact zone between the two taxa (Fig. 2).
Two discrete taxa with limited genomic intermediacy
Genomic analysis of more than 8000 SNPs showed that White-faced and Kentish Plovers are clearly differentiated. This differentiation is at a level higher than that observed between Japanese nihonensis and Chinese alexandrinus Kentish Plovers in this region, suggesting that dealbatus and alexandrinus populations have already diverged further (Fig. 1; Fig. 3; Fig. S1). The deepest genomic divisions within our sampling regime were exclusively between White-faced Plovers and Kentish Plovers (K = 2 in Fig. 1; PC1 in Fig. 3), even though our geographic sampling encompassed Spanish individuals from the far western end of the Kentish Plover’s breeding distribution roughly 11,000 km from the sampling sites on the Chinese coast. This confirms earlier results26 showing that across continental Eurasia, Kentish Plovers exhibit very low differentiation. Our results show that genomic divisions between White-faced and Kentish Plovers from the same areas in Fujian Province (Fig. 1) appear much deeper than those between members of opposing ends of the Kentish Plover continental distribution (Table 1).
What is the nature of gene flow dynamics between Kentish and White-faced Plovers in areas of potential sympatry? To answer this question, we relied on fine-scale sampling especially on the coast of northern Fujian and southern Zhejiang provinces, where their breeding ranges come close to each other (Fig. 1). Despite our intensive sampling of 68 genotyped individuals across China’s continental coastline, we only clearly identified a single individual ( < 1.5%) near Fuzhou that exhibited a signature of genomic admixture, with roughly ¼ White-faced and ¾ Kentish Plover ancestry in its overall make-up. All other individuals sampled, including all other 60 birds from Fujian and Zhejiang, had genotypes that were fully assigned to pure Kentish or White-faced clusters based on over 8000 SNPs (e.g., Fig. 1; Fig. 2). This near-dichotomous pattern suggests that although White-faced and Kentish Plovers still may have the capacity to interbreed, they probably only do so infrequently. Such reproductive isolation is typical for young avian sister species meeting along a narrow hybrid zone27,28,29. From our transect data, the area where the breeding ranges of Kentish and White-faced Plovers meet appears to be in southern and central Zhejiang. Between Taizhou and Wenzhou along Zhejiang’s shoreline, our sampling during the breeding season revealed only two White-faced Plovers and seven Kentish Plovers (Fig. 1). Further south, in Fujian, 83% of our 35 samples from May and June were White-faced Plovers (excluding the one hybrid). The majority (16; i.e., 80%) of the 20 samples from non-breeding months (January, March, November) in Fujian and further south were pure Kentish Plovers based on their genomic SNP profiles, while the remainder were White-faced Plovers (Fig. 1). This pattern is consistent with the Kentish Plover’s higher global abundance, and with the notion that Kentish Plovers use the breeding areas of White-faced Plovers during winter and during migration.
On the island of Taiwan, we detected another three out of eight individuals with a similarly admixed profile as the one hybrid from Fujian, with the remainder being pure Kentish Plovers (Fig. 1). This suggests a fundamentally different interaction between these two taxa on Taiwan compared to the mainland, and in agreement with our own observations that White-faced Plovers seem to be a marginally occurring bird on Taiwan’s west coast that is greatly outnumbered by Kentish Plovers (per. obs.). In situations where two hybridizing species co-occur, hybridization is typically promoted by the rarer species10,30,31,32. On Taiwan, consequently, the two plovers may find themselves in a situation that favours hybridization more than on the mainland, where the two seem to hybridize much more rarely. Future investigations into the dynamics of breeding populations on Taiwan will shed more light on this finding.
Phenotypic differentiation in the Kentish Plover complex
White-faced Plovers are known to differ greatly in breeding plumage coloration and by a number of biometric measurements from other members of the Kentish Plover complex19,20,25. In this study, we corroborated these phenotypic differences based on unequivocal genomic assignments and examined further differences between the sexes. Regardless of sex, White-faced Plovers had a significantly longer bill than Kentish Plovers based on bill only and head plus bill measurements (Table S3). This is consistent with other recent results25. As for tarsus, tail and full length measurements, we detected a significant sex-specific interaction whereby male White-faced Plovers were larger than male Kentish Plovers, even though female White-faced Plovers were of equal size or even marginally smaller than Kentish Plover females (Table S3; Fig. 4). This phenomenon may hint towards differences in the intensity of sexual selection16 or male competitiveness between species33, which could play a role in reproductive isolation34 or even the dynamics of speciation. Based on observed differences in size, it is plausible that White-faced Plovers displace Kentish Plovers during the breeding season on the southern Chinese coast. Particularly, male Kentish Plovers, with their smaller overall body size (Fig. 4), may be less competitive than White-faced Plovers when it comes to establishing and defending territories.
The White-faced Plover’s distinctly longer bill is also consistent with a continent-wide trend within the Kentish Plover complex towards increasing bill length towards the east. The Japanese subspecies nihonensis has been hitherto defined almost solely on account of its distinctly longer beak in comparison with alexandrinus, even though there are also differences in breeding plumage24. Consistent with previous studies26, Japanese Kentish Plovers formed a fairly distinct genetic cluster in both PCA and STRUCTURE analyses and showed high levels of differentiation from samples collected from the adjacent northern Chinese mainland (Fig. 1; Table 1), substantiating their division as an independent subspecies, although their differentiation is much shallower than that of White-faced Plovers.
In the future, comparative behavioural studies should focus on the interactions of the two taxa in the contact zone during the breeding season in order to relate patterns of sexual dimorphism and taxon differences to sexual reproductive traits. In Charadrius plovers, breeding systems are known to influence the propensity of a species to differentiate, with polygamous species showing lower population differentiation than monogamous species5,26, but great variation in behaviour and plumage characteristics exists even within a single species such as the Kentish Plover35.
Conclusion
We have shown that White-faced and Kentish Plovers are two genomically diverged taxa. Sampling across a transect spanning their continental contact zone and adjacent areas, we identified only few (<1.5%) hybrids among our samples of breeding birds, indicating that gene flow, while present, is rare between these two taxa. Our results favour a narrow contact zone resulting in two lineages with deep genomic differentiation, rather than a genetic cline from one form to the other (Fig. 2B).
Their pronounced differences in breeding plumage coloration are matched by different bill sizes and sex-specific differences in tarsus, wing and full length measurements, hinting at differences in the intensity of sexual selection between the two taxa. Future research into differences in the reproductive behaviour of these two young taxa may shed light on the evolution of sexual traits in these two shorebirds.
Methods
Field sampling
We sampled blood from 122 individuals belonging to the Kentish Plover Charadrius alexandrinus complex (including White-faced Plover C. dealbatus), mainly in China across a coastal transect spanning the breeding distribution of the two forms (n = 87), but also including samples from Taiwan (n = 18), Japan (n = 10) and Spain (n = 7) (Table S1). We captured plovers either using walk-in funnel traps placed on nests or with mist nets. Approximately 30–50 µL of blood were collected from each bird via brachial venipuncture before releasing the bird. We stored blood in ethanol or Queen’s Lysis Buffer36 until further processing in the laboratory. Sampling protocols were approved by Jiangsu Normal University and the National Natural Science Foundation of China.
ddRADSeq library preparation
The following procedures were approved by National University of Singapore’s Office of Safety, Health, and Environment. We extracted DNA either with DNEasy Blood & Tissue Kits (Qiagen) using the manufacturer’s recommended protocol or using an ammonium salt method37. We used a Qubit® 2.0 Fluorometer to quantify DNA concentrations of extracts.
We performed double digest restriction enzyme associated DNA sequencing (ddRADseq38) as per Tang et al.23. We split samples into total DNA yield bands between 170–500 ng, based on post-extraction concentrations. We then calculated input volumes for the first restriction step based on these cut-off values. We used the restriction enzymes EcoRI and Msp1 (New England Biolabs Inc.) to double digest the samples for 3.5 hours at 37 °C, followed by clean up using a 1.1X ratio of Sera-Mag SpeedBead Carboxylate-Modified Magnetic Particles (Thermo Scientific). We then quantified samples again and ligated unique barcodes (PIE adaptors) to the fragments using T4 DNA Ligase (New England Biolabs Inc.) at 16 °C for 16 hours.
For subsequent library preparation, we split the samples into seven pools for distribution across two Illumina HiSeq 4000 lanes according to their post-restriction concentrations. We pooled samples with similar post-restriction concentrations to avoid either over or under-representation of individual samples. We cleaned up each pool with AMPure XP beads (Agencourt) using a 1.5X bead ratio, and then carried out size selection for the seven pools using a Pippin Prep Gel Electrophoresis system (Sage Science) to isolate fragments for a sample peak of 420 base-pairs in length. After another subsequent AMPure XP clean up, we amplified size-selected fragments using a polymerase chain reaction (PCR) for 12 cycles, followed by a final AMPure XP clean up step. We screened pools for quality control on a Fragment Analyzer (Advanced Analytical) and then quantified pools on a Qubit 2.0 Fluorometer before creating two final libraries of pools in equimolar proportions. We submitted the DNA libraries to Novogene (Tianjin, China) for 150 bp paired-end sequencing using the Illumina HiSeq X Ten platform.
Data processing and SNP calling
We used FastQC (Babraham Bioinformatics) to analyze sequence quality across all base positions. Demultiplexing was then performed using the process_radtags command in STACKS v1.3439. We removed samples with less than one million reads from subsequent analysis. We aligned the sequence reads of the remaining 115 samples against the congeneric Killdeer (Charadrius vociferus) genome assembly40 using BWA-MEM41.
We used the pipeline ref_map.pl in STACKS to call single nucleotide polymorphic markers (SNPs), employing a minimum stack depth of 10 in our analyses. We ran the ‘populations’ module to retain loci present in more than 90% of individuals, whilst assigning all individuals to a single population. To reduce the effects of linkage disequilibrium in subsequent analyses, we only accepted one SNP from each locus using the –write_single_snp option provided.
We ran PLINK 2.042 to remove any individuals exhibiting more than 30% missing data, and then to further filter SNPs to account for missing data and linkage disequilibrium. We checked for SNPs under selection using BayeScan43.
We used the R package, SNPRelate44 to estimate pairwise kinship coefficients using maximum likelihood estimation. We removed a single individual from each pair which displayed a kinship coefficient above 0.125 (second order kinship). We then re-ran the populations module in Stacks v1.34 and re-ran PLINK 2.0 without filtered individuals/loci to generate the final dataset.
Population genetic analysis
We assessed population subdivision using a model-based clustering approach implemented in STRUCTURE v2.3.445. First, we removed any singleton or doubleton alleles46 using a minimum allele count filter in PLINK 2.0. We then implemented STRUCTURE runs using the remaining 5639 SNPs without a priori hypotheses of cluster membership. We ran STRUCTURE from K = 1 to K = 10 with five iterations per K. For each iteration we implemented a burn-in of 100,000 generations and MCMC for 500,000 generations. We used STRUCTURE Harvester Web v0.6.9447 to process the output and subsequently averaged the results across replicates by evaluating individual ancestry coefficients (q values) with CLUMPP v1.1.248. Averaged population subdivision over changing values of K is depicted in Fig. S2, while the results of K = 3, the optimum K value as determined by the Evanno method, which calculates the most statistically likely number of genetic clusters47, are additionally incorporated into Fig. 1.
We then used Netview, a network theory-based approach, to construct networks of individuals illustrating the connectivity and information flow within and between populations based on genetic similarity49,50. Different network topologies were explored by specifying different values of k, which dictates the number of genetically nearest neighbors each sample is connected to. We ran Netview for k values of 1–35, and plotted the resultant graphs using the Kamada-Kawai force-directed graph drawing algorithm51 as implemented in iGraph52. We explored population structure using a PCA in SNPRelate44 using a genetic covariance matrix calculated from genotypes.
Among our samples we identified hybrids between Kentish and White faced Plover based on their consistent genotypic assignment indicating mixed genotypes based on both STRUCTURE and PCA analyses.
We calculated pairwise population summary statistics for Kentish and White-faced Plover populations using the R packages Hierfstat53 and Adegenet54,55. We divided our Kentish Plover samples into four populations (Spain, western China, eastern China and Japan) and retained White-faced Plovers as a single population. We excluded any hybrids we identified from this analysis. We utilized Nei’s genetic distance in Adegenet to calculate \({{\rm{d}}}_{{\rm{xy}}}\) and calculated pairwise \({{\rm{F}}}_{{\rm{ST}}}\) using Hierfstat’s ‘Nei87’56.
Morphological analysis
We recorded morphological measurements for a total of 54 birds sampled along the Chinese coastline (Table S2), including the lengths of wings, tarsi, bills, as well as body weight. We measured tarsus lengths from the outer bend of the tibiotarsal articulation to the base of the toes, and recorded bill length as the distance of the tip of the rostrum to the base of the forehead. We did not have access to morphological measurements for many of the samples from other localities, and in cases where we did we chose not to include them to reduce recorder bias from influencing our data analyses.
We obtained ddRADSeq genotypes for 41 of the 54 measured individuals. These comprised 13 males and 28 females, of which we identified 11 birds based on their genotypes as Kentish Plovers, 29 as White-faced Plovers and one as a hybrid (see Results). We assessed normality of the morphological data using Shapiro-Wilk tests57. For traits that adhered to a normal distribution we examined differences between sexes and taxa using two-way ANOVAs. We utilized the R package ggplot258 to generate boxplots comparing morphological measurements between the two genomically-prescribed groups for the measurements which showed significant interactions between taxa and sex, and plotted relevant significance bars with the R package ggsignif59. We used R version 3.4.1 for all statistical analyses60.
Ethics statement
Research protocols were approved by Jiangsu Normal University and National Natural Science Foundation of China. All lab work was conducted in accordance with regulations outlined by the National University of Singapore’s Office of Safety, Health, and Environment.
Data Availability
The Kentish and White-faced Plover genomic data are pending accession on Genbank NCBI. Raw Stacks output and custom scripts are available from the corresponding authors upon request. All other data analysed during this study are included in the Supplementary information files.
References
Darwin, C. On the origin of species by means of natural selection. (Murray, 1859).
Coyne, J. A. & Orr, H. A. Speciation. (Sinauer Associates Sunderland, 2004).
Mayr, E. Systematics and the origin of species, from the viewpoint of a zoologist. (Columbia University Press, 1942).
Seddon, N. et al. Sexual selection accelerates signal evolution during speciation in birds. Proc. R. Soc. B 280, 20131065 (2013).
D’Urban Jackson, J. et al. Polygamy slows down population divergence in shorebirds. Evolution 71, 1313–1326 (2017).
Lamichhaney, S. et al. Rapid hybrid speciation in Darwin’s finches. Science 359, 224–228 (2018).
Martin, S. H. et al. Genome-wide evidence for speciation with gene flow in Heliconius butterflies. Genome Res. gr, 159426 (2013).
Ng, N. S. et al. The effects of Pleistocene climate change on biotic differentiation in a montane songbird clade from Wallacea. Mol. Phylogenet. Evol. 114, 353–366 (2017).
Irestedt, M. et al. The spatio-temporal colonization and diversification across the Indo-Pacific by a ‘great speciator’(Aves, Erythropitta erythrogaster). Proc. R. Soc. Lond. 280, 20130309 (2013).
vonHoldt, B. M., Kays, R., Pollinger, J. P. & Wayne, R. K. Admixture mapping identifies introgressed genomic regions in North American canids. Mol. Ecol. 25, 2443–2453 (2016).
Bulla, M. et al. Unexpected diversity in socially synchronized rhythms of shorebirds. Nature 540, 109–113 (2016).
Piersma, T. & Lindström, Å. Migrating shorebirds as integrative sentinels of global environmental change. Ibis 146, 61–69 (2004).
Thomas, G. H., Szekely, T. & Reynolds, J. D. Sexual conflict and the evolution of breeding systems in shorebirds. Adv. Study Behav. 37, 279–342 (2007).
Küpper, C. et al. A supergene determines highly divergent male reproductive morphs in the ruff. Nat. Genet. 48, 79–83 (2016).
Dos Remedios, N., Lee, P. L., Burke, T., Székely, T. & Küpper, C. North or south? Phylogenetic and biogeographic origins of a globally distributed avian clade. Mol. Phylogenet. Evol. 89, 151–159 (2015).
Székely, T., Reynolds, J. D. & Figuerola, J. Sexual size dimorphism in shorebirds, gulls, and alcids: the influence of sexual and natural selection. Evolution 54, 1404–1413 (2000).
McCormack, J. E., Hird, S. M., Zellmer, A. J., Carstens, B. C. & Brumfield, R. T. Applications of next-generation sequencing to phylogeography and phylogenetics. Mol. Phylogenet. Evol. 66, 526–538 (2013).
Küpper, C. & dos Remedios, N. Defining species and populations: molecular genetic studies in plovers. Stud. Avian Biol (CRC Press, 2019).
Kennerley, P. R., Bakewell, D. N. & Round, P. D. Rediscovery of a long-lost Charadrius plover from South-East. Asia. Forktail 24, 63–79 (2008).
Rheindt, F. E. et al. Conflict between genetic and phenotypic differentiation: the evolutionary history of a ‘lost and rediscovered’shorebird. PLoS One 6, e26995 (2011).
del Hoyo, J., Collar, N., Kirwan, G. M. & Sharpe, C. J. White-faced Plover (Charadrius dealbatus). In: del Hoyo, J., Elliott, A., Sargatal, J., Christie, D. A. & de Juana, E. (eds). Handbook of the Birds of the World Alive. https://www.hbw.com/node/467300 (2018).
Craik, R. C. & Lê, M. Q. Birds of Vietnam. (Lynx Edicions, 2018).
Tang, Q., Low, G. W., Lim, J. Y., Gwee, C. Y. & Rheindt, F. E. Human activities and landscape features interact to closely define the distribution and dispersal of an urban commensal. Evol. Appl. 11, 1598–1608 (2018).
Deignan, H. G. Remarks on the Kentish Plovers of the extreme Orient, with separation of a new subspecies. J. Washington Acad. Sci. 31, 105–107 (1941).
Wang, X. et al. Divergence in the face of gene flow in two Charadrius plovers along the Chinese coast. BioRxiv, 406041 (2018).
Küpper, C. et al. High gene flow on a continental scale in the polyandrous Kentish plover Charadrius alexandrinus. Mol. Ecol. 21, 5864–5879 (2012).
Aguillon, S. M., Campagna, L., Harrison, R. G. & Lovette, I. J. A flicker of hope: Genomic data distinguish Northern Flicker taxa despite low levels of divergence. Auk 135, 748–766 (2018).
Carling, M. D., Serene, L. G. & Lovette, I. J. Using historical DNA to characterize hybridization between Baltimore Orioles (Icterus galbula) and Bullock’s Orioles (I. bullockii). Auk 128, 61–68 (2011).
Mettler, R. D. & Spellman, G. M. A hybrid zone revisited: molecular and morphological analysis of the maintenance, movement, and evolution of a Great Plains avian (Cardinalidae: Pheucticus) hybrid zone. Mol. Ecol. 18, 3256–3267 (2009).
Yang, J. et al. Genetic evidence of hybridization of the world’s most endangered tern, the Chinese Crested Tern Thalasseus bernsteini. Ibis 160, 900–906 (2018).
Clarke, R. H., Gordon, I. R. & Clarke, M. F. Intraspecific phenotypic variability in the black-eared miner (Manorina melanotis); human-facilitated introgression and the consequences for an endangered taxon. Biol. Conserv. 99, 145–155 (2001).
Wallis, G. Genetic status of New Zealand black stilt (Himantopus novaezelandiae) and impact of hybridisation. (Wellington Department of Conservation, 1999).
Qvarnström, A., Vallin, N. & Rudh, A. The role of male contest competition over mates in speciation. Curr. Zool. 58, 493–509 (2012).
Price, T. Speciation in Birds. (Roberts & Company, 2008).
Argüelles-Ticó, A. et al. Geographic variation in breeding system and environment predicts melanin-based plumage ornamentation of male and female Kentish plovers. Behav. Ecol. Sociobiol. 70, 49–60 (2016).
Seutin, G., White, B. N. & Boag, P. T. Preservation of avian blood and tissue samples for DNA analyses. Can. J. Zool. 69, 82–90 (1991).
Nicholls, J. A., Double, M. C., Rowell, D. M. & Magrath, R. D. The evolution of cooperative and pair breeding in thornbills Acanthiza (Pardalotidae). J. Avian Biol. 31, 165–176 (2000).
Peterson, B. K., Weber, J. N., Kay, E. H., Fisher, H. S. & Hoekstra, H. E. Double digest RADseq: an inexpensive method for de novo SNP discovery and genotyping in model and non-model species. PLoS One 7(5), e37135 (2012).
Catchen, J., Hohenlohe, P. A., Bassham, S., Amores, A. & Cresko, W. A. Stacks: an analysis tool set for population genomics. Mol. Ecol. 22, 3124–3140 (2013).
Zhang, G. et al. Comparative genomics reveals insights into avian genome evolution and adaptation. Science 346, 1311–1320 (2014).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Chang, C. C. et al. Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience 4, 7 (2015).
Foll, M. & Gaggiotti, O. E. A genome scan method to identify selected loci appropriate for both dominant and codominant markers: a Bayesian perspective. Genetics (2008).
Zheng, X. SNPRelate: parrallel computing toolset for genome-wide association studies. R package version 95 (2012).
Pritchard, J. K., Stephens, M. & Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 155, 945–959 (2000).
Linck, E. & Battey, C. J. Minor allele frequency thresholds strongly affect population structure inference with genomic data sets. Mol. Ecol. Resour. 19, 639–647 (2019).
Earl, D. A. STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 4, 359–361 (2012).
Jakobsson, M. & Rosenberg, N. A. CLUMPP: a cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics 23, 1801–1806 (2007).
Neuditschko, M., Khatkar, M. S. & Raadsma, H. W. NetView: a high-definition network-visualization approach to detect fine-scale population structures from genome-wide patterns of variation. PLoS One 7, e48375 (2012).
Steinig, E. J., Neuditschko, M., Khatkar, M. S., Raadsma, H. W. & Zenger, K. R. netview p: a network visualization tool to unravel complex population structure using genome-wide SNPs. Mol. Ecol. Resour. 16, 216–227 (2016).
Kamada, T. & Kawai, S. An algorithm for drawing general undirected graphs. Inf. Process. Lett. 31, 7–15 (1989).
Csardi, G. & Nepusz, T. The igraph software package for complex network research. Inter. Complex Syst. 1695, 1–9 (2006).
Goudet, J. Hierfstat, a package for R to compute and test hierarchical F‐statistics. Mol. Ecol. Notes 5, 184–186 (2005).
Jombart, T. adegenet: a R package for the multivariate analysis of genetic markers. Bioinformatics 24, 1403–1405 (2008).
Jombart, T. & Ahmed, I. adegenet 1.3-1: new tools for the analysis of genome-wide SNP data. Bioinformatics 27, 3070–3071 (2011).
Nei, M. Molecular Evolutionary Genetics. (Columbia University Press, 1987).
Shapiro, S. S. & Wilk, M. B. An analysis of variance test for normality (complete samples). Biometrika 52, 591–611 (1965).
Wickham, H. ggplot2: elegant graphics for data analysis. J Stat Softw 35, 65–88 (2010).
Ahlmann-Eltze, C. ggsignif: Significance Brackets for ‘ggplot2’. https://cran.r-project.org/web/packages/ggsignif/index.html (2017).
R Core Team. R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. http://www.R-project.org (2016).
Acknowledgements
We thank N. Ng and W.Q. Ng for field work support, F. Lei, J. Amat and Y. Qu for providing samples, E.Y.X. Ng for lab support and J. Tolliver for comments on statistical analysis. K.R.S. acknowledges funding from the Deutsche Akademische Austauschdienst. C.K. is supported by the Max Planck Society. This work was supported by the National Natural Science Foundation of China (31772441 to S.W. and F.E.R., 31470111 to S.W.), and Jiangsu Provincial Natural Science Foundation (17KJA180003 to S.W.).
Author information
Authors and Affiliations
Contributions
S.W. and F.E.R. designed the study. S.W. organized the fieldwork and S.W., F.E.R., G.W.L., C.-T.Y., Y.L. and T.X. performed the field sampling. K.R.S. conducted the laboratory work and bioinformatic analysis, with assistance from G.W.L. and F.E.R., G.W.L. and C.K. carried out the statistical analyses on the morphological traits. K.R.S., F.E.R., C.K. and S.W. interpreted the results. S.W., Y.L. and T.X. coordinated the sequencing. K.R.S. and F.E.R. wrote the manuscript with input from C.K. and S.W. All authors approved the final manuscript.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sadanandan, K.R., Küpper, C., Low, G.W. et al. Population divergence and gene flow in two East Asian shorebirds on the verge of speciation. Sci Rep 9, 8546 (2019). https://doi.org/10.1038/s41598-019-44996-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-44996-5
This article is cited by
-
Dispersal in Kentish Plovers (Charadrius alexandrinus): adult females perform furthest movements
Journal of Ornithology (2024)
-
The allocation between egg size and clutch size depends on local nest survival rate in a mean of bet-hedging in a shorebird
Avian Research (2020)
-
The conservation value of admixed phenotypes in a critically endangered species complex
Scientific Reports (2020)
-
Population differentiation and historical demography of the threatened snowy plover Charadrius nivosus (Cassin, 1858)
Conservation Genetics (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
