
Abstract
Within the Canidae, the African wild dog (Lycaon pictus) is the most specialized with regards to cursorial adaptations (specialized for running), having only four digits on their forefeet. In addition, this species is one of the few canids considered to be an obligate meat-eater, possessing a robust dentition for taking down large prey, and displays one of the most variable coat colorations amongst mammals. Here, we used comparative genomic analysis to investigate the evolutionary history and genetic basis for adaptations associated with cursoriality, hypercanivory, and coat color variation in African wild dogs. Genome-wide scans revealed unique amino acid deletions that suggest a mode of evolutionary digit loss through expanded apoptosis in the developing first digit. African wild dog-specific signals of positive selection also uncovered a putative mechanism of molar cusp modification through changes in genes associated with the sonic hedgehog (SHH) signaling pathway, required for spatial patterning of teeth, and three genes associated with pigmentation. Divergence time analyses suggest the suite of genomic changes we identified evolved ~1.7 Mya, coinciding with the diversification of large-bodied ungulates. Our results show that comparative genomics is a powerful tool for identifying the genetic basis of evolutionary changes in Canidae.
Similar content being viewed by others


Introduction
Among the living species of Canidae, the African wild dog (hereafter, AWD) is considered to be the most specialized with regard to adaptations for cursoriality, diet, and coat coloration1. Along with a gracile appendicular skeleton, the most notable characteristic of AWDs is the loss of the first digit on the forefeet. This trait increases their stride length and speed allowing them to pursue prey for long distances in open plain habitats and is unique among living canids2. The dentition of the AWD is also exceptional, as the teeth are generally sectorial in shape and the premolars are the largest relative to body size of any living carnivoran except spotted hyenas3. AWDs also show a transformation of the talonid on the lower first molar (carnassial) from a basin-like crushing depression into a trenchant heel or cutting blade for slicing flesh, which also occurs independently in two other hypercarnivorous canids, the bush dog (Speothos venaticus) and the dhole (Cuon alpinus). This feature is accompanied by the reduction or loss of post-carnassial molars, also a characteristic of hypercarnivorous canids4. Fossil evidence suggests that the reduction of the first digit and transformation of the carnassial in AWDs evolved gradually during the Plio-Pleistocene5. Finally, AWDs exhibit one of the most variegated coats among mammals, with individuals uniquely differing in pigmentation pattern and color6 (Fig. 1), which suggests the expression of a diversity of genes. The function of this highly individualistic coat pattern is uncertain but may represent an adaptation for concealment, communication or thermoregulation7,8,9,10.

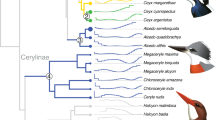
Evolutionary history and adaptation in the African wild dog (AWD) and nine other species of canids. (a) A species tree was inferred by applying ASTRAL-III to 8,117 25 kb-windows (light gray, in background), with internal nodes placed according to average genomic divergence estimated via MCMCTree and calibrated using two fossil priors (see Methods and Fig. S1 for details). A demographic model was inferred for the same species (excluding gray fox) by applying G-PhoCS to 11,112 putative neutral 1 kb windows (dark gray, in foreground). The same phylogenetic tree topology was assumed, augmented with 44 directed migration bands (see Methods and Table S2 for details). Block arrows depict the eight migration bands inferred with total rates higher than 0.05, with arrow widths scaled proportionally to the estimated total rate. The widths of branches in the demographic model are scaled proportionally to inferred effective population sizes (see scale bar at top-left), and their lengths are scaled proportionally to inferred species divergence times. Both scales assume an average per-generation mutation rate of μ = 4.0 × 10−9 and an average generation time of three years88. Species divergence times are much smaller than the average genomic divergence times. Divergence times associated with AWD are indicated at the bottom with 95% Bayesian credible intervals (note the change in time scale between 2.5–5 Mya). Genes with signals of positive selection are specified on the branches leading to the AWD and the dhole. Different phenotypic categories are indicated by color; genes marked with an asterisk had in-frame deletions and genes marked with a cross were pseudogenized. Note that CREBBP has undergone parallel adaptation in both lineages. (b) Venn diagrams showing shared positively-selected genes (left) and pathways (right) obtained from different analytical approaches. Among the seven genes that resulted in significant scores from both the HKA-like and branch-site tests, only HPS6 was associated with AWD adaptations. Primary cilium was the only pathway that was identified by both G-profiler and polysel.
We investigated the genetic origins of adaptations associated with the evolution of cursoriality, hypercarnivory, and coat color variation in the AWD in the context of their evolutionary and demographic history. To accomplish these goals, we sequenced one high coverage AWD genome and utilized three previously reported AWD genomes11,12. Additionally, we used three de novo AWD reference genomes13. Coverage depths are provided in the Supplementary Table S1. These genomes were compared with existing genomes from Canis (wolves, coyote and golden jackal) and Cuon alpinus (dhole)11,12,14,15,16,17,18. The genome of the bush dog was not included in this study because it is part of an ongoing research investigation on comparative genomics of South American canids13. We hypothesized that genes showing signals of positive selection and other molecular changes in AWDs are associated with digit reduction, tooth morphology, and pigmentation. Furthermore, we aimed to investigate the possibility of convergent evolution at the genetic level, exploring shared signals of selection among the wolf-like canids that have a trenchant heel (AWDs and dholes).
Results and Discussion
Evolutionary history
To provide an accurate evolutionary framework for the comparative genomic analyses of AWDs relative to other wolf-like canids, we first reconstructed the phylogenetic relationships among species of Canis, Cuon, and Lycaon. Species tree analysis using ASTRAL-III19 produced 102 distinct gene trees from 8,117 25 kb alignments sampled from 38 autosomes (~203 Mb), with the final species tree being the consensus topology of the 100 replicates from the analysis (Fig. 1). The resulting topology shows the Ethiopian wolf basal relative to the rest of Canis, with dhole and AWDs as successive sister lineages having diverged earlier (Fig. 1). Within Canis, the golden jackal and African wolf are independent lineages sister to the clade comprised of the coyote, gray wolf and domestic dog. These patterns of relationship are consistent with previous analyses based on nuclear DNA sequences12,15,20.
We next estimated the age of divergence of the AWD lineage using the inferred tree and two methods. First, we computed average genomic divergence times using MCMCTree21, in which two fossil priors were used to calibrate nodes of the phylogeny (see Methods for details). Our estimates suggest an average genomic divergence of 3.91 mya (95% HPD = 3.30–4.50 mya) between Lycaon and the clade containing Cuon and Canis, approximately two million years earlier than the earliest fossil evidence recorded for the lineage1. This more ancient divergence is feasible because average genomic divergence captures not only the time since species divergence, but also the time for lineages to coalesce in ancestral populations, which are known to be very large in canids18. To better address this discrepancy, we thus jointly inferred a complete demographic model using G-PhoCS22, taking into consideration gene flow between 44 possible pairs of branches in the phylogeny (see Methods for details). Indeed, the species divergence time for Lycaon is then estimated at 1.72 mya (95% HPD = 1.70–1.74 mya; Table S2 and Fig. 1), which is much closer to estimates from both the fossil record and recent analyses of whole-genome data1,12,17. Importantly, while our inferred model suggests prevalent gene flow between divergent canid species, Lycaon is inferred to be largely isolated from genetic exchange with other canid lineages. This isolation provided more time for unique genomic adaptations to evolve.
African wild dogs are uniquely enriched in positively-selected genes related to primary cilia
To identify positive selection events that occurred on protein-coding genes during the evolution of the AWD lineage, the sequencing reads for four AWDs and eight other canid species were mapped to the domestic dog reference assembly (CanFam3.1) to take advantage of the high-quality annotation of the dog reference genome (Table S1). The mapping process was based on the GATK Best Practices pipeline (Methods). For almost all canids, we found that more than 97% of reads successfully mapped to the dog genome. The only exception was a low coverage (12.1x) AWD that had ~93% of the reads mapped to the dog. To avoid potential reference bias from aligning reads to a different species, we further confirmed our results on three recently published de novo AWD reference genomes13.
After calling genotypes with SAMtools and filtering with GATK 3.723 as well custom python scripts, we identified ~19,000 orthologous protein-coding genes. Among these genes, 18,327 passed our quality filters (no internal stop codon, permissible length, and longest transcript) and were used to identify genes under positive selection using the branch-site model21. This test was conducted on each multi-species gene alignment generated with PRANK v.15080324 and using the topology in Fig. 1 as the guide tree. AWD, dhole, and gray wolf were specified as different foreground branches. A gene was considered positively selected if the value obtained from the likelihood-ratio test comparing a model where the ratio of nonsynonymous substitutions (dN) to synonymous substitutions (dS) was greater than 1 (dN/dS > 1) against a null model where dN/dS = 1. Significant differences were determinated with a chi-square distribution with 1 degree of freedom25.
One issue with the branch-site model is that it is highly sensitive to alignment errors. Therefore, we conducted an extensive filtering process on our data, first using SWAMP26 and then visually inspecting the alignments of genes with p < 0.05. After masking regions with unusual enrichment of amino acid changes, we conducted three independent runs for each foreground branch and gene family, and retained the one with the best likelihood-ratio score of each run27. This guaranteed that large log-likelihood ratios depicted from the branch-site model were not the result of convergence problems of the test27. Another concern in the exploration for genes under positive selection is the role of multiple nucleotide changes28. Although these changes may occur simultaneously, the branch-site model assumes that they occur in a successive manner. The result will be unrealistically high likelihood-ratio scores at a codon were nucleotide changes occurred at the same time28. Among our 12 candidate genes (Fig. 1), we identified three genes with multiple nucleotide changes (CC2d2a, TMEM67, PAH) and thus support for the positive selection on these genes should be interpreted with caution. Although it is challenging to elucidate the order of multiple nucleotide changes, our main conclusions are not affected even if we take a conservative approach and do not include such genes in the analysis.
We found 43 genes (Table S3) that were significant at a false discovery rate (FDR) of 20%, after conducting multiple hypothesis testing of 18,327 genes along the three foreground branches (AWD, dhole, and gray wolf). Since only a few genes passed the genome-wide significance threshold, we used all genes with a p-value ≤ 0.01 to test for enrichment of gene functions with G-profiler29. Ensembl identification of genes with a p-value ≤ 0.01 were input as query lists and the 18,327 total gene set was used as the background list. We allowed a minimum of two genes to overlap between query genes and genes belonging to a gene ontology (GO) term. This resulted in genes with specific signals of selection in AWDs overrepresented in terms related to primary cilia (Fig. 1), which are significantly involved in coordinating signaling pathways during mammalian development30.
The disadvantage of common tests for gene ontology enrichment like G-profiler29 is that an arbitrary significance cutoff must be specified, and data below that cutoff is expected to be lost. Therefore, instead of just focusing on some outlier genes with high likelihood ratios, we used polysel31 to conduct analyses of polygenic selection across the full set of tested genes. We looked for pathways that were overrepresented with genes having low or moderate likelihood ratios more than would be expected by chance. This model takes likelihood ratio test statistics estimated from the branch-site test and finds weak to moderate polygenic selection within biological pathways. Then, p-values are generated from an empirical null distribution obtained by randomly sampling gene sets in specified pathways. Using this approach, we also found that the “primary cilia” GO category was significantly enriched with a variety of levels of positive selection (Fig. 1b). To rule out the possibility that this GO category could be enriched just by chance due to the large number of genes, we tested for overrepresentation of significant genes in primary cilia in the gray wolf and dhole and found no evidence of enrichment. Finally, to account for possible errors generated from mapping short reads of the AWD to a different species (domestic dog), we verified every mutation reported in this study (e.g., nucleotide and amino acid deletions and substitutions) with a consensus sequence of the three recently published de novo AWD reference genomes (NCBI Bioproject PRJNA488046; Table S1)13.
Digit reduction through apoptosis
Two developmental mechanisms of digit reduction from the ancestral five-digit morphology have been characterized in mammals. One is related to a complete absence of a digit during development through regulation of the transduction of sonic hedgehog (SHH) signaling and the other involves apoptosis of digits during early development32. The loss of the first digit, as found in AWDs, has been shown to be independent of SHH signaling33. Therefore, we focused our analyses on genes associated with apoptosis pathways, particularly those related to digit development.
We used the Variant Effect Predictor annotation tool34 to identify amino acid-changing substitutions unique to the AWD that could have a significant impact on the associated proteins but will be ignored by the branch-site model test. We identified 403 genes with both high and moderate impact. High impact indicates a disruptive substitution that could cause truncation, loss of function, or nonsense-mediated decay of a protein whereas moderate impact indicating a non-disruptive substitution that might change protein functional efficiency. The substitutions we identified were categorized as in-frame indels, frameshift variations, and stop codon gains (see Methods for details).
Strikingly, we found 596 genes with in-frame-deletions, with moderate impact, unique to AWDs. These amino acid deletions were tested for enrichment of GO categories using G-profiler29. We found that this type of mutation was overrepresented in digit-loss categories with a false discovery rate of 5%. Specifically, the term, “abnormality of the thumb,” was over-represented by the genes FANCC, FANCD2, and FANCM, which are associated with the Fanconi anemia (FA) pathway35. We also observed an overrepresentation of 33 genes with frameshift variation mutations in the olfactory receptor (OR) GO category. Twenty eight of these genes were also enriched in the olfactory transduction KEGG category, in accordance with the dynamic evolution of OR gene families36.
Our results implicate amino acid deletions in genes associated with the FA pathway in the loss of the first digit in AWDs through an apoptosis pathway that typically directs interdigital cell death (Fig. 2). Specifically, in the development of the ancestral five-digit foot, the primary function of apoptosis is to eliminate excessive cells on the interdigital webs which trims the dimension of the digit32,37. When this digit individualization occurs during development, apoptosis has only a small effect on digit dimension38. Studies have shown that FA proteins form a complex with the CtBP1 protein that results in the repression of the DKK1 gene. As expression of this gene is restricted to the interdigital area during the early development of digits, its repression prevents apoptosis from extending into the digits37,39. When the FA-CtBP1 complex fails to properly form, inhibition of DKK1 expression is removed, thereby permitting apoptosis to extend to the digits. We suggest that the amino acid deletions in FANCC, FANCD2, and FANCM found exclusively in AWDs may reduce the binding affinity of the FA protein complex to CtBP1, thus allowing the loss of the first digit through apoptosis (Fig. 2). Our findings are strongly supported by the fact that the mutations we have identified in the FA genes in AWDs are responsible for a condition commonly associated with the absence of the first digit in humans35,40 and expression of DKK1 is related with absence of the first digit in mice41. Moreover, apoptosis as a mode of evolutionary digit has also has been shown in horses, jerboas, and camels32.

Apoptosis of the first digit in the African wild dog. (a) Primitive condition of five digits in the Canidae; note the small first digit in gray wolf called the “dewclaw”. The absence of the first digit is shown in the African wild dog (AWD). (b) Schematic representation of digit reduction and separation of digits. In the normal five-digit pattern scenario shown at the top, apoptosis (blue circles) is restricted to interdigital regions. The first digit, enclosed by a rectangle, is protected from apoptosis by FANCC and FANCM that form a complex with CtBP1 and repress DKK139. The stability of this complex is regulated by the FANCD2-FANCI association100. In the scenario shown below, amino acid deletions (red stars) observed in FA genes may reduce both the affinity of FA genes to CtBP1 and the stability of the protein complex. Consequently, the FANCC-FNACM-CtBP1 complex is not formed and DKK1 is not suppressed (indicated by empty arrow). Deficiency of the FA complex activity increases DKK1 expression. As a result, apoptosis expands to the first digit; note blue circles (apoptosis) on the region of the thumb. (c) Effect of the mutations in the FA and DKK1 genes in humans (Reprinted from ref.35© 2009 with permission from Elsevier) and mice (Reprinted from ref.41 © 2004 with permission from Elsevier). (d) Multiple sequence alignments of mammalian FANCD2, FANCM, and FANCC amino acid sequences showing deletions specific to AWDs. Five AWDs are shown; “RWK481” and “SAMN04312208” are individuals from Kruger National Park, South Africa; “CN3669” and “SAMN04312209” are individuals from Kenya and “Dnv Lycaon pictus” is the consensus sequence of three de novo reference AWD genomes13. The top panel also shows a 3D protein-model of FANCD2 with the location of the observed amino acid deletion, which is important for the association with FANCI.
Hypercarnivory through sonic hedgehog (SHH) signaling
The primary cilium is a hair-like structure that projects from the surface of cells and serves as a sensory organelle, transmitting signals from the extracellular space into the nucleus42 (Fig. 3a). This structure is located at the dental epithelium and mesenchyme during the formation of dental cusps43,44. As the primary cilium regulates numerous signaling pathways necessary for odontogenesis, ciliary defects can alter the process of cusp patterning45.

Possible mode of evolutionary molar cusp modification in the African wild dog through SHH signaling. (a) The figure at the top right shows genes found to be enriched in primary cilium with polysel; genes above dashed line are those found to be enriched with G-profiler as well (see Methods for details). Only gene names with known function in tooth development are shown. The figure at the top left shows a schematic of the primary cilium and the role of candidate genes in SHH transduction. SHH, represented by red circles, reaches the ciliary membrane. Then, transport of transcriptional factors GLIs such as GLI1, through the axoneme, is promoted by WDR35/IFT121 as well as the efficient docking of the axoneme in the plasma membrane, which is conducted by CC2d2a and TMEM67. Together, these components of primary cilium cause rapid accumulation of GLI into the basal body. Ultimately, GLIs will enter the nucleus and promote the expression of SHH-dependent genes. In the case of CREBBP, the observed amino acid deletion (red star) may increase the affinity of this cofactor with smad genes and increase the expression of TGF-β and BMP dependent genes involved in molar cusp development. (b) Left-bottom figure showing the single cuspid talonid of the lower first molar (carnassial) in the AWD as opposed to a bi-cusped talonid carnassial in the gray wolf. Right-bottom figures show the effect that mutations in CREBBP have on humans53 (reprinted by permission from Wiley-Liss, Inc.; American Journal of Medical Genetics53 © 2007); talon cusp condition is observed; cusps that protrude from the anterior region of incisors on the left and extra cusps on molars on the right. (c) Amino acid alignment of the glutamine-rich region of CREBBP for 51 species of mammals showing the deletions specific to AWDs and the dhole. Five AWDs are shown; “RWK481” and “SAMN04312208” are individuals from South Africa; “CN3669” and “SAMN04312209” are individuals from East Africa; “Dnv Lycaon pictus” is the consensus sequence of three de novo reference AWD genomes13. The ancestral condition in canids is inferred as 15 glutamine residues.
We found AWD-specific amino acid substitutions in components of the primary cilium, specifically the genes WDR35, TMEM67, CC2d2a and GLI1. These substitutions suggest a combined regulatory effect, through GLI transcription, on SHH-dependent genes (Fig. 3a). Specifically, the role of primary cilia depends on the retrograde (to the basal body region) and antegrade (to the tip of the ciliary membrane) intraflagellar transport (IFT) of the transcriptional factor GLI143,46 (Fig. 3a). This transportation occurs through a microtubule structure called the axoneme. The efficacy of GLI mobilization through the axoneme is dependent upon protein-mediated transport by WDR35 (also known as IFT121) as well as the proper docking of the axoneme into the basal body region, which is mediated by TMEM67 and CC2d2a proteins43,47,48 (Fig. 3a). Mutations within genes encoding primary cilium components alter mobilization of GLI to the basal body, and hence result in gain or loss of GLI function in this region. An increase in GLI will result in the gain of SHH phenotypes such as growth of molar cusps. In contrast, a decrease in GLI will cause loss of SHH phenotypes49 such as inhibition of molar cusp development44. We suggest that the amino acid changes observed in AWDs in WDR35, TMEM67, CC2d2a, and GLI1 may cause rapid transportation of GLI to the basal body, and consequently overexpression of SHH target genes43,50. Variants in the candidate genes reported in this study have been associated with abnormal tooth shape and may thus be related to the exaggerated hypercarnivorous dentition of AWDs, which includes the development of a lower trenchant carnassial43,50,51. Most of the amino acid sites from these genes were unique to AWD even when compared to other placental mammals and marsupials, including two South American canids, Speothos venaticus and Chrysocyon brachyurus (Chavez et al., unpublished data, Fig. S2). Also, specific sites in CC2d2a had a relatively high probability of being affected by positive selection as suggested by a Bayes empirical Bayes test (BEB > 0.90). Although nucleotide changes were not recorded for GLI1 with BEB, mutations in this gene were located within significant windows as suggested by the \(\frac{\theta }{D}\) estimate (p < 0.01; see Methods section) as estimated from the HKA-like test52, after verifying that the amount of information within windows of 25 kb in length was not driving higher differences between diversity and divergence (Fig. S3). Our results suggest that the transduction of SHH through primary cilium may have promoted the modification of a primitive molar with a posterior crushing basin into a trenchant sectorial single cusp in AWDs (Fig. 3b).
Another gene associated with spatial patterning of the tooth, CREBBP, was found to have an amino acid deletion in AWDs. Interestingly, we also observed a two amino acid deletion in the same region in the hypercarnivorous dhole, a canid that also possesses a lower trenchant carnassial heel (Fig. 3b)4. CREBBP is a strong candidate for the modified carnassial observed in these two hypercarnivorous canids. Notably, this gene is associated with abnormal numbers or features (talon cusps) of molar cusps in humans53 (Fig. 3b). Even though CREBBP is ubiquitously expressed, the shared amino acid deletions were in the glutamine-rich region of the protein (Fig. 3c). This region is highly conserved in eukaryotes and serves as the binding site for Smad proteins54,55. These proteins are transcriptional factors that regulate the expression of genes located at the dental lamina and mesenchyme, and play important roles in regulating differentiation and proliferation of cells during tooth development56. Specifically, Smad transcription factors enter the nucleus and bind to the coactivator CREBBP and regulate the expression of target genes (Fig. 3a). We suggest that the observed amino acid deletions observed in CREBBP in AWDs and dholes may alter the formation of the Smad-CREBBP complex. This ultimately will have a regulatory effect on the expression of TGF-β and BMP dependent genes56. In Smad knockout mice, dental cusp formation is affected56. Our results suggest that the formation of the trenchant heel of AWD, initially guided by primary cilium components, may be reinforced by a regulatory effect of Smad transcriptional factors on genes involved in tooth development56. Also, our findings suggest that this may be the regulatory pathway that also determines the blade-like cusps in the dhole.
The shared amino acid deletions in the CREBBP gene observed in AWDs and dholes (Fig. 3c) could have arisen through different routes. First, the changes could have evolved independently in each species. Alternatively, the changes could have resulted from past adaptive introgression between species, as has been found among species within the Panthera lineage57. To account for the latter possibility, we conducted tests of admixture in the context of the evolutionary and demographic history of the sampled canids22. Results from models with and without gene flow among different lineages suggested a history of extensive admixture among species of Canis (Supplementary Discussion; Table S2 and Fig. 1), consistent with recent findings12. However, the genomic data for AWDs and dholes suggest little or no gene flow between these lineages and those leading to Canis species. Although the lack of post-speciation gene flow between the dhole and AWD could suggest that the amino acid deleting changes in CREBBP evolved independently, we do not entirely rule out the possibility of shared post-divergence ancestry. Particularly, we found a low proportion of divergent sites between the AWD and dhole (only 9 divergent sites out of 3,000 flanking sites analyzed) around CREBBP that suggest a plausible shared ancestry. Regardless of the mode of evolution of CREBBP, the amino acid deletions shared between the AWD and the dhole may reflect similar selective forces favoring hypercarnivory4.
Positively-selected genes associated with AWD pelage coloration
Assuming that positive selection could have occurred in genes associated with the unique pelage coloration and patterning seen in AWDs, we tested a set of 151 genes that have been shown to be involved in mammalian pigmentation58,59,60,61,62 using the branch-site test21. We found six genes with AWD-specific signals of positive selection, three of which are known to have relevant function in coat coloration: MYO5A, HPS6, and PAH (Fig. 4a). The resulting amino acid changes were confirmed in a consensus sequence (“Dnv Lycaon pictus” in Fig. 4b) of three high-coverage de novo AWD genomes. Moreover, most amino acid changes in coat color were unique to AWDs when compared to other species of placental mammals, marsupials, and monotremes (Fig. 4b).

Candidate genes and possible mechanisms associated with coat pigmentation and patterning in African wild dogs. (a) Plot showing Q-values depicted from the branch-site test after a multiple hypothesis correction of 151 different coat color genes with the AWD as the foreground branch (see methods). Significant genes (Q-value < 0.20) are shown above the horizontal blue line; for illustration purposes, Q-values shown on the Y-axis were transformed to -log10. Only names of genes with relevant functions are shown. Illustration at the bottom showing coat color in the AWD; genes are shown in red circles and are placed on the type of color they regulate (e.g., PAH regulates brown and yellow colors). (b) Amino acid alignment of coat color candidate genes for an average of 49 species of mammals showing changes specific to AWDs. Five AWDs are shown: “RWK481” and “SAMN04312208” are individuals from Kruger National Park, South Africa; “CN3669” and “SAMN04312209” are individuals from Kenya and “Dnv Lycaon pictus” is the consensus sequence of three de novo reference AWD genomes13. (c) A schematic showing the role of candidate genes in color pattern. (I) shows that black coat color will need both proper cargo of melanin by HPS6 to melanosomes and its transport by MYO5A to the bulk of the hair. (II) In the case of white blotches or spots, they could be the result of either proper transport of melanosomes but containing no melanin or melanosomes containing melanin that fail to reach the bulk of the hair. The figure on the bottom-right shows the effect of MYO5A mutations on the pelage of a domestic mouse69. (d) 3D model of the PAH protein depicted with SWISS-model (see methods) that shows a mutation on the cofactor of the enzyme (B\({{\rm{H}}}_{4}\)). The scheme at the bottom illustrates the role of PAH and its cofactor. The right figure shows a gradual recovery of the black color of yellow mice (PKU) with deficiency of PAH65 (Reprinted by permission from Springer Nature: Springer Nature, Gene Therapy ref.65 © 2006).
Considering that positively-selected substitutions related to the pigmentary system might fall outside protein-coding regions and could have also occurred during recent evolutionary history, we used the Hudson-Kreitman-Aguadé (HKA) test to examine regions with high divergence and low diversity for signals of selective sweeps. To conduct this test, we first called genotypes using Haplotype Caller in GATK 3.823. We then calculated polymorphism within 100 kb windows among the four AWDs genomes that were mapped to the domestic dog reference genome. At the same time, per-site divergence was calculated between the AWD with the highest coverage (LPI_RKW 4881) and the domestic dog. Considering that demographic effects are expected to have an effect across the entire genome, we used empirical p-values to identify loci with extreme values of high divergence and low diversity as evidence for positive selection.
After verifying that the amount of information within 100 kb windows was not driving higher differences between diversity and divergence (Fig. S3), a total of 159 genes were located within windows with a magnitude of differentiation greater than expected by chance (empirical p-value < 0.01). Among the genes identified in the HKA test, seven were also observed in the branch-site test (Fig. 1). From this set of genes, we identified HPS6 as a candidate gene that may have been recently selected. Among the set of genes with AWD-specific signals of positive selection, we did not observe four genes (ASIP, MITF, MLPH, PMEL) that were previously shown to have elevated ratios of non-synonymous/synonymous substitutions in two lower coverage AWD genomes11. For example, these authors reported a stop codon-gain in PMEL, but we found this was due to a misorientation in the codon translation frame (Fig. S4).
The positively-selected genes associated with pigmentation we detected have notable functions that strongly suggest regulation of the variegated pelage of AWDs (Fig. 4a). Although phenylalanine hydroxylase (PAH) has several pleiotropic effects, phenylalanine levels are closely related to melanin deposition63,64,65 (Fig. 4d). Levels of phenylalanine are determined by the PAH gene, whose protein catalyzes hydroxylation of phenylalanine to tyrosine64,66. Using available information to construct a 3D structure of the PAH protein, we located the AWD-specific mutation in the biopterin H domain (Fig. 4d), which contains a binding domain for the PAH cofactor67. This finding suggests that the mutation observed in the protein domain of PAH could be in part responsible for the AWD coat color pattern, as this gene and its cofactor are known to regulate the proportion of yellow and black fur64 through the conversion of phenylalanine to tyrosine66 (Fig. 4d). The observed amino acid change in MYO5A (myosin 5 A) is located at the myosin head region (Fig. S5), which is relevant to the motor function of the protein and the transportation of melanosomes68. Mutations in this gene result in patchy color patterns in mice69 (see Fig. 4c) and dilute pigment in dogs61. The AWD-specific mutations observed in MYO5A could be associated with white and black patches by regulating melanosome transport to the bulk of the hair70,71 (Fig. 4c). Similarly, sites under selection in HPS6 may be responsible for the white and black patches by regulating melanin deposition in melanosomes59,72 (Fig. 4c). Mutations in this gene are associated with dilution of melanin in mice72 and Hermansky-Pudlak syndrome in humans (oculocutaneous albinism), a group of autosomal recessive disorders that cause abnormally light coloring of the hair and skin73. As all AWDs are born black2 and develop pigmentation patterns as puppies, longitudinal studies of gene expression will help corroborate the function of these genes in AWD pigmentation.
Conclusions
A commonly observed trend among large mammalian predators that are cursorial is the elongation of limbs and the reduction or loss of digits which allows increased speed and improved pursuit and capture of increasingly faster prey74. Among canids, the AWD displays the most specialized morphological changes associated with cursoriality, including a unique reduction of the number of digits on the forepaws75. Similarly. AWDs have specialized carnassial molars that enhance the slicing of flesh. This decreases the consumption time of prey and therefore, the likelihood of encounters with competitors4. Also, while the function of the conspicuous and individual-specific coat coloration patterns of AWDs is uncertain, it may help confuse both prey and competing predators76, or it may not be the direct object of natural selection77,78.
Our comparative genomic analyses suggest that the evolution of cursoriality in AWDs has been driven by a known apoptotic pathway implicated in evolutionary digit loss in other mammals32 and involved a single major gene pathway. We also found evidence of substitutions and amino acid deletions in genes possibly associated with the hypercarnivorous dentition of AWDs and that changes in one of these genes (CREBBP) are also found in the Asiatic dhole. Consequently, our data support the idea that convergent phenotypic evolution can result from genetic changes in the same genes. Our study provides a unique example of genome-scale adaptive evolution analysis of one of the most successful pack-hunters, the African wild dog, and exemplifies molecular pathways which can iteratively adapt organisms to the challenges of prey capture and consumption.
The unique adaptations observed in African wild dogs were likely facilitated by their unique demographic history. Most large canid lineages have experienced gene flow from divergent species, whereas our inferred demographic model suggests that African wild dogs were genetically isolated from other species. Furthermore, divergence dating analyses provide a temporal framework for understanding the general rate of evolution of the molecular changes that underlie the morphological adaptations in AWDs. The earliest fossils of Lycaon (L. sekowei n. sp.) were described from sites in South Africa and dated to ca. 1.0–1.9 mya and suggest that the development of the hypercarnivorous dentition preceded the morphological changes associated with cursoriality in the modern AWD1. Divergence times estimated using a model that considers ancestral population size and post-divergence gene flow suggest that AWDs split from their common ancestor ~1.7 Mya, which is consistent with episodes of faunal turnover and the evolution of faster-moving ungulates during the Pleistocene that likely influence the adaptations of carnivores in sub-Saharan Africa79,80. Our study demonstrates that comparative analyses of genomes provide a powerful approach to investigate the genetic basis of unique adaptations in an evolutionary context.
Methods
DNA sample and sequencing
Genomic DNA from a female African wild dog (LPI_RKW 4881) was pair-end sequenced (100 bp) to ~27.9X coverage using an Illumina HiSeq2000 (Illumina, USA). The library preparation and genome sequencing was performed by the Vincent J. Coates Genomics Sequencing Laboratory at University of California, Berkeley. The individual that we sequenced originally belonged to the Skukuza pack in Kruger National Park, South Africa, and was originally identified as SF581. This sample was selected based on sufficient quantities of high molecular weight DNA using a DNA fluorometer (Qubit 2.0), the NanoDrop spectrophotometer (ThermoFisher, USA) and gel electrophoresis. Genome sequences from 11 other canid species, including three African wild dogs, were obtained from previous studies11,12,14,15,16,17,18 and are detailed in Table S1.
Alignment to the dog reference genome and annotation
An initial quality control of raw reads of LPI_RKW 4881, as well as those from 11 other canids obtained from the literature, was performed with FastQC (http://www.bioinformatics.babraham.ac.uk/projects/fastqc). Reads were then trimmed and filtered for adapters, short reads, and low-quality bases (Q < 20) with Trim Galore (http://www.bioinformatics.babraham.ac.uk/projects/trim_galore/)
before being aligned to the domestic dog genome reference assembly (canFam3.1) using Bowtie282. The percentage of aligned reads to the domestic dog for most species was more than 97 and resulted in different coverage depths per species (Table S1). Variant calling was performed with HaplotypeCaller using the Genome Analysis Toolkit 3.7 (GATK)23 with a series of filtering steps to minimize the presence of false genotypes (Supplementary Methods).
Species tree estimation
To reconstruct a phylogenetic tree of the the African wild dog and nine other species of canids, 8,177 sliding-window fragments of 25 kb were generated (Supplementary methods) and further aligned with PRANK v.15080324 using one iteration (-F once option) and the topology shown in Fig. 1a as the guide tree. Then, windows were trimmed using GBlocks83 with default parameters. Trimmed alignments were run with RAxML v8.2.984 under the GTR model for each locus to infer individual maximum likelihood (ML) gene trees with 100 bootstrap replicates. For each locus, the best tree was selected from the RAxML output, while the 100 bootstrap trees were merged into a single file per locus. Additional alignment trimming and tree generation was done using a modified script from the SqCL pipeline85 (phylogeny_align_genetrees_prank.py). The best tree files were concatenated into one file with only 10% missing data tolerated, collapsing branch lengths shorter than 1e-05 substitution per site, and collapsing clades with support less than one using a script from the SqCL pipeline (phylogeny_prep_astrid_astral.py).
The species tree was estimated using ASTRAL-III v.5.5, which models the discordance between gene trees and species trees using a coalescent model19. We used both the concatenated best-tree and bootstrap tree files as inputs. The analysis was conducted with 100 bootstrap replicates and the best multi-locus tree was selected with ML support values. The best tree was then scored to obtain a posterior probability and quartet support values for each node/clade. The gray fox (Urocyon cinereoargenteus) was used as the outgroup to root the tree, based on results from previous molecular systematic investigations of canid relationships20.
Estimation of divergence times
To estimate the ages of divergence among species of Canis, Cuon, and Lycaon (10 species, including the Andean fox and gray fox as outgroups) we first generated alignments for 1,183 single-copy coding orthologues. From the codon alignments, 166,182 four-fold degenerate sites (clock-like) were extracted and concatenated into a data matrix with 6,155 missing sites (3.7%) across the 10 species. Average genomic divergence times were estimated using MCMCTree from the software package PAML421 with the HKY + G model of nucleotide substitution, the topology of the species tree obtained from the ASTRAL-III analyses as input, and 2,200,000 MCMC generations, of which the first 200,000 generations were discarded as burn-in. Other parameter settings used in the analysis are detailed in the Supplementary File. We applied two calibration priors with associated distributions and densities based on the fossil record of the Canidae to calibrate node ages, as previously described14. The first prior was set at the root with an age distribution of 9.0–11.9 million years ago (Mya), which provides an approximate age for the split between the tribes Canini and Vulpini, based on the first appearance of Eucyon, thought to be an early member of the Canini86,87. The second prior had an age range of 1.1–3.0 Mya ago, based on the earliest fossils of the modern gray wolf, Canis lupus, and the earliest known fossils of Canis, specifically, Canis edwardsii86.
Phylogenetic analyses suggest Canis edwardsii is sister to a clade that includes Canis aureus (golden jackal) and Canis latrans (coyote)86. However, since Canis lupaster (African wolf) and Canis simensis (Ethiopian wolf) are also contained in the genus Canis12,15,20, we assumed that the earliest known age of Canis edwardsii bracketed all extant species of Canis. The MCMC analysis was repeated twice, as recommended in MCMCTree manual, and no discordance was observed between runs.
Demographic history and admixture
Twelve canid individuals were used in the demographic analysis, including the domestic dog reference and excluding the gray fox (see Table S1 for the remaining 11 genomes). Sequence alignments were obtained for these 12 genomes at 13,647 putatively neutral noncoding loci computed in previous studies to be short (1 kb long) interspersed (>30 kb apart) and distant from protein-coding genes (>10 kb)18. Of these alignments, 2,535 had more than 10% genotypes missing due to a sequencing depth below four reads or above the 95th coverage percentile. These loci were removed and the remaining 11,112 loci were analyzed using the Generalized Phylogenetic Coalescent Sampler or G-PhoCS22. We assumed a population phylogeny consistent with the topology of the species tree inferred by ASTRAL-III. After labeling ancestral populations, we modeled gene flow by augmenting this phylogeny with 44 directional migration bands (Supplementary methods). An additional analysis was done assuming a species tree obtained by switching the position of the golden jackal and Ethiopian wolf in the species tree inferred by ASTRAL-III with the same 44 migration bands (see Supplementary Discussion).
We ran the multi-threaded version of G-PhoCS V1.3.2 (https://github.com/gphocs-dev/G-PhoCS) using five threads per run and a standard MCMC setup. Namely, we assumed an exponential distribution with mean of 0.0001 as the prior of all the mutation-scaled population sizes (θ) and divergence times (τ), and a Gamma (α = 0.002, β = 0.00001) distribution for the prior of migration rates (m). Because of the large number of migration bands, the Monte Carlo Markov chain was let to converge for 200,000 burn-in iterations, after which parameters were sampled every 50 iterations, for the next 400,000 iterations, resulting in a total of 8,000 samples from the approximate posterior distribution. For each parameter, we recorded the mean sampled value and the 95% Bayesian credible interval (CI). Population size estimates (Ne) were obtained from the mutation-scaled samples (θ) by assuming a mutation rate per-generation of μ = 4.0 × 10−9 88, and divergence times (T) were calibrated by assuming the same rate and an average generation time of three years. Migration rates were scaled by the duration of time of the migration band, resulting in total rates, which approximate the probability that a lineage experienced migration. Parameter estimates are summarized in Table S2 and Fig. 1.
Positive selection
Coordinates of ~19,000 orthologous genes were obtained using the domestic dog reference genome (canFam3.1) available in the Ensembl database89. To exclude paralogous genes, we filtered sites following previous recommendations90. Specifically, we filtered out sites according to the following criteria: (1) coverage higher than a 95th percentile of distribution; (2) sites that occurred in more than one locus (with fix mate in GATK); and (3) duplicated sites likely generated from PCR libraries (with PCR duplicates in GATK). We also manually checked sequences for signals of duplication events and kept only sites that were bi-alellic.
To reduce the inclusion of false signals of positive selection caused by errors in the alignment process, short regions enriched with unreasonably high rates of nonsynonymous substitutions (dN) sites were masked with the Sliding Window Alignment Masker for PAML (SWAMP) tool26. Specifically, a two-step masking procedure was conducted. First, with dN ≥ 10 in a 15-codon window, followed by dN ≥ 3 in a 5-codon. This approach has been proven to effectively remove most of the problematic sequences associated with misalignment26.
Genes that passed our filters (no internal stop codon, permissible length and longest transcript) were then tested for signals of positive selection, based on the reconstructed species tree (Fig. 1a), using the branch-site model in PAML 4.821 (Supplementary Methods). We run this model on the AWD and the other two pack-hunting species (dhole and gray wolf) were each used as foreground branches. Model A (allowing sites to be under positive selection; fix omega = 0) was compared to the null model A1 (sites may evolve neutrally or under purifying selection; fix omega = 1 and omega = 1). We included only genes with omega values > 1, since genes lower than this threshold could be driven by relaxed selection. Statistical significance (P < 0.05) was assessed using likelihood ratio tests (LRTs) and chi-square tests. Multiple hypothesis correction for 18,327 protein-coding genes and three foreground branches was conducted with a 20% false discovery rate (FDR) criterion using QVALUE in R91.
To detect recent signals of selection that include non-coding regions such as promoters and enhancers, we aimed to detect selective sweeps through an HKA-like approach52,92,93. To determine interspecific variation, genotypic variants were called with Haplotype Caller from the Genome Analysis Toolkit 3.823. Independent gVCF files were created for the four AWD genomes (Table S1) and then joined with the option “CombineGVCFs” from GATK. On the multiple-samples gVCF, per-site polymorphism among the four AWDs was calculated across non-overlapping 100 kb windows with 10k steps between windows. At the same time, per-site divergence was calculated between the high coverage AWD (LPI_RKW 4881) and the domestic dog. These estimates were calculated with the following equation:
where p is the frequency of one allele in the four AWD genomes, L is the total number of callable sites with good quality in the window, n is number of sampled chromosomes (n = 8 for 4 diploid individuals), Xi is the number of derived alleles in the AWD (LPI_RKW 4881) with respect to the dog; and Yi is the number of heterozygous sites.
Windows were required to have at least 10 kb of sequence and sites were filtered for low coverage (less than 3x and no more than a 95th percentile of distribution), low quality variant sites (QUAL < 50), missing genotype, non-bi-allelic sites, low quality genotype scores (Q < 20) and regions with high GC content. The windows with the lowest θ/D ratio were candidates for a selective sweep92,94. Empirical p-values were obtained for a total of 22,269 windows. To ensure that outliers were not driven by less sequencing data, we plotted the divergence and diversity ratio (θ/D) vs. the number of called bases per window (Fig. S3).
To further investigate candidate genes detected to be under positive selection, protein structure information from the relevant gene available in the literature was used. Nucleotide sequences of orthologous genes in FASTA format were translated into amino acids using “vespa.py translate” from the VEPSA tool95. The AWD protein sequences were then aligned to human annotated versions of the orthologous genes within Geneious v11.1.196 to identify amino acid changes that occur within a given protein domain (see Fig. S4). When information was available, the effect of amino acid changes on protein structure and function was evaluated using three-dimensional models depicted from SWISS-MODEL97. Finally, to evaluate the degree of conservation (constraint) of the AWD-specific amino acid changes and deletions, AWD alignments were compared with available orthologous coding sequences from: (1) 12 other canid species, including corresponding sequences from the genomes Speothos venaticus (bush dog) and Chrysocyon brachyurus (maned wolf) (unpublished data); and (2) 29 to 41 other species of placental, marsupial and/or monotreme mammals, using the tool OrthoMaM v998. Some amino acid changes in the PAH as well as the WDR35 genes described in the result section were not observed in all samples of AWDs. However, they were confirmed on a consensus sequences of three high-coverage de novo ADW reference genomes (“Dnv Lycaon pictus” in Figs 4b and S2). This suggests that these mutations may be heterozygotes for PAH and WDR35 genes, failing to be detected in some samples due to insufficient coverage.
Enrichment test
Information about the functional impact of amino acid changes was obtained from the Ensembl Variant Effect Predictor tool34 with the domestic dog (Ensembl’s release89) and further used to identify indels (frameshift variation, in-frame insertions and in-frame deletions), loss-of-function mutations (stop codons) and regulatory variants (5′ UTR, 3′ UTR) that were unique to AWDs. Genes within these categories were then tested for enrichment of Gene Ontology (GO) categories using G-profiler version r1732_e89_eg3629, with a Benjamini-Hochberg (BH) false discovery rate (FDR) to correct for multiple testing.
To identify overrepresentation of genes with AWD specific-signals of positive selection depicted from the branch-site model, a GO analysis using G-profiler version r1732_e89_eg3629 was performed. Ensembl identification numbers of genes that resulted from the branch-site model (p < 0.05) were input as query lists, and all the genes tested (18,327 filtered genes) were employed as the background gene list. Multiple hypothesis correction was conducted with the Benjamini-Hochberg FDR method99. Likewise, Ensembl identification numbers of genes within windows that had empirical p-values < 0.01 from the HKA-like test were input as query lists, and all the genes tested were used as the background gene list.
Polygenic selection
To detect biological pathways overrepresented by weak to moderate signals of selection on the AWD, dhole, and gray wolf, the program polysel (Detection of polygenic selection in gene sets) was employed31. Polysel uses information from all genes depicted by the branch-site model test to find low-level polygenic selection across many genes within a pathway. Significant pathways were identified with an FDR < 0.20, after removal of the overlap between pathways with the pruning procedure implemented in polysel (see Supplementary methods for details).
Accession codes
A description of all custom scripts used for the analyses in this paper can be found at https://github.com/dechavezv/African_wild_dog_Project.git.
Data Availability
Numbers for the sequencing reads of canids genomes are listed in Table S1. The sequences of the red fox were obtained from BioMart - Ensembl (Ensembl Genes 95 release). Speothos venaticus (bush dog) and Chrysocyon brachyurus (maned wolf) sequences are available upon request from the authors. Gene sequences of other species of placental, marsupial and/or monotreme mammals, were obtained from OrthoMaM v998.
References
Hartstone-Rose, A., Werdelin, L., de Ruiter, D. J., Berger, L. R. & Churchill, S. E. The Plio-Pleistocene ancestor of wild dogs, Lycaon sekowein. sp. Journal of Paleontology 84, 299–308 (2010).
Creel, S. & Creel, N. M. The African wild dog: behavior, ecology and conservation. (Princeton University Press, 2002).
Van Valkenburgh, B. Iterative evolution of hypercarnivory in canids (mammalia, carnivora) - evolutionary interactions among sympatric predators. Paleobiology 17, 340–362 (1991).
Van Valkenburgh, B. & Koepfli, K. P. Cranial and dental adaptations to predation in canids. Mammals as Predators 65, 15–37 (1993).
Martinez-Navarro, B. & Rook, L. Gradual evolution in the African hunting dog lineage - Systematic implications. Comptes Rendus Palevol 2, 695–702, https://doi.org/10.1016/j.crpv.2003.06.002 (2003).
Shumba, T., Montgomery, R. A., Sillero-Zubiri, C. & Rasmussen, G. S. A. Morphological variation of wild dogs across Africa. International Journal of Zoology and Applied Biosciences 2, 145–154 (2017).
Walsberg, G. E. Coat color and solar heat gain in animals. Bioscience 33, 88–91, https://doi.org/10.2307/1309169 (1983).
Gerald, M. S. Primate colour predicts social status and aggressive outcome. Animal Behaviour 61, 559–566, https://doi.org/10.1006/anbe.2000.1648 (2001).
Steiner, C. C., Weber, J. N. & Hoekstra, H. E. Adaptive variation in beach mice produced by two interacting pigmentation genes. Plos Biology 5, 1880–1889, https://doi.org/10.1371/journal.pbio.0050219 (2007).
Candille, S. I. et al. A beta-defensin mutation causes black coat color in domestic dogs. Science 318, 1418–1423, https://doi.org/10.1126/science.1147880 (2007).
Campana, M. G. et al. Genome sequence, population history, and pelage genetics of the endangered African wild dog (Lycaon pictus). Bmc Genomics 17, https://doi.org/10.1186/s12864-016-3368-9 (2016).
Gopalakrishnan, S. et al. Interspecific Gene Flow Shaped the Evolution of the Genus Canis. Current Biology 28, 3441–3449, https://doi.org/10.1016/j.cub.2018.08.041 (2018).
Armstrong, E. E. et al. Cost-effective assembly of the African wild dog (Lycaon pictus) genome using linked reads (2018).
Robinson, J. A. et al. Genomic Flatlining in the Endangered Island Fox. Current Biology 26, 1183–1189, https://doi.org/10.1016/j.cub.2016.02.062 (2016).
Koepfli, K. P. et al. Genome-wide Evidence Reveals that African and Eurasian Golden Jackals Are Distinct Species. Current Biology 25, 2158–2165, https://doi.org/10.1016/j.cub.2015.06.060 (2015).
Auton, A. et al. Genetic Recombination Is Targeted towards Gene Promoter Regions in Dogs. Plos Genetics 9, https://doi.org/10.1371/journal.pgen.1003984 (2013).
vonHoldt, B. M. et al. Whole-genome sequence analysis shows that two endemic species of North American wolf are admixtures of the coyote and gray wolf. Science Advances 2, https://doi.org/10.1126/sciadv.1501714 (2016).
Freedman, A. H. et al. Genome Sequencing Highlights the Dynamic Early History of Dogs. Plos Genetics 10, https://doi.org/10.1371/journal.pgen.1004016 (2014).
Zhang, C., Rabiee, M., Sayyari, E. & Mirarab, S. ASTRAL-III: polynomial time species tree reconstruction from partially resolved gene trees. Bmc Bioinformatics 19, https://doi.org/10.1186/s12859-018-2129-y (2018).
Lindblad-Toh, K. et al. Genome sequence, comparative analysis and haplotype structure of the domestic dog. Nature 438, 803–819, https://doi.org/10.1038/nature04338 (2005).
Yang, Z. H. PAML 4: Phylogenetic analysis by maximum likelihood. Molecular Biology and Evolution 24, 1586–1591, https://doi.org/10.1093/molbev/msm088 (2007).
Gronau, I., Hubisz, M. J., Gulko, B., Danko, C. G. & Siepel, A. Bayesian inference of ancient human demography from individual genome sequences. Nature Genetics 43, 1031–U1151, https://doi.org/10.1038/ng.937 (2011).
McKenna, A. et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Research 20, 1297–1303, https://doi.org/10.1101/gr.107524.110 (2010).
Loytynoja, A. & Goldman, N. An algorithm for progressive multiple alignment of sequences with insertions. Proceedings of the National Academy of Sciences of the United States of America 102, 10557–10562, https://doi.org/10.1073/pnas.0409137102 (2005).
Zhang, J. Z., Nielsen, R. & Yang, Z. H. Evaluation of an improved branch-site likelihood method for detecting positive selection at the molecular level. Molecular Biology and Evolution 22, 2472–2479, https://doi.org/10.1093/molbev/msi237 (2005).
Harrison, P. W., Jordan, G. E. & Montgomery, S. H. SWAMP: Sliding Window Alignment Masker for PAML. Evolutionary Bioinformatics 10, https://doi.org/10.4137/ebo.s18193 (2014).
Yang, Z. H. & dos Reis, M. Statistical Properties of the Branch-Site Test of Positive Selection. Molecular Biology and Evolution 28, 1217–1228, https://doi.org/10.1093/molbev/msq303 (2011).
Venkat, A., Hahn, M. W. & Thornton, J. W. Multinucleotide mutations cause false inferences of lineage-specific positive selection. Nature Ecology & Evolution 2, 1280–1288, https://doi.org/10.1038/s41559-018-0584-5 (2018).
Reimand, J., Kull, M., Peterson, H., Hansen, J. & Vilo, J. g: Profiler - a web-based toolset for functional profiling of gene lists from large-scale experiments. Nucleic Acids Research 35, W193–W200, https://doi.org/10.1093/nar/gkm226 (2007).
Venkatesh, D. Primary cilia. J Oral Maxillofac Pathol 21, 8–10 (2017).
Daub, J. T., Moretti, S., Davydov, I. I., Excoffier, L. & Robinson-Rechavi, M. Detection of Pathways Affected by Positive Selection in Primate Lineages Ancestral to Humans. Molecular Biology and Evolution 34, 1391–1402, https://doi.org/10.1093/molbev/msx083 (2017).
Cooper, K. L. et al. Patterning and post-patterning modes of evolutionary digit loss in mammals. Nature 511, 41–U537, https://doi.org/10.1038/nature13496 (2014).
Scherz, P. J., McGlinn, E., Nissim, S. & Tabin, C. J. Extended exposure to Sonic hedgehog is required for patterning the posterior digits of the vertebrate limb. Developmental Biology 308, 343–354, https://doi.org/10.1016/j.ydbio.2007.05.030 (2007).
McLaren, W. et al. The Ensembl Variant Effect Predictor. Genome Biology 17, https://doi.org/10.1186/s13059-016-0974-4 (2016).
Auerbach, A. D. Fanconi anemia and its diagnosis. Mutation Research 668, 4–10 (2009).
Nei, M., Niimura, Y. & Nozawa, M. The evolution of animal chemosensory receptor gene repertoires: roles of chance and necessity. Nature Reviews Genetics 9, 951–963, https://doi.org/10.1038/nrg2480 (2008).
Grotewold, L. & Ruther, U. The Wnt antagonist Dickkopf-1 is regulated by Bmp signaling and c-Jun and modulates programmed cell death. Embo Journal 21, 966–975, https://doi.org/10.1093/emboj/21.5.966 (2002).
Zakeri, Z., Quaglino, D. & Ahuja, H. S. Apoptotic cell-death in the mouse limb and its suppression in the hammertoe mutant. Developmental Biology 165, 294–297, https://doi.org/10.1006/dbio.1994.1255 (1994).
Huard, C. C., Tremblay, C. S., Magron, A., Levesque, G. & Carreau, M. The Fanconi anemia pathway has a dual function in Dickkopf-1 transcriptional repression. Proceedings of the National Academy of Sciences of the United States of America 111, 2152–2157, https://doi.org/10.1073/pnas.1314226111 (2014).
Webb, M. L. et al. Incidence of Fanconi Anemia in Children with Congenital Thumb Anomalies Referred for Diepoxybutane Testing. Journal of Hand Surgery-American Volume 36A, 1052–1057, https://doi.org/10.1016/j.jhsa.2011.02.018 (2011).
Adamska, M., MacDonald, B. T., Sarmast, Z. H., Oliver, E. R. & Meisler, M. H. En1 and Wnt7a interact with Dkk1 during limb development in the mouse. Developmental Biology 272, 134–144, https://doi.org/10.1016/j.ybdio.2004.04.026 (2004).
Bangs, F. & Anderson, K. V. Primary Cilia and Mammalian Hedgehog Signaling. Cold Spring Harbor Perspectives in Biology 9, https://doi.org/10.1101/cshperspect.a028175 (2017).
Hampl, M. et al. Role of Primary Cilia in Odontogenesis. Journal of Dental Research 96, 965–974, https://doi.org/10.1177/0022034517713688 (2017).
Jernvall, J. & Thesleff, I. Reiterative signaling and patterning during mammalian tooth morphogenesis. Mechanisms of Development 92, 19–29, https://doi.org/10.1016/s0925-4773(99)00322-6 (2000).
Thivichon-Prince, B. et al. Primary Cilia of Odontoblasts: Possible Role in Molar Morphogenesis. Journal of Dental Research 88, 910–915, https://doi.org/10.1177/0022034509345822 (2009).
Liem, K. F. et al. The IFT-A complex regulates Shh signaling through cilia structure and membrane protein trafficking. Journal of Cell Biology 197, 789–800, https://doi.org/10.1083/jcb.201110049 (2012).
Veleri, S. et al. Ciliopathy-associated gene Cc2d2a promotes assembly of subdistal appendages on the mother centriole during cilia biogenesis. Nature Communications 5, https://doi.org/10.1038/ncomms5207 (2014).
Abdelhamed, Z. A. et al. The Meckel-Gruber syndrome protein TMEM67 controls basal body positioning and epithelial branching morphogenesis in mice via the non-canonical Wnt pathway. Disease Models & Mechanisms 8, 527–541, https://doi.org/10.1242/dmm.019083 (2015).
Goetz, S. C. & Anderson, K. V. The primary cilium: a signalling centre during vertebrate development. Nature Reviews Genetics 11, 331–344, https://doi.org/10.1038/nrg2774 (2010).
Ohazama, A. et al. Primary cilia regulate Shh activity in the control of molar tooth number. Development 136, 897–903, https://doi.org/10.1242/dev.027979 (2009).
Hoffer, J. L., Fryssira, H., Konstantinidou, A. E., Ropers, H. H. & Tzschach, A. Novel WDR35 mutations in patients with cranioectodermal dysplasia (Sensenbrenner syndrome). Clinical Genetics 83, 92–95, https://doi.org/10.1111/j.1399-0004.2012.01880.x (2013).
Hudson, R. R., Kreitman, M. & Aguade, M. A test of neutral molecular evolution based on nucleotide data. Genetics 116, 153–159 (1987).
Bloch-Zupan, A. et al. Oro-dental features as useful diagnostic tool in Rubinstein-Taybi syndrome. American Journal of Medical Genetics Part A 143A, 570–573, https://doi.org/10.1002/ajmg.a.31622 (2007).
Kraus, W. L., Manning, E. T. & Kadonaga, J. T. Biochemical analysis of distinct activation functions in p300 that enhance transcription initiation with chromatin templates. Molecular and Cellular Biology 19, 8123–8135 (1999).
Janknecht, R., Wells, N. J. & Hunter, T. TGF-beta-stimulated cooperation of Smad proteins with the coactivators CBP/p300. Genes &. Development 12, 2114–2119, https://doi.org/10.1101/gad.12.14.2114 (1998).
Huang, X. & Chai, Y. TGF-β signaling and tooth development. Chinese Journal of Dental Research 13, 7–13 (2010).
Figueiro, H. V. et al. Genome-wide signatures of complex introgression and adaptive evolution in the big cats. Science Advances 3, https://doi.org/10.1126/sciadv.1700299 (2017).
Sturm, R. A., Teasdale, R. D. & Box, N. F. Human pigmentation genes: identification, structure and consequences of polymorphic variation. Gene 277, 49–62, https://doi.org/10.1016/s0378-1119(01)00694-1 (2001).
Raposo, G. & Marks, M. S. Melanosomes-dark organelles enlighten endosomal membrane transport. Nature Reviews Molecular Cell Biology 8, 786–797, https://doi.org/10.1038/nrm2258 (2007).
Lamoreux, M. L., Véronique, D., Lionel, L. & Dorothy, B. The colors of mice: a model genetic network. (John Wiley & Sons, 2010).
Kaelin, C. B. & Barsh, G. S. Genetics of Pigmentation in Dogs and Cats. Annual Review of Animal Biosciences 1, 125–156, https://doi.org/10.1146/annurev-animal-031412-103659 (2013).
Crawford, N. G. et al. Loci associated with skin pigmentation identified in African populations. Science 358, 887–+, https://doi.org/10.1126/science.aan8433 (2017).
Nagasaki, Y. et al. Reversal of hypopigmentation in phenylketonuria mice by adenovirus-mediated gene transfer. Pediatric Research 45, 465–473, https://doi.org/10.1203/00006450-199904010-00003 (1999).
Ding, Z. et al. Correction of murine PKU following AAV-mediated intramuscular expression of a complete phenylalanine hydroxylating system. Molecular Therapy 16, 673–681, https://doi.org/10.1038/mt.2008.17 (2008).
Ding, Z., Georgiev, P. & Thony, B. Administration-route and gender-independent long-term therapeutic correction of phenylketonuria (PKU) in a mouse model by recombinant adeno-associated virus 8 pseudotyped vector-mediated gene transfer. Gene Therapy 13, 587–593, https://doi.org/10.1038/sj.gt.3302684 (2006).
Thony, B., Auerbach, G. & Blau, N. Tetrahydrobiopterin biosynthesis, regeneration and functions. Biochemical Journal 347, 1–16, https://doi.org/10.1042/0264-6021:3470001 (2000).
Erlandsen, H. et al. Crystal structure of the catalytic domain of human phenylalanine hydroxylase reveals the structural basis for phenylketonuria. Nature Structural Biology 4, 995–1000, https://doi.org/10.1038/nsb1297-995 (1997).
Yao, L. L., Shen, M., Lu, Z. K., Ikebe, M. & Li, X. D. Identification of the Isoform-specific Interactions between the Tail and the Head of Class V Myosin. Journal of Biological Chemistry 291, 8241–8250, https://doi.org/10.1074/jbc.M115.693762 (2016).
Meehan, T. F. et al. Disease model discovery from 3,328 gene knockouts by The International Mouse Phenotyping Consortium. Nature Genetics 49, 1231–1238, https://doi.org/10.1038/ng.3901 (2017).
Wu, X. F., Bowers, B., Rao, K., Wei, Q. & Hammer, J. A. Visualization of melanosome dynamics within wild-type and dilute melanocytes suggests a paradigm for myosin V function in vivo. Journal of Cell Biology 143, 1899–1918, https://doi.org/10.1083/jcb.143.7.1899 (1998).
Evans, R. D. et al. Myosin-Va and Dynamic Actin Oppose Microtubules to Drive Long-Range Organelle Transport. Current Biology 24, 1743–1750, https://doi.org/10.1016/j.cub.2014.06.019 (2014).
Gautam, R. et al. The Hermansky-Pudlak syndrome 3 (cocoa) protein is a component of the biogenesis of lysosome-related organelles complex-2 (BLOC-2). Journal of Biological Chemistry 279, 12935–12942, https://doi.org/10.1074/jbc.M311311200 (2004).
Di Pietro, S. M., Falcón‐Pérez, J. M. & Dell’Angelica, E. C. Characterization of BLOC-2, a complex containing the Hermansky-Pudlak syndrome proteins HPS3. HPS5 and HPS6. 5, 276–283 (2004).
Van Valkenburgh, B. Skeletal indicators of locomotor behavior in living and extinct carnivores. 7, 162–182 (1987).
Swann, R. L. In Adaptations to aquatic, arboreal, fossorial and cursorial habits in mammals. - IV Cursorial adaptations Vol. 38 (ed The American Naturalist) 1–11 (1904).
Van Valkenburgh, B. Feeding behavior in free-ranging, large African carnivores. Journal of Mammalogy 77, 240–254, https://doi.org/10.2307/1382725 (1996).
Anderson, T. M. et al. Molecular and Evolutionary History of Melanism in North American Gray Wolves. Science 323, 1339–1343, https://doi.org/10.1126/science.1165448 (2009).
Stahler, D. R., MacNulty, D. R., Wayne, R. K., vonHoldt, B. & Smith, D. W. The adaptive value of morphological, behavioural and life-history traits in reproductive female wolves. Journal of Animal Ecology 82, 222–234, https://doi.org/10.1111/j.1365-2656.2012.02039.x (2013).
Turner, A. The evolution of the guild of larger terrestrial carnivores during the Plio-Pleistocene in. Africa. Geobios 23, 349–368, https://doi.org/10.1016/0016-6995(90)80006-2 (1990).
Bobe, R. & Behrensmeyer, A. K. The expansion of grassland ecosystems in Africa in relation to mammalian evolution and the origin of the genus Homo. Palaeogeography Palaeoclimatology Palaeoecology 207, 399–420, https://doi.org/10.1016/j.palaeo.2003.09.033 (2004).
Girman, D. J., Mills, M. G. L., Geffen, E. & Wayne, R. K. A molecular genetic analysis of social structure, dispersal, and interpack relationships of the African wild dog (Lycaon pictus). Behavioral Ecology and Sociobiology 40, 187–198, https://doi.org/10.1007/s002650050332 (1997).
Langmead, B., Trapnell, C., Pop, M. & Salzberg, S. L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biology 10, https://doi.org/10.1186/gb-2009-10-3-r25 (2009).
Castresana, J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Molecular Biology and Evolution 17, 540–552, https://doi.org/10.1093/oxfordjournals.molbev.a026334 (2000).
Stamatakis, A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313, https://doi.org/10.1093/bioinformatics/btu033 (2014).
Singhal, S., Grundler, M., Colli, G. & Rabosky, D. L. Squamate Conserved Loci (SqCL): A unified set of conserved loci for phylogenomics and population genetics of squamate reptiles. Molecular Ecology Resources 17, e12–e24, https://doi.org/10.1111/1755-0998.12681 (2017).
Tedford, R. H., Wang, X. & Taylor, B. E. Phylogenetic systematics of the North American fossil Caninae (Carnivora: Canidae). Bulletin of the American Museum of Natural History 325, 1–218, https://doi.org/10.1206/574.1 (2009).
Slater, G. J., Harmon, L. J. & Alfaro, M. E. Integrating fossils with molecular phylogenies improves inference of trait evolution. Evolution 66, 3931–3944, https://doi.org/10.1111/j.1558-5646.2012.01723.x (2012).
Skoglund, P., Ersmark, E., Palkopoulou, E. & Dalen, L. Ancient Wolf Genome Reveals an Early Divergence of Domestic Dog Ancestors and Admixture into High-Latitude Breeds. Current Biology 25, 1515–1519, https://doi.org/10.1016/j.cub.2015.04.019 (2015).
Zerbino, D. R. et al. Ensembl 2018. Nucleic Acids Research 46, D754–D761, https://doi.org/10.1093/nar/gkx1098 (2018).
McCormack, J. E., Hird, S. M., Zellmer, A. J., Carstens, B. C. & Brumfield, R. T. Applications of next-generation sequencing to phylogeography and phylogenetics. Molecular Phylogenetics and Evolution 66, 526–538, https://doi.org/10.1016/j.ympev.2011.12.007 (2013).
Storey, J., Bass, A. J., Dabney, A. & Robinson, D. qvalue: Q-value estimation for false discovery rate control version 2.10.0, http://github.com/jdstorey/qvalue (2017).
Huber, C. D., Degiorgio, M., Hellmann, I. & Nielsen, R. Detecting recent selective sweeps while controlling for mutation rate and background selection. Molecular Ecology 25, 142–156, https://doi.org/10.1111/mec.13351 (2016).
Johnson, K. E. & Voight, B. F. Patterns of shared signatures of recent positive selection across human populations. Nature Ecology &. Evolution 2, 713–720, https://doi.org/10.1038/s41559-018-0478-6 (2018).
Zhai, W. W., Nielsen, R. & Slatkin, M. An Investigation of the Statistical Power of Neutrality Tests Based on Comparative and Population Genetic Data. Molecular Biology and Evolution 26, 273–283, https://doi.org/10.1093/molbev/msn231 (2009).
Webb, A. E., Walsh, T. A. & O’Connell, M. J. VESPA: Very large-scale Evolutionary and Selective Pressure Analyses. PeerJ Computer Science 3, 1–16 (2017).
Kearse, M. et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28, 1647–1649, https://doi.org/10.1093/bioinformatics/bts199 (2012).
Biasini, M. et al. SWISS-MODEL: modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Research 42, W252–W258, https://doi.org/10.1093/nar/gku340 (2014).
Douzery, E. J. P. et al. OrthoMaM v8: A Database of Orthologous Exons and Coding Sequences for Comparative Genomics in Mammals. Molecular Biology and Evolution 31, 1923–1928, https://doi.org/10.1093/molbev/msu132 (2014).
Benjamini, Y. & Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. Journal of the Royal Statistical Society. Series B (Methodological) 57, 289–300 (1995).
Joo, W. et al. Structure of the FANCI-FANCD2 Complex: Insights into the Fanconi Anemia DNA Repair Pathway. Science 333, 312–316, https://doi.org/10.1126/science.1205805 (2011).
Acknowledgements
We thank Tom Gilbert from the University of Copenhagen for sharing some of the genome sequences analyzed in this study prior to publication. We thank Kirk Lohmueller, Jacqueline Robinson and Annabel Beichman for guidance in data analysis and improvement of the manuscript. We thank Tina del Carpio for proofreading the manuscript. We thank Duyen Tran from UCLA for making the illustrations of canid species as well as molars and paws presented in the Figures 1–4. We thank Nadia Falconi for making the schemes about primary cilium, digit reduction and melanocyte pigmentation presented in the Figures 2–4. We acknowledge the Vincent J. Coates Genomics Sequencing Laboratory at the University of California, Berkeley for DNA sequencing of samples. We thank SENESCYT for the financial support received during D.E.C.’s doctoral studies at UCLA. We acknowledge support from NSF grant DEB20150429 to R.K.W. K.P.K and S.K. were partially supported by funding from St. Peterburg State University, Russia (Genome Russia Grant no. 1.52.1647.2016). S.K. was also supported by a grant from the Russian Foundation for Basic Research (RFBR), Grant No. 17-00-00144.
Author information
Authors and Affiliations
Contributions
D.E.C., K.-P.K. and R.K.W. designed and performed the experiment. Sequence and adaptive evolution analysis was primarily carried out by D.E.C. Species tree analysis was conducted by T.H. and K.-P.K. Divergence time estimation was conducted by S.K. and K.-P.K. Demographic inference analysis was performed by I.G. The manuscript was written by D.E.C., K.-P.K. and R.K.W. with the final approval by all authors.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chavez, D.E., Gronau, I., Hains, T. et al. Comparative genomics provides new insights into the remarkable adaptations of the African wild dog (Lycaon pictus). Sci Rep 9, 8329 (2019). https://doi.org/10.1038/s41598-019-44772-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-44772-5
This article is cited by
-
African wild dogs (Lycaon pictus) from the Kruger National Park, South Africa are currently not inbred but have low genomic diversity
Scientific Reports (2022)
-
Analysis of the microbial diversity in the fecal material of the critically endangered African wild dog, Lycaon pictus
Archives of Microbiology (2022)
-
The early hunting dog from Dmanisi with comments on the social behaviour in Canidae and hominins
Scientific Reports (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
