
Abstract
Maculinea (=Phengaris) are endangered butterflies that are characterized by a very complex biological cycle. Maculinea larvae behave as obligate parasites whose survival is strictly dependent on both particular food plants and species-specific Myrmica ants. In this interaction, Maculinea caterpillars induce Myrmica workers to retrieve and rear them in the nest by chemical and acoustic deception. Social insect symbiotic microorganisms play a key role in intraspecific and interspecific communication; therefore, it is possible that the Maculinea caterpillar microbiome might be involved in the chemical cross-talk by producing deceptive semiochemicals for host ants. To address this point, the microbiota of Maculinea alcon at different larval stages (phytophagous early larvae, intermediate larvae, carnivorous late larvae) was analyzed by using 16S rRNA-guided metabarcoding approach and compared to that of the host ant Myrmica scabrinodis. Structural and deduced functional profiles of the microbial communities were recorded, which were used to identify specific groups of microorganisms that may be involved in the chemical cross-talk. One of the most notable features was the presence in all larval stages and in the ants of two bacteria, Serratia marcescens and S. entomophila, which are involved in the chemical cross-talk between the microbes and their hosts.
Similar content being viewed by others


Introduction
The endangered Maculinea butterflies (Lepidoptera, Lycaenidae) are perceived as “umbrella species” in many grassland ecosystems and have been chosen to raise support to conserve biodiversity in grassland habitats in Europe1. Maculinea butterflies are characterized by a complex life-history2. They are obligate parasites, and the survival of local populations is largely dependent on both specific food plants for the early instars and Myrmica ant species providing food and protection for the final instars3. Females of Maculinea lay eggs on a specific food plant, early larvae hatch and live feeding inside flowers for two to three weeks. As they reach their final instar, they drop to the ground and wait until they are collected by foraging workers of the genus Myrmica that return them to their nests. Inside the nests, the caterpillars of the five European species act either as predators, Ma. arion and Ma. teleius, preying directly on ant brood4 or adopt a “cuckoo” strategy, Ma. alcon and Ma. rebeli (which are considered as separate taxa according to Bonelli et al.5), as they mimic ant larvae and are fed directly by worker ants3. Ma. nausithous adopts both strategies6. Inside the ant nests, Maculinea last instars spend 11 (or 23 two-year developer7) months increasing their body mass before pupating inside the ant colony from which adults emerge the following summer.
Two steps are critical for the larval survival in Maculinea species: (i) the choice of suitable food plants for oviposition; and (ii) the interaction with the worker ants that adopt the larvae. Indeed, there is evidence that while Maculinea caterpillars can be rapidly adopted by any Myrmica species they encounter on the ground underneath the host plant, they tend to only survive in the nests of a single or a few species of Myrmica8. Several Maculinea species including Ma. alcon, the focus species of our study, may use different ant hosts in different European geographical regions9.
As Maculinea larvae leave their host plant, their profile of surface hydrocarbons is sufficiently similar to that of Myrmica spp. workers to ensure their adoption only by ants belonging to this genus10. Once they have been moved inside the host colony, the full integration of the parasites, particularly in “cuckoo” feeding species, is facilitated by both queen-like acoustic signals11 and more complex chemical mimicry matching the chemical profile of the primary Myrmica host ant species12. In colonies of non-host Myrmica species the Maculinea larvae are recognized as intruders and often killed12.
Besides the chemical and acoustic signaling, there is increasing evidence from social or non-social insects that symbiotic microorganisms may contribute to their nutrition, immune system modulation, and protection by the production of antibiotics or metabolites crucial for food digestion, detoxification, and nitrogen fixation13. Termites, ants, and bees harbor specialized communities of gut bacteria with beneficial functions14,15,16. Social interactions among individuals, such as the exchange of food and allogrooming, provide the opportunity to transfer and share these specialized gut communities13. In ants, horizontal transmission of microbes by trophallaxis might also occur between distinct species involved in tight interactions, as described for Solenopsis invicta and Solenopsis richteri parasitized by their inquiline ants, Solenopsis daguerrei17.
Gut microorganisms, through biosynthetic or catabolic pathways, may produce semiochemicals that function in the host as pheromones or kairomones. For instance, in the desert locust Schistocerca gregaria, Pantoea agglomerans and other common gut bacteria such as Klebsiella pneumoniae and Enterobacter cloacae metabolize plant-derived vanillic acid producing guaiacol and other components of the locust cohesion pheromone that is responsible for forming dense migrating swarms18. In contrast, in aphid guts Staphylococcus sciuri is the source of kairomones that are released into honeydew and act as attractants to aphid predators19. In the model organism Drosophila melanogaster, the diet-associated gut microbiome oversees important developmental, physiological and behavioral processes including mating attractiveness. Flies mate preferentially with individuals harboring the same microbiome assemblage, and mating preferences have been manipulated by introduction of selected gut microorganisms such as Lactobacillus plantarum, which is predominant in flies feeding on starch-rich substrates, into axenic flies20. Here mating preference was associated with the ability of the gut bacteria to affect the scent profile used by Drosophila in choosing mates21.
The interactions between microbiome assemblage and insect feeding strategy and behaviors have potential evolutionary implications, and may be considered as an important driver for insect speciation20. Recent studies have clearly shown that these interactions can be much more complex than imagined when they involve multiple trophic levels22,23,24,25. These studies prompted us to investigate whether the M. alcon caterpillar microbiome may be involved in the production of semiochemicals used to communicate with the Myrmica ants. Although the biology of Maculinea butterflies has been extensively studied, information on the associated microbial community is entirely lacking. We predict that in Maculinea “cuckoo” species, where chemical mimicry is crucial to obtain a full integration in the host colony, the stages associated with the ants possess characteristic microbial communities that are different from those found while they are feeding on the host plant. Due to the occurrence of food exchange (trophallaxis), we also expect some similarity in the microbiota of ants and fully-grown Maculinea larvae. To address these points, the microbiota of caterpillars at different larval stages were analyzed using 16S rRNA amplicon metabarcoding followed by microbiomes functional prediction, and compared to that of the host ant My. scabrinodis.
Results and Discussion
16S rRNA-based profiling of bacterial communities from Myrmica scabrinodis and Maculinea alcon larvae
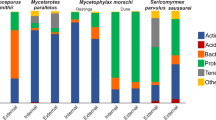
Comparative 16S rRNA-based profiling of microbiota of Ma. alcon instars at different life history stages (phytophagous early larvae [EL], intermediate larvae [IL], carnivorous late larvae [LL]) show small differences between samples in terms of detected phyla. Phyla with a relative abundance equal or higher than 0.001%, are 14, 16 and 12 in EL, IL and LL, respectively (Fig. 1 and Supplementary Dataset S1 and S2). The number of detected phyla is higher, i.e. 20, in the host ant My. scabrinodis. Several phyla can be found in all samples, while others are detected exclusively in Ma. alcon (Thermotogae) or in specific larval stages of Ma. alcon and in My. scabrinodis (Fig. 1). In particular, Gemmatimonadetes, Nitrospirae and Chlamydiae are detected in both EL and IL (and M. scabrinodis), whereas Thermi and Synergistetes are only found in LL and My. scabrinodis. This result indicates dynamic variations of the bacterial community during the larval development of Ma. alcon from phytophagous EL to carnivorous LL, and possible interactions with the microbiota of the omnivorous ants. During metamorphosis, diet-induced dynamic variability of the “adaptive” microbiota and the presence of common microbiota are reported in other lepidoptera26,27,28.

Detected phyla by 16S rRNA gene-based metabarcoding.
Normalized percentages (equal or higher than 1%) of the most representative taxa of bacteria detected are shown in Fig. 2. Proteobacteria dominate the microbiota of Ma. alcon larvae with normalized percentages of 94.2, 96.8 and 99.8 in EL, IL and LL, respectively (Figs 1 and 2A). Yet in both EL and IL stages Alphaproteobacteria dominate (69% and 91%, respectively), while Gammaproteobacteria dominate the microbiota in the LL stage (92%) (Fig. 2A). Most Alphaproteobacteria in EL and IL stages are Rickettsiales of the genus Wolbachia (family Anaplasmataceae) (55.5% and 88.7%, respectively). These maternally transmitted bacterial endosymbionts are well known as reproductive manipulators altering sex ratios in several arthropods29 (Figs 2B–D and 3). The presence of Wolbachia is well documented in both ants and butterflies30,31.

Relative percentages of main phyla (A), orders (B), families (C) and genera (D) associated with distinct larval stages and host ants, as deduced by 16S rRNA gene-based metabarcoding. Relative percentages of Proteobacteria sub-phyla are also reported in A.

Heatmap showing the distribution of different genera detected by 16S rRNA gene-based metabarcoding among analyzed samples.
Notably, among Enterobacterales, only bacteria of the genus Serratia (Yersiniaceae) are detected in all larval stages of the butterfly and in the host ants (Figs 2B–D and 3). Erwinia (Erwiniaceae) is found in both IL and LL, whereas Enterobacter and Citrobacter (both Enterobacteriaceae) are only detected in LL (Figs 2B–D and 3).
Other less represented groups of bacteria are reported in Fig. 3. From IL and LL microbiota show a substantial reduction in the relative abundance of Wolbachia (7.6% in LL), and an increasing prevalence of Gammaproteobacteria of the genus Rickettsiella (facultative insect endosymbionts belonging to the family Coxiellaceae of the order Legionallales), which attains normalized percentage of 86.8% (Figs 2B–D and 3).
The prevalence of Rickettsiella spp. in LL is noteworthy, because these facultative insect endosymbionts are demonstrated to be responsible for a red to green color shift within a population of pea aphids, that camouflages aphids with leaves of different colors making them less visible to predators and parasites32,33. It would be interesting to investigate whether similar a red to yellow color shift, which has been observed during Ma. alcon larval development is involved in the integration of butterfly larvae in host colonies considering that most ant species have a dichromatic color vision system, which are insensitive to red light34.
The metabarcoding analysis of the My. scabrinodis microbiota shows a slight prevalence of Tenericutes (48.9%) of the Mesoplasma genus (Entomoplasmatales, Entomoplasmataceae) with respect to Proteobacteria (42.4%) mostly belonging to Alphaproteobacteria of the genus Wolbachia (27%) (Figs 1 and 2). The prevalence of Mesoplasma is intriguing because recent evidence suggests that a monophyletic clade of Mesoplasma has been found associated with eight Attine, Leaf-Cutter ant species in Brazil and Atta texana in the US, but also with Army ants, Aenictus spp. and Eciton spp.35,36.
Structure similarity of the bacterial communities
Ordination plots of bacterial phylotypes and relative within-sample abundance reveal a complex structure of the bacterial community. NM-MDS (Fig. 4A–C and Supplementary Dataset S3A) indicates a high similarity between EL and IL, which occupy an intermediate position in the plot with respect to My. scabrinodis and LL. Chart plots are used to obtain a global view of orders, families and genera based on within-sample abundance (Supplementary Fig. S1). The results illustrate that EL exhibits the highest levels of phylogenetic diversity and variance in the relative abundance of the different phylotypes, followed by ANT, IL and LL.

Similarities in the microbiomes of larval stages and host ants. Non-metric Multidimensional Scaling (NM-MDS) analysis of order (A), family (B) and genus (C) data (within-sample abundance). The results of the analysis show global similarity between M. scabrinodis (ANT) and M. alcon larvae in early (EL), intermediate (IL) and late (LL) stages. IL and EL stages have a large global similarity. LL is distant as ANT from EL/IL.
Figure 5 shows ordination plots of a correspondence analysis that highlight groups of taxa exclusive or predominant in a given sample, shared by two or three samples, or common to all samples (Supplementary Dataset S3C). At the level of genera (Fig. 5C), obligate intracellular bacteria are excluded, because they are overrepresented in some samples masking other relevant differences between samples. Only few taxa are found common to all samples (Common), ANT (ANT) or EL, IL and LL (Larvae), while major groups of orders, families and genera are specifically associated with EL and IL (EL/IL), or EL, IL and ANT (EL/IL-ANT). In contrast to our initial hypothesis, we find a very limited overlap between LL and ANT samples. The overlap is restricted to Synergistales (Fig. 5A), which are typically characterized by strong amino acid fermenting capability37. Among families, Vibrionaceae is enriched in IL/LL-ANT (Fig. 5B) and genus Vibrio is prevalent in IL/LL-ANT (Fig. 5C).

Correspondence analysis of the microbial communities visualized by ordination plot. Data of orders (A), families (B), genera (C) and predicted KEGG pathways by PICRUSt (D) were used as input. Orders, families, genera and KEGG predictions that were exclusive or largely predominant in a given sample, were shared between two or three samples, or were common to all samples are shown.
In terms of alpha-diversity, the Maculinea microbial community becomes increasingly simpler during larval development (Figs 1–4). This could indicate a secondary loss of endosymbiotic bacteria that in a protected, stable and food rich environment, such as the nest, are not necessary anymore. This might explain why we do not find a close match in the composition between LL and Myrmica ant microbiota (Fig. 5), as hypothesized. However, this general pattern does not exclude that bacteria belonging to Thermi or amino acid-fermenting Synergistetes phyla, which are only shared by LL and ants, could be key factors in promoting the digestion of molecules present in the ant regurgitations. Together with further behavioral and morphological adaptations, the ability to share the same diet of Myrmica ants would have been essential for Maculinea to evolve such a peculiar lifestyle (trophallaxis), which enables the parasite to have this most intimate interaction with its host.
With regard to obligate intracellular genera, which are separately computed, correspondence analysis indicates that some genera are common to all categories sampled and prevalent in EL (Wolbachia, Neorickettsia, Ehrlichia), while other genera are mostly associated with one or two categories (Rickettsia in EL and IL, Rickettsiella in LL, Mesoplasma in ANT) (Supplementary Fig. S2 and Dataset S3B). In insects, endosymbiont shifts or density fluctuations are commonly observed across the different life stages38. The endosymbiont plasticity along with the tropism of endosymbiotic bacteria for the different developing tissues may cope with distinct (metabolic and non-metabolic) requirements during the insect development38,39.
Functional microbiomes predictions of bacterial communities
Predicted KEGG pathways by PICRUSt are distributed more evenly among the samples than phylotypes (Supplementary Dataset S4). In many cases, orders, genera and families appear to be enriched or depleted in several samples, while KEGG predictions have a more even distribution among them (Fig. 5D, Supplementary Dataset S3C). This is rather expected, since different bacteria can provide the same or similar functions. PICRUSt predicts KEGG pathways common to all samples, others shared by ANT and EL, and a group of pathways mostly associated with LL (Fig. 5D, Supplementary Dataset S3C). This last group could be split in two subgroups (LL 70 and LL 40–50) based on their relative within-sample abundance (expressed as percentage) in LL with respect to the other samples (Supplementary Dataset S3C). Five pathways, i.e., lipopolysaccharide biosynthesis, lipopolysaccharide biosynthesis protein, glycan biosynthesis and metabolism, steroid biosynthesis, ether lipid metabolism belong to the LL 70 group, and five pathways, i.e., galactose metabolism, alpha-linolenic acid metabolism, tetracycline biosynthesis, caprolactam degradation belong to the LL 40–50 group. The group of KEGG pathways enriched in LL (Fig. 5D) is consistent with the group of the ten genera (Fig. 5C) that, in addition to endosymbiotic Rickettsiella, are similarly enriched in LL.
PICRUSt analysis predicts a number of pathways involved with lipid biosynthesis and metabolism that are highly enriched in LL, for instance the ether lipid/fatty acid metabolism and alpha-linolenic acid metabolism (Supplementary Fig. S3A). These functional predictions are noteworthy in light of a possible involvement of the LL-specific microbial community in the synthesis of compounds mimicking ant cuticular hydrocarbons. A number of semiochemicals are identified in mandibular gland and Dufour gland secretions of My. scabrinodis40,41 and the chemical profile of cuticular hydrocarbons (CHCs) of this ant species is described in several studies42,43.
The CHC profile of My. scabrinodis sampled in different European countries is dominated by alkenes (accounting for >50% of hydrocarbon classes), followed by dienes, mono-methyl alkanes, n-alkanes and mono-methyl dienes43. Alkane and alkene biosynthesis has been reported in some microorganisms although the biosynthetic pathways are not fully understood44. Four major microbial pathways that convert free fatty acids or fatty acid derivatives into alkanes/alkenes have been proposed involving (i.) fatty aldehyde decarbonylation, (ii.) fatty acid decarboxylation, (iii.) head-to-head hydrocarbon biosynthesis, or (iv.) polyketide synthase reactions44. Notably, none of these pathways is present in the KEGG database that is used for PICRUSt analysis. Particularly the two-step biosynthetic pathway for alkane/alkene biosynthesis starting from acyl-CoA, and involving acyl-CoA reductase and aldehyde decarbonylase has been better documented than the others44. Fatty acid and acyl-CoA reductases are widely distributed in microbial world. In contrast, until a few years ago, decarbonylation of fatty aldehydes is consistently reported only in Cyanobacteria. However, a systematic screening exercise has led recently to the identification of a wide range of microbial species possessing this activity, and in particular, the genus Klebsiella is found to have a common ability to produce alkanes from aldehydes via enzyme catalyzed reaction45. It would be interesting to test whether this ability is related to the specific association of Klebsiella with LL samples. CHCs are the key elements for nestmate recognition processes in social insects. Members of the same colony share a bouquet of CHCs which functions as a template and enables ants to identify intruders46 and references therein. Therefore, the ability to synthesize CHCs similar to those of the host ant is crucial for the parasite to achieve a full integration in the ant colony, to such an extent that Ma. alcon larvae are not only accepted, but also treated like ant brood and fed by trophallaxis43.
The mechanism underlying the ability of post-adoption Maculinea larvae to synthesizes new CHCs peculiar of the host chemical profile is far from understood. The presence of endosymbiotic microbes involved in the biosynthesis and metabolism of lipids suggests that the microbiome could potentially be involved in the Maculinea chemical mimicry.
PICRUSt predictions of secondary metabolism pathways also highlight differences between analyzed sample categories. The most marked difference is a group of metabolic pathways that are depleted in IL and more so in LL (Supplementary Fig. S3B). This group included the following pathways: butyrosin and neomycin biosynthesis, penicillin and cephalosporin biosynthesis, carotenoid biosynthesis, stilbenoid, diarylheptanoid and gingerol biosynthesis, beta-lactam resistance, biosynthesis of siderophore group nonribosomal peptides, flavonoid biosynthesis. Another group of secondary metabolism pathways is highly enriched in ants (and highly depleted in LL); this group is composed of the following pathways: caffeine metabolism, sesquiterpenoid biosynthesis, biosynthesis of type II poliketide products, biosynthesis of 12-, 14- and 16-membered macrolides, biosynthesis of vancomycin group antibiotics, betalain biosynthesis, isoflavonoid biosynthesis, indole alkaloid biosynthesis, melanogenesis. Other pathways are specifically depleted in IL and partially enriched in LL, i.e, tetracycline biosynthesis, caprolactam degradation, geraniol degradation. The relative enrichment in pathways for ubiquinone and other terpenoid-quinone biosynthesis and terpenoid backbone biosynthesis in IL is also noteworthy, because they may provide precursors for biosynthesis of farnesene family compounds that are used as semiochemicals by My. scabrinodis47,48.
Symbiotic Serratia spp. as possible sources of deceptive pyrazines
The metabarcoding data indicate the presence of bacteria belonging to the genus Serratia in all larval stages of Ma. alcon as well as in the host ants, My. scabrinodis. At the species level, Serratia marcescens and S. entomophila are the predominant species, and the relative abundance between them is almost constant in all larval stages of Ma. alcon (Supplementary Fig. S4). There is experimental evidence that S. marcescens, a microorganism that is more often recorded in plants and has been associated with growth-promoting activity49, is capable of producing volatile pyrazines, including 2,5-dimethylpyrazine and 3-ethyl-2,5-dimethylpyrazine50, which are used as pheromones by ants51. Production of pyrazines responsible for potato-like odor is also demonstrated in other Serratia species including S. rubidaea, S. odorifera, and S. ficaria52. Also note that these Serratia species cluster together with S. marcescens and S. entomophila in the phylogenetic tree53. It is possible that the colonization of EL and IL by plant-associated S. marcescens and S. entomophila, which is consistent with the feeding behavior of EL and IL eating the host plant, may play a specific role in the production of deceptive pyrazines.
If confirmed, the ability of Maculinea larvae to emit the foraging trail markers of its host ant (3-ethyl-2,5-dimethylpyrazine) thanks to the association with Serratia spp. would require to re-interpret the observations on the Maculinea adoption rituals, which are generally based on caterpillar behavior and tactile signals (CHC).
Concluding remarks
Our work provides the first description of the microbiota of Maculinea alcon and reports how they change during the larval development. The composition and the variation of the microbial community along with larval growth suggest a still uninvestigated role of microbes in driving not only the shift from phytophagous to an omnivorous diet but also in mediating the chemical deception through which the butterfly parasite achieves the integration and exploitation of the host ant society. The microbial communities associated with multicellular organisms have been shown to significantly influence how the latter interact with their environment. Ant and plant evolutionary pathways are tightly related54 and provide the basis for the evolution of ant-butterflies symbiosis55. Altogether our results suggest the need to consider the structure and function of microbiota as a potential evolutionary force shaping complex and multilevel interactions with the final aim of contributing to their conservation, which is a priority in the case of Maculinea butterflies.
Methods
Insect collection
Butterflies and ants were collected at Caselette (45°070N, 07°290E), about 20 km far from Turin (NW Italy). In this wet grassland, Maculinea alcon exploits Myrmica scabrinodis colonies as host and Gentiana pneumonanthe as larval food plants. Adult butterflies are on the wing from late July to middle August56. Phytophagous early larvae [EL] are first instar caterpillars feeding inside the plant buds. To collect EL, gentian buds bearing eggs were gently dissected in the field at the end of the Ma. alcon flight period. At the beginning of September, five plants with eggs were gathered and maintained in laboratory conditions (26 °C, 60% humidity and 120 mmol m−2 s−1 light under a 16 L: 8 D regime) until IV instar larvae, called intermediate larvae [IL], spontaneously dropped off the plant and were collected. In June, about one month before the adult emergence, My. scabrinodis nests were excavated and examined for the presence of Ma. alcon carnivorous late larvae [LL]. LLs are overwintering, fully-grown caterpillars, which have been reared by worker ants in the brood chambers for about ten months. Worker ants were collected from the same colonies where Ma. alcon LLs were found. All samples were immediately soaked in 50% glycerol and stored at −4 °C.
Sample processing
Whole, surface-sterilized insects were utilized in this study to analyze the dominant microbial taxa mostly associated with the internal body regions. Insects were rinsed in sterile water, soaked in 70% ethanol for 30 s followed by 10% bleach for 30 s, and rinsed again in sterile water. Sterility of insect surfaces was checked by cultural analysis. After surface washing and sterilization, samples were homogenized using sterilized WheatonTM Dounce tissue grinder in order to release and partially lyse the microorganisms. To minimize inter-individual variations, pools of insects were used for metabarcoding analysis, namely: 20 EL, 40 IL, 5 LL, and 30 ANT.
DNA procedures
Total DNA extractions from insect homogenized suspensions were performed by the QIAamp DNA Stool isolation Mini Kit (Qiagen) following the manufacturer’s instructions. Eluted DNA was precipitated in ice-cold 100% ethanol and sodium acetate and then resuspended in 10 mM TrisHCl, pH 8. Extracted DNA was sent to Genomix4life S.R.L. (Baronissi, Salerno, Italy) for library preparation and sequencing of the V3 and V4 region of the 16S rRNA gene.
16S rRNA gene-based metabarcoding
Next generation sequencing experiments, comprising samples quality control and Bioinformatics analysis, were performed by Genomix4life S.R.L. (Baronissi, Salerno, Italy). Final yield and quality of extracted DNA were determined by using NanoDrop ND-1000 spectrophotometer (Thermo Scientific, Waltham, MA) and Qubit Fluorometer 1.0 (Invitrogen Co., Carlsbad, CA). PCR amplifications were performed with primers: Forward: 5′-CCTACGGGNGGCWGCAG-3′ and Reverse: 5′-GACTACHVGGGTATCTAATCC-3′57, which target the hypervariable V3 and V4 region of the 16S rRNA gene. Each PCR reaction was assembled according to 16 S Metagenomic Sequencing Library Preparation (Illumina, San Diego, CA). Libraries were quantified used Qubit fluorometer (Invitrogen Co., Carlsbad, CA) and pooled to an equimolar amount of each index-tagged sample to a final concentration of 4 nM, including the Phix Control Library (Illumina; expected 25%). Pooled samples were subject to cluster generation and sequenced on MiSeq platform (Illumina, San Diego, CA) in a 2 × 250 paired-end format at a final concentration of 10 pmol. The raw sequence files generated (fasta files) underwent quality control analysis with FastQC. The 16S rRNA gene-based metabarcoding analysis performs taxonomic classification of 16S rRNA targeted amplicon reads using the last available version (May 2013) of the GreenGenes taxonomic database. The algorithm is a high-performance implementation of the Ribosomal Database Project (RDP) Classifier described in Wang et al.58 (http://rdp.cme.msu.edu/).
Structure similarity and imputed functional predictions of the bacterial communities
For bioinformatic analysis raw data (Supplementary Dataset S1–S4) were refined by calculating WS-A (relative abundance), the resulting data are shown in Supplementary Dataset S3. Starting from WS-A of phylotypes (orders, families and genera) (Supplementary Dataset S1 and S2) and KEGG (PICRUSt59 predictions (Supplementary Dataset S4) we clustered the data. To obtain multivariate plots (CA analysis, NM-MDS) and Box-Plots we used PAST 3.18 software60. This software package was developed to perform static analysis on paleontological dataset (PAleontological STatistics) but is also useful to analyze biological data. In ordination plots (CA analysis) the tags “EL”, “IL” and “LL” were used to indicate M. alcon EL, IL and LL larvae respectively, and “ANT” for M. scabrinodis. For “EL” and “IL” samples, dots in the following plots were very close, so we represented EL and IL in a common cluster tagged with “EL/IL”. To cluster the results and generate plots, BS-A was also determined. In this case, for each genus (or family or KEGG) we summed the values in all samples and divided the value of each genus by the total value. Some order, family and genus that were only present (or largely predominant BS-A ≥80%) in one sample were marked with the sample tag. One group of data showed lower values for “ANT” (BS-A < 10%) and were indicated with tag “Larvae”. Another group of data showed lower value for “LL” (BS-A < 10%) and was labeled “EL/IL-ANT”. Some data were present in all samples (BS-A > 10% for each sample), and marked with the tag “Common”. In the plot of orders (Fig. 5A) one order had a BS-A = 0% for EL and IL and was reported with a “LL-ANT” tag. In the plot of families (Fig. 5B) one family had a BS-A of 0% in EL column and was labeled as “IL-LL-ANT” and another family with BS-A of 0% in EL and ANT columns and were reported as “IL-LL” tag. In the plots of genera (Fig. 5C), two genera had a BS-A = 0 for LL and ANT data (“EL-LL” tag), one had a BS-A = 0 for EL and ANT (“IL-LL” tag) and, finally, one genus had a BS-A = 0 for EL (“IL-LL-ANT” tag). These tags were used to mark clusters of dots in ordination plots. To analyze all data in a global manner we performed a Non-Metric Multidimensional Scaling (NM-MDS) on family and genus data using Bray-Curtis similarity index with samples in rows, and phylotypes (or KEGG pathway) in columns (Supplementary Dataset S3A). In resulting ordination plots samples are represented as dots.
Vice versa, Correspondence Analysis (CA) were performed with samples in columns and phylotype (or KEGG) in rows (Supplementary Dataset S3C). The WS-A of Wolbachia, Rickettsia, Rickettsiella and Mesoplamsa were exceptionally dominant compared to other genera (resulting in low-resolution plots) so the data of intracellular obligate bacterial were excluded and the remaining family and genus data were used to generate ordination plots by CA (Supplementary Dataset S3B).
Regarding KEEG predictions, the spread between samples was lower, probably because different strains contribute to predicted KEGG functions, so different thresholds were chosen for clustering (according BS-A: >20, >30, >40, >50 and >70) and the following tagged clusters (with thresholds in parenthesis) were generated: “Larvae”, “Common”, “ANT-EL-LL(20)”, “ANT-EL-IL(20–30)”, “EL-IL(30)”, “EL-LL(30)”, “ANT-EL(30)”, “ANT-LL(30)”, “EL-IL(40–50)”, “ANT(40–50)”, “EL(40–50)”, “LL(40–50)”, “EL(70)”, “LL(70)” and “ANT(70)”. This set of data (Supplementary Dataset S3C) was used to generate CA plots. To simplify plots, groups that showed higher similarity were aggregated: “Larvae” and “Common” were grouped together and indicated as “Common/Larvae”, “EL-LL(30)”, “ANT-EL-LL(20)” and “ANT-LL(30)” were grouped together into “LL-EL/ANT”, “EL-IL(30)” and “EL-IL(40–50)” into “EL-IL”, EL(70) and EL(40–50) into “EL”, “ANT(70)” and “ANT(40–50)” into “ANT”, “EL-IL(30)” and “EL-IL(40–50)” into “EL-IL”, and the remaining groups were reported without threshold value. Data of relevant bacterial orders were extracted and used to construct bar plots and perform CA multivariate analysis.
Information about resources and reagents are provided in the Supplementary Table S1.
Data Availability
All data generated or analyzed during this study are included in Supplementary Information files.
References
Settele, J. & Kuhn, E. Insect Conservation. Science 325, 41–42 (2009).
Thomas, J. A. & Settele, J. Evolutionary biology - Butterfly mimics of ants. Nature 432, 283–284 (2004).
Elmes, G., Wardlaw, J. & Thomas, J. Larvae of Maculinea rebeli, a large‐blue butterfly and their Myrmica host ants: patterns of caterpillar growth and survival. J. Zool. 224, 79–92 (1991).
Thomas, J. & Wardlaw, J. The capacity of a Myrmica ant nest to support a predacious species of Maculinea butterfly. Oecologia 91, 101–109 (1992).
Bonelli, S. et al. The first red list of Italian butterflies. Insect Conserv. Divers. 11, 506–521 (2018).
Patricelli, D. et al. Evidence of high larval host ant (Hymenoptera: Formicidae) specificity in the first post-adoption phase for the myrmecophilous butterfly Phengaris (Maculinea) nausithous (Lepidoptera: Lycaenidae). Sociobiology 55, 861–869 (2010).
Schönrogge, K., Wardlaw, J., Thomas, J. & Elmes, G. Polymorphic growth rates in myrmecophilous insects. Proc. R. Soc. Lond. B 267, 771–777 (2000).
Casacci, L. P. et al. Host specificity pattern and chemical deception in a social parasite of ants. Sci. Rep. 9, 1619, https://doi.org/10.1038/s41598-018-38172-4 (2019).
Tartally, A. et al. Patterns of host use by brood parasitic Maculinea butterflies across. Europe. Philos. Trans. Royal Soc. B 374, 20180202, https://doi.org/10.1098/rstb.2018.0202 (2019).
Akino, T., Knapp, J. J., Thomas, J. A. & Elmes, G. W. Chemical mimicry and host specificity in the butterfly Maculinea rebeli, a social parasite of Myrmica ant colonies. Proc. R. Soc. Lond. B 266, 1419–1426 (1999).
Barbero, F., Thomas, J. A., Bonelli, S., Balletto, E. & Schönrogge, K. Queen ants make distinctive sounds that are mimicked by a butterfly social parasite. Science 323, 782–785 (2009).
Schönrogge, K. et al. Changes in chemical signature and host specificity from larval retrieval to full social integration in the myrmecophilous butterfly Maculinea rebeli. J. Chem. Ecol. 30, 91–107 (2004).
Zientz, E., Feldhaar, H., Stoll, S. & Gross, R. Insights into the microbial world associated with ants. Arch. Microbiol. 184, 199–206 (2005).
Engel, P. & Moran, N. A. The gut microbiota of insects–diversity in structure and function. FEMS Microbiol. Rev. 37, 699–735 (2013).
Kwong, W. K. & Moran, N. A. Gut microbial communities of social bees. Nat. Rev. Microbiol. 14, 374 (2016).
Peterson, B. F. & Scharf, M. E. Metatranscriptome analysis reveals bacterial symbiont contributions to lower termite physiology and potential immune functions. BMC Genom. 17, 772 (2016).
Dedeine, F., Ahrens, M., Calcaterra, L. & Shoemaker, D. D. Social parasitism in fire ants (Solenopsis spp.): a potential mechanism for interspecies transfer of Wolbachia. Mol. Ecol. 14, 1543–1548 (2005).
Dillon, R., Vennard, C. & Charnley, A. A note: gut bacteria produce components of a locust cohesion pheromone. J. Appl. Microbiol. 92, 759–763 (2002).
Leroy, P. D. et al. Microorganisms from aphid honeydew attract and enhance the efficacy of natural enemies. Nat. Commun. 2, 348 (2011).
Sharon, G., Segal, D., Zilber-Rosenberg, I. & Rosenberg, E. Symbiotic bacteria are responsible for diet-induced mating preference in Drosophila melanogaster, providing support for the hologenome concept of evolution. Gut microbes 2, 190–192 (2011).
Lewis, Z., Heys, C., Prescott, M. & Lizé, A. You are what you eat: gut microbiota determines kin recognition in Drosophila. Gut microbes 5, 541–543 (2014).
Casteel, C. L. & Hansen, A. K. Evaluating insect-microbiomes at the plant-insect interface. J. Chem. Ecol. 40, 836–847 (2014).
Pizzolante, G. et al. Cultivable gut bacteria provide a pathway for adaptation of Chrysolina herbacea to Mentha aquatica volatiles. BMC Plant Biol 17, 30 (2017).
Tamborindeguy, C., Huot, O. B., Ibanez, F. & Levy, J. The influence of bacteria on multitrophic interactions among plants, psyllids, and pathogen. Insect Sci. 24, 961–974 (2017).
Shikano, I., Rosa, C., Tan, C.-W. & Felton, G. W. Tritrophic interactions: microbe-mediated plant effects on insect herbivores. Ann. Rev. Phytopathol. 55, 313–331 (2017).
Hammer, T. J., McMillan, W. O. & Fierer, N. Metamorphosis of a butterfly-associated bacterial community. PloS one 9, e86995 (2014).
Shao, Y. et al. Symbiont-derived antimicrobials contribute to the control of the Lepidopteran gut microbiota. Cell Chem. Biol. 24, 66–75 (2017).
Tang, X. et al. Complexity and variability of gut commensal microbiota in polyphagous lepidopteran larvae. PloS one 7, e36978 (2012).
Stouthamer, R., Breeuwer, J. A. & Hurst, G. D. Wolbachia pipientis: microbial manipulator of arthropod reproduction. Ann. Rev. Microbiol. 53, 71–102 (1999).
Hilgenboecker, K., Hammerstein, P., Schlattmann, P., Telschow, A. & Werren, J. H. How many species are infected with Wolbachia?–A statistical analysis of current data. FEMS Microbiol. Lett. 281, 215–220 (2008).
Russell, J. A. et al. A veritable menagerie of heritable bacteria from ants, butterflies, and beyond: broad molecular surveys and a systematic review. PLoS One 7, e51027 (2012).
Tsuchida, T. et al. Symbiotic bacterium modifies aphid body color. Science 330, 1102–1104 (2010).
Tsuchida, T., Koga, R., Fujiwara, A. & Fukatsu, T. Phenotypic effect of “Candidatus Rickettsiella viridis,” a facultative symbiont of the pea aphid (Acyrthosiphon pisum), and its interaction with a coexisting symbiont. Appl. Environ. Microbiol. 80, 525–533 (2014).
Menzel, R. & Backhaus, W. Colour vision in insects. In Vision and visual dysfunction (ed. Gouras, P.) 262–293. (Mac-Millan, 1991).
Meirelles, L. A. et al. Bacterial microbiomes from vertically transmitted fungal inocula of the leaf‐cutting ant Atta texana. Environ. Microbiol. Rep. 8, 630–640 (2016).
Funaro, C. F. et al. Army ants harbor a host-specific clade of Entomoplasmatales bacteria. Appl. Environ. Microbiol. 77, 346–350 (2011).
Bhandari, V. & Gupta, R. S. Molecular signatures for the phylum Synergistetes and some of its subclades. Antonie Van Leeuwenhoek 102, 517–540 (2012).
Wernegreen, J. J. & Wheeler, D. E. Remaining flexible in old alliances: functional plasticity in constrained mutualisms. DNA Cell. Biol. 28, 371–382 (2009).
Skidmore, I. H. & Hansen, A. K. The evolutionary development of plant‐feeding insects and their nutritional endosymbionts. Insect Sci. 24, 910–928 (2017).
Morgan, E., Inwood, M. & Cammaerts, M. C. The mandibular gland secretion of the ant, Myrmica scabrinodis. Physiol. Entomol. 3, 107–114 (1978).
Morgan, E., Parry, K. & Tyler, R. The chemical composition of the Dufour gland secretion of the ant Myrmica scabrinodis. Insect Biochem. 9, 117–121 (1979).
Elmes, G., Akino, T., Thomas, J., Clarke, R. & Knapp, J. Interspecific differences in cuticular hydrocarbon profiles of Myrmica ants are sufficiently consistent to explain host specificity by Maculinea (large blue) butterflies. Oecologia 130, 525–535 (2002).
Guillem, R. M., Drijfhout, F. P. & Martin, S. J. Species-specific cuticular hydrocarbon stability within European Myrmica ants. J. Chem. Ecol. 42, 1052–1062 (2016).
Wang, W. & Lu, X. Microbial synthesis of alka(e)nes. Front. Bioeng. Biotech. 1, 10 (2013).
Ito, M., Kambe, H., Kishino, S., Muramatsu, M. & Ogawa, J. A search for microorganisms producing medium-chain alkanes from aldehydes. J. Biosci. Bioeng. 125, 87–91 (2018).
Barbero, F. Cuticular lipids as a cross-talk among ants, plants and butterflies. Int. J. Mol. Sci. 17, 1966 (2016).
Cammaerts, M., Evershed, R. & Morgan, E. D. Comparative study of the Dufour gland secretions of workers of four species of Myrmica ants. J. Insect. Physiol 27, 59–65 (1981).
Cammaerts, M., Evershed, R. & Morgan, E. D. Comparative study of the mandibular gland secretion of four species of Myrmica ants. J. Insect. Physiol 27, 225–231 (1981).
Abreo, E. & Altier, N. Pangenome of Serratia marcescens strains from nosocomial and environmental origins reveals different populations and the links between them. Sci. Rep. 9, 46, https://doi.org/10.1038/s41598-018-37118-0 (2019).
Silva, E. A. et al. Pyrazines from bacteria and ants: convergent chemistry within an ecological niche. Sci. Rep. 8, 2595, https://doi.org/10.1038/s41598-018-20953-6 (2018).
Evershed, R., Morgan, E. D. & Cammaerts, M. Identification of the trail pheromone of the ant Myrmica rubra L., and related species. Naturwissenschaften 68, 374–376 (1981).
Gallois, A. & Grimont, P. A. D. Pyrazines responsible for the potato-like odor produced by some serratia and cedecea strains. Appl. Environ. Microbiol. 50, 1048–1051 (1985).
Van Houdt, R., Moons, P., Jansen, A., Vanoirbeek, K. & Michiels, C. W. Genotypic and phenotypic characterization of a biofilm-forming Serratia plymuthica isolate from a raw vegetable processing line. FEMS Microbiol. Lett. 246, 265–272 (2005).
Moreau, C. S., Bell, C. D., Vila, R., Archibald, S. B. & Pierce, N. E. Phylogeny of the ants: Diversification in the age of angiosperms. Science 312, 101–104 (2006).
DeVries, P. J. Evolutionary and Ecological Patterns in Myrmecophilous-Riodinid Butterflies In Ant-Plant Interactions (eds Huxley, C. R. & Cutler, D. F.) 143–156 (Oxford University Press, 1991).
Nowicki, P., Bonelli, S., Barbero, F. & Balletto, E. Relative importance of density-dependent regulation and environmental stochasticity for butterfly population dynamics. Oecologia 161, 227–239 (2009).
Klindworth, A. et al. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 41, https://doi.org/10.1093/nar/gks808 (2013).
Wang, Q., Garrity, G. M., Tiedje, J. M. & Cole, J. R. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 73, 5261–5267 (2007).
Langille, M. G. I. et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nature Biotech. 31, 814–821 (2013).
Hammer, Ø., Harper, D., Ryan, P. PAST: Paleontological Statistics Software Package for Education and Data Analysis. Palaeontol. Electronica 4: 1–9, http://palaeo-electronica.org/2001_1/past/issue1_01.htm (2001).
Author information
Authors and Affiliations
Contributions
F.B., M.E.M., K.S. and P.A. were involved in early experimental design. F.B. collected the insect samples. A.T., M.D.S. and S.M.T. conceived and conducted the metagenomic experiments. M.C. and M.D.S. performed bioinformatic analysis. F.B. and P.A. wrote the paper. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Di Salvo, M., Calcagnile, M., Talà, A. et al. The Microbiome of the Maculinea-Myrmica Host-Parasite Interaction. Sci Rep 9, 8048 (2019). https://doi.org/10.1038/s41598-019-44514-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-44514-7
This article is cited by
-
Ectoparasitic fungi Rickia wasmannii infection is associated with smaller body size in Myrmica ants
Scientific Reports (2021)
-
Ectoparasitic fungi of Myrmica ants alter the success of parasitic butterflies
Scientific Reports (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
