
Abstract
Bacteria of the genus Paracoccus inhabit various pristine and anthropologically-shaped environments. Many Paracoccus spp. have biotechnological value and several are opportunistic human pathogens. Despite extensive knowledge of their metabolic potential and genome architecture, little is known about viruses of Paracoccus spp. So far, only three active phages infecting these bacteria have been identified. In this study, 16 Paracoccus strains were screened for the presence of active temperate phages, which resulted in the identification of five novel viruses. Mitomycin C-induced prophages were isolated, visualized and their genomes sequenced and thoroughly analyzed, including functional validation of their toxin-antitoxin systems. This led to the identification of the first active Myoviridae phage in Paracoccus spp. and four novel Siphoviridae phages. In addition, another 53 prophages were distinguished in silico within genomic sequences of Paracoccus spp. available in public databases. Thus, the Paracoccus virome was defined as being composed of 66 (pro)phages. Comparative analyses revealed the diversity and mosaicism of the (pro)phage genomes. Moreover, similarity networking analysis highlighted the uniqueness of Paracoccus (pro)phages among known bacterial viruses.
Similar content being viewed by others

Introduction
Paracoccus spp. (Alphaproteobacteria) are metabolically versatile bacteria, that have been isolated from a wide range of environments in various geographical locations, e.g.: biofilters for the treatment of waste gases from an animal rendering plant in Germany (P. alkenifer DSM 11593), contaminated soil in Japan (P. aminophilus JCM 7686 and P. aminovorans JCM 7685), rhizospheric soil of an Indian tropical leguminous plant (P. bengalensis DSM 17099), sea water from South Korea (P. haeundaensis LGM P-21903), marine sediments of the South China Sea (P. halophilus JCM 14014T) and marine bryozoan Bugula plumosa from North Sea in Germany (P. seriniphilus DSM 14827)1,2,3,4,5,6,7. Some Paracoccus spp. have also been recognized as causative agents of human disease8. The metabolic flexibility of Paracoccus spp. relies mostly on the wide variety of respiratory processes employed by these bacteria, including the usage of nitrate, nitrite, nitrous oxide and nitric oxide as alternative electron acceptors in denitrification, and the ability to use substrates that lack carbon-carbon bonds (e.g. methylamine) as electron donors to respiratory chains1.
Paracoccus spp. have substantial biotechnological potential, especially in bioremediation, since they can conduct denitrification (e.g. P. denitrificans)9 and utilize various toxic organic compounds, e.g. N,N-dimethylformamide3 and herbicides10.
Paracoccus spp. have multipartite genomes composed of a chromosome plus extrachromosomal replicons, including essential chromids and diverse plasmids8. As of June 25th 2018, when data were retrieved for this study, the following DNA sequences had been submitted to NCBI databases: (i) nine complete genomes of Paracoccus spp., i.e. P. denitrificans PD1222 (GenBank acc. nos. CP000489-CP000491), P. aminophilus JCM 768611, P. aminovorans JCM 768512, P. contaminans RKI 16-01929T13, P. yeei FDAARGOS_252 (GenBank acc. nos. NZ_CP020440-NZ_CP020447), P. yeei TT1314, P. zhejiangensis J615, Paracoccus sp. BM15 (GenBank acc. nos. NZ_CP025408-NZ_CP025411) and Paracoccus sp. CBA4604 (GenBank acc. nos. NZ_CP025583-NZ_CP025585), (ii) 54 draft genome sequences, and (iii) 52 plasmids of Paracoccus spp.
Although much is known about the metabolic properties and genome architecture of Paracoccus spp., there is very little information about phages of these bacteria. To date, only three active phages infecting Paracoccus spp. have been identified and described: two lytic phages vB_PmaS-R3 (vB_PmaS_IMEP1)16 and Shpa17, plus one temperate virus ϕPam-6 of P. aminophilus JCM 768611. Moreover, five other prophages (ϕPam-1−ϕPam-5, respectively) were identified within the genome of P. aminophilus11.
In this study, we identified five novel active temperate phages and 53 prophages in the available genome sequences of Paracoccus spp., and performed a thorough comparative analysis of the Paracoccus virome.
Results and Discussion
Identification and morphology of novel active temperate Paracoccus phages
The occurrence of active temperate phages was examined in 16 species of the genus Paracoccus: P. alcaliphilus JCM 7364, P. aminovorans JCM 7685, P. alkenifer DSM 11593, P. bengalensis DSM 17099, P. ferroxidans NCCB 1300066, P. haeudaensis LGM P-21903, P. halophilus JCM 14014T, P. homiensis DSM 17862, P. kondratievae NCIMB 13773T, P. pantotrophus DSM 11072, P. seriniphilus DSM 14827, P. solventivorans DSM 11592, P. sulfuroxidans JCM 14013, P. thiocyanatus JCM 20756, P. versutus UW1R and P. yeei CCUG 32053. In each case, an exponentially growing culture was exposed to mitomycin C and released phage particles were concentrated using PEG/NaCl solution. This approach resulted in the induction of five phages, named vB_PbeS_Pben1 (P. bengalensis), vB_PkoS_Pkon1 (P. kondratievae), vB_PsuS_Psul1 (P. sulfuroxidans), vB_PthS_Pthi1 (P. thiocyanatus) and vBPyeM_Pyei1 (P. yeei). It is important to mention, that the term “active” is used in this work for describing mitomycin C-induced and lytic viruses of Paracoccus spp., while it is still possible that other, in silico distinguished, prophages may respond to another stimuli (e.g. temperature or nutrient deprivation/excess) and therefore they may also be in fact active.
All of the aforementioned strains, plus P. aminophilus JCM 7686, were then tested as potential hosts for the induced phages using a spot test. None of the tested strains supported detectable lytic growth of any phage. It was concluded that all of the identified phages are species-specific, with a narrow host range that is possibly confined to their natural host strain.
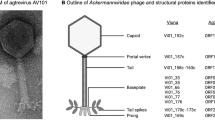
TEM analysis was then performed to visualize virions of the identified phages. This revealed that all the phages have icosahedral heads and tails of the sizes presented in Table 1. The morphological features of these phages indicated that four of them (vB_PbeS_Pben1, vB_PkoS_Pkon1, vB_PsuS_Psul1 and vB_PthS_Pthi1) belong to the Siphoviridae family, while vB_PyeM_Pyei1 represents the Myoviridae family (Fig. 1A). It is noteworthy that vB_PyeM_Pyei1 is the first representative of the Myoviridae family to be identified in Paracoccus spp.

Particle morphology and genome organization of phages vB_PbeS_Pben1, vB_PkoS_Pkon1, vB_PsuS_Psul1, vB_PthS_Pthi1 and vB_PyeM_Pyei1. (A) Transmission electron micrographs of the phage particles. A scale bar is shown below each micrograph. (B) Phage genome organization. Arrows indicate the transcriptional orientation of the genes. The distinguished genetic modules are indicated by black boxes.
Genomic analysis of identified Paracoccus phages
General features of active phage genomes
The genomes of the identified active phages were sequenced. After digestion of the Paracoccus phage DNAs with the restriction enzymes, no alteration in the banding pattern was observed after heating the DNA to 70 °C (data not shown), which indicated that the ends of their genomes did not form complementary overhangs and the phage DNAs was packaged by a headful mechanism (pac type). The headful mechanism is characteristic for circularly permuted genomes18. General characteristics and features of the Paracoccus phage genomes are summarized in Table 2.
Thorough manual sequence annotation of the phage genomes revealed modular structures that are typical for temperate bacteriophages19. The distinguished gene clusters determine functions crucial for the phage life cycle, such as integration/excision, DNA recombination, early transcriptional regulation, DNA replication, packaging, capsid and tail assembly, and lysis (Fig. 1B). Specific functions of predicted phage-encoded proteins were assigned on the basis of their similarity to known phage proteins. Features of the distinguished genes are summarized in Supplementary Table S1. Only one putative tRNA gene was identified within the genome of phage vB_PbeS_Pben1 – tRNAVal(TAC). No obvious biological function could be attributed to 62% of the predicted phage gene products, so these were assigned as hypothetical proteins.
Although the aforementioned genetic modules with predicted functions show conservation of their order within the analyzed genomes, only two regions of sequence similarity were found in the DNA sequences of phages vB_PbeS_Pben1 and vB_PsuS_Psul1. The first region contains 13 predicted genes encoding proteins (Pben1_p36-p48 and Psul1_p26-p38, respectively) sharing at least 46% amino acid (aa) identity (Supplementary Table S2). Within this region there are genes encoding terminase and phage structural proteins. The second conserved region is shorter and composed of three genes encoding a putative Cro-like protein and two hypothetical proteins (Pben1_p20-p22 and Psul1_p17-p18, respectively) (Supplementary Table S2).
Prophage attachment sites and integration modules
In the lysogenic state a prophage is usually flanked by short directly repeated sequences – attL and attR20. To identify putative attachment sites, bacterial genomic sequences (of the hosts from which the phages have been induced) adjacent to the identified prophages were screened for the presence of direct repeats. This analysis revealed that the vB_PsuS_Psul1, vB_PthS_Pthi1 and vB_PyeM_Pyei1 phages integrated at the 3′ ends of tRNA genes [tRNATrp(CCA), tRNASer(GGA) and tRNAPro(TGG), respectively], and their integration reconstituted an intact copy of the target genes. Introduction of vB_PkoS_Pkon1 into the host chromosome disrupted a gene encoding a putative OmpR transcriptional regulator, while vB_PbeS_Pben1 integrated within an intergenic region between genes encoding a putative oxidoreductase and formyl-CoA transferase (Table 2).
Site-specific recombination between attB and attP is mediated by the phage integrases, i.e. tyrosine or serine recombinases20. The predicted integrases of all analyzed phages belong to the tyrosine recombinase (XerC/XerD) family, but they share little sequence similarity.
Lysogeny control regions
The switch between the lytic and lysogenic cycles of temperate phages is dependent on the expression of divergently transcribed genes encoding functional homologues of λ regulatory proteins CI and Cro21. In all analyzed Paracoccus phages, predicted CI- and Cro-like repressors, belonging to the XRE family of transcriptional regulators (COG2932), were identified (Supplementary Table S1, Fig. 1B). Each regulatory protein contains a helix-turn-helix domain (HTH, pfam01381), but only one CI-like protein, Pyei1_p19 of vB_PyeM_Pyei1, has an S24 signal peptidase domain (pfam00717).
Replication modules
DNA segments adjacent to the lysis/lysogeny switch modules of tailed phages usually contain a cluster of genes involved in DNA replication. Genes encoding predicted replication initiation proteins could be distinguished in the genomes of four phages, i.e. vB_PbeS_Pben1 (pben1_p32), vB_PkoS_Pkon1 (pkon1_p28), vB_PthS_Pthi1 (pthi1_p19-p20) and vB_PyeM_Pyei1 (pyei1_p23) (Table S1, Fig. 1B). None of the proteins encoded by vB_PsuS_Psul1 exhibit sequence similarity to previously described phage replication proteins. However, the Psul1_p21 protein of this phage contains a predicted HTH DNA-binding domain NUMOD1 and we hypothesize that it may be involved in the virus replication process.
Toxin-antitoxin systems
Toxin-antitoxin (TA) operons are commonly found within bacterial genomes. They encode two components: a stable toxin, which recognizes a specific cellular target and evokes a bactericidal or bacteriostatic effect, and a labile antitoxin that counteracts the toxin. These loci play important roles in bacterial growth, physiology and pathogenicity. They can also stabilize mobile genetic elements (MGEs) by elimination of MGE-less cells from a bacterial population22.
TA systems were identified in two prophages – vB_PbeS_Pben1 (pben1_p24-p25) and vB_PkoS_Pkon1 (pkon1_p43-p44). In both cases the TA system genes are oriented oppositely to the surrounding genes. The pben1_p24-p25 locus encodes a HicA-type toxin (Pben1_p25; COG1724), possibly involved in mRNA cleavage23, while pkon1_p43-p44 encodes an mRNA-degrading toxin of the RelE/ParE family (Pkon1_p44; COG2026)24.
We tested the functionality of these systems in a heterologous host – P. versutus UW225. For this purpose, both TA systems were cloned into the stability test vector pABW3, yielding pABW3-TA_PBE (TA of vB_PbeS_Pben1) and pABW3-TA_PKO (TA of vB_PkoS_Pkon1). Plasmid pABW3-TA_PBE was stably maintained in strain UW225 (no plasmid-less cells were detected after 30 generations of growth under non-selective conditions), but this was not the case for pABW3-TA_PKO (9% of cells carried the plasmid following growth without selection). The “empty” vector pABW3 was present in 4% of cells after the same period of non-selective growth. These results indicate that pben1_p24-p25 comprises an active stabilizing system, while pkon1_p43-p44 seems to be non-functional, at least in this host. However, this TA system might be active in other hosts, as was previously observed for tad-ata-type systems25.
Lysis modules
Many dsDNA bacteriophages use a holin-endolysin system for host cell lysis to release progeny virions26. Endolysins are responsible for the degradation of the bacterial cell wall, causing the release of newly formed viral particles. These enzymes are synthesized without a signal sequence and thus accumulate in the cytosol during the viral life cycle26. Holins accumulate in the cell membrane and then perforate it, causing lesions that allow endolysin to access the cell wall peptidoglycan27.
A BLASTp search indicated that predicted proteins Pben1_p67, Pkon1_p73, Psul1_p45, Pthi1_p48 and Pyei1_p72 (Supplementary Table S1) are endolysins. Pkon1_p73 and Pthi1_p42 share 92% aa identity, while Pben1_p67 and Pyei1_p72 share 74.3% identity. These four proteins were classified as N-acetylmuramoyl-L-alanine amidases, i.e. enzymes that cleave the amide bond between N-acetylmuramic acid (MurNAc) and the first highly conserved stem L-alanine residue26. Psul1_p45, the predicted endolysin of phage vB_PsuS_Psul1, is a glycosidase (or muramidase), that presumably cleaves the linkage between MurNAc and N-acetylglucosamine26.
The identification of holin-encoding genes within the genomes of Paracoccus phages was challenging. Only Pkon1_p70 and Pthi1_p46 (64% aa sequence identity) share significant sequence similarities with known holins. In addition, membrane-spanning domains were detected in Psul1_p43 and Pyei1_p73 using the programs TMHMM and TMPRED. However, confirmation that these proteins are indeed holins will require further experimental study.
DNA methyltransferase genes
Lytic and lysogenic phages often encode multi- and monospecific solitary DNA methyltransferases (MTases), not associated with restriction endonucleases28. They may also have complete restriction-modification (RM) systems29.
Three of the analyzed phages (vB_PbeS_Pben1, vB_PkoS_Pkon1 and vB_PyeM_Pyei1) possess genes encoding orphan DNA MTases. Protein Pyei1_p05 exhibits similarity to several well characterized C5-methylcytosine (m5C) MTases, e.g. JCM7686_0772 and JCM7686_2655 of the prophages ФPam-1 and ФPam-5 of P. aminophilus JCM768 (~69% aa identity), respectively11. It was previously demonstrated that DNA modified by m5C MTases homologous to Pyei1_p05 is protected from cleavage by a wide variety of cytosine methylation-sensitive restriction endonucleases11. Therefore, it may be assumed that the Pyei1_p05 MTase also has a relaxed substrate specificity. The predicted MTases of the phages vB_PbeS_Pben1 and vB_PkoS_Pkon1 exhibit similarity to N6-adenine (m6A) modification enzymes. Protein Pben1_p29 is similar to DNA MTases encoded by viruses infecting Alphaproteobacteria (e.g. GenBank acc. no. YP_009146999 of Aurantimonas phage AmM-1; 50% aa identity). This group of viral MTases targets a sequence (5′-GANTC-3′) that is also recognized by Alphaproteobacteria-specific cell cycle-regulated MTase CcrM30,31. The pben1_p29 gene is located adjacent to the predicted replication module of phage vB_PbeS_Pben1. A putative m6A MTase was also identified in phage vB_PkoS_Pkon1. The pkon1_p75 gene is located at the end of the right arm of this genome (Supplementary Table S1).
Auxiliary metabolic genes
Temperate bacteriophages can contain auxiliary genes that modulate and augment host cell metabolism during infection and facilitate production of new viruses. A presumed auxiliary metabolic gene was found only within the genome of vB_PyeM_Pyei1. The pyei1_p13 gene encodes a homolog of a tellurite-resistance protein TerB. TerB is encoded within a tellurite resistance operon (terZABCDEF) found in e.g. E. coli APEC O1 plasmid pAPEC-O1-R32. Homologous (56.6% aa identity with the Pyei1_p13 protein), TerB protein is encoded by Sinorhizobium phage ФM5 (GenBank acc. no. ARV77549).
Identification and characterization of (pro)phages occurring in Paracoccus spp. genomes
Paracoccus spp. genomes (nine complete and 54 drafts) and 52 complete plasmid sequences (present in NCBI databases on June 25th, 2018) were inspected for the presence of prophage regions using the PhiSpy tool33. Obtained results were afterwards manually curated. Only the regions comprising complete prophage genomes were included in further analyses. This was determined based on the presence of phage integration, replication, packaging, structural and lysis modules. Additionally, boarders of prophages were indicated based on the presence of predicted attB and attP sequences or, when not distinguishable, differences in %GC content between the prophage region and the surrounding host genome. As a result, 53 novel prophages were identified (Supplementary Dataset 1). These were detected in 29 Paracoccus strains; thus nearly half of the tested bacteria were lysogens. All of the identified prophages were classified within the order Caudovirales using VIRFAM34. Fifty were classified as members of the Siphoviridae, eight as Podoviridae (the first Podoviridae-like phages specific to Paracoccus) and only one as Myoviridae (Table 3). Interestingly, among the 29 lysogenic strains, 14 are polylysogens, carrying multiple prophages within their genomes: 6 strains have two prophages, 3 have three prophages, 3 have four prophages, and single strains have five and six prophages, respectively (Table 3).
The integration modules of the identified prophages encode tyrosine recombinases (38 prophages), serine recombinases (16) or Mu-like transposases (5) (Supplementary Table S3). Putative integration sites were identified for the majority of the tyrosine recombinase-encoding viruses (Supplementary Table S3). For 31 prophages these sites were various tRNA genes of which the most commonly targeted were (i) tRNAMet(CAT) used by 10 prophages and (ii) tRNAPro(TGG) by four prophages, including active phage vB_PyeM_Pye1. These observations corroborate previous findings regarding the preferential integration of phages (and other integrative elements) within tRNA genes35.
With regard to phage structural proteins, the presence of the coding sequence for a nearly 700-amino acid-long protein in 17 (34%) of the Siphoviridae prophages (indicated as fused in Supplementary Table S4) is noteworthy. The best BLASTp hits for these proteins are annotated as peptidases of the U35 or U37 families, but characteristic domains for these could not be identified. Instead, a caseinolytic protease domain (ClpP; peptidase S14) cd07016 was always present in the N-terminal region. In addition, a Mu-like prophage major head subunit gpT domain (pfam10124) was identified in the C-terminal region of all these proteins. These observations strongly suggests that the proteins encoded by Paracoccus (pro)phages evolved via fusion of genes encoding the protease and major capsid protein. This is also in accordance with previous reports, e.g. regarding Lactococcus phage c2 structural proteins36. Such protein products were also predicted in the genomes of two of the active phages identified in this study: vB_PbeS_Pben1 (pben1_p41) and vB_PsuS_Psul1 (psul1_p30).
Paracoccus prophages encode endolysins which were classified as N-acetylmuramyol-L-alanine amidases (16 prophages), muramidases (18), peptidases M15 (15) and chitinases (4) (Supplementary Table S4). Interestingly, three prophages (vB_PsaS_PD29, vB_PsaS_PD48, and vB_PspS_PD38) also encode tail-associated peptidoglycan-degrading enzymes, that presumably facilitate phage DNA injection into the host cell in the initial stages of infection. Only in case of vB_PpaP_PD14 any protein resembled similarity to known endolysins was found.
It was also revealed that Paracoccus prophages encode an extensive repertoire of DNA modification proteins. In the genomes of 48 (out of 59) prophages, at least one DNA MTase gene was identified (Supplementary Table S4). Of the 88 predicted DNA MTases, 58 were classified as m6A or m4C DNA MTases and 30 as m5C DNA MTases. Although homologues of these MTases are abundant in sequence databases, their activity has yet to be confirmed by experimental data. Only in the case of Pd42_p05, encoded by vB_PspS_PD42, its specificity can be presumed (i.e. YGGCCR), based on its similarity (78% aa identity) to Pami1_p55 of vB_PamS_Pami111.
The most numerous subgroup of DNA m6A/m4C MTases (36 examples) is comprised of enzymes containing the ParB domain within their N-terminal region (Fig. 2A). These genes were all found upstream of genes encoding the terminase subunits (PAC module) (Fig. 2B). The presence of MTase genes (and other DNA modifying genes) within a specified region of the phage genome (named the ParB-Tls locus), sandwiched between genes encoding a ParB-like protein and terminase large subunit has been reported previously37. It was suggested that phage-encoded ParB-like proteins may be involved in directing the DNA-modification apparatus to specific sites within the virus genome during packaging37. It was also shown that ParB proteins may be fused with the MTases37, as was observed in the case of the Paracoccus phages. Interestingly, in the predicted ParB-Tls loci of the Paracoccus phages, an additional 21 genes encoding m5C DNA MTases (lacking ParB domains) were found. Far fewer MTase genes were found at other locations, including downstream (12 genes) and upstream (2) of the integrase gene, or downstream of the lysis module (7) (Fig. 2B). Interestingly, MTase genes were located in the proximity of the replication modules in only four phages (including three related prophages of polylysogenic P. aminophilus), whereas such genomic localization of these genes was common in previously analyzed Sinorhizobium (also representatives of Alphaproteobacteria) prophages11,31.

Diversity of DNA MTases of Paracoccus (pro)phages and genomic location of their genes. (A) General diversity of identified MTases as a network of MTases (nodes) connected with lines (edges) that reflect at least 80% amino acid sequence identity over at least 75% sequence coverage. The colours of the nodes representing single MTases, reflect the target base of their methylation, except the half blue-half green nodes, which additionally indicates the presence of a ParB domain at the N-terminus. Several nodes are also marked with stars to indicate experimental verification of their specificity. The labels/numbers give the names of the phage from which each MTase originates (e.g. Pami4 from vB_PamS_Pami4, 11 from vb_PdeS_PD11). This corresponds to data presented in Table 3. Where more than one MTase gene is present within a (pro)phage genome, the suffixes “a”, “b” or “c” are added to the label/number, corresponding to their order in that genome. (B) Simplified schematic representation of phage genomes showing virus-specific gene modules and the location of the MTase genes. MT blocks are coloured according to MTase target base specificity. The prophages that share each genome arrangement are listed on the right side of the genome diagrams.
Bacteriophages can also encode RM systems that may restrict the entry of other phage or plasmid DNA during lysogeny or reprogram gene expression38. In the Paracoccus prophage genomes, 13 RM systems (four type I, six type II and three type III) were identified (Supplementary Table S4).
As mentioned above, phages can carry auxiliary metabolic genes that may benefit their hosts. Interestingly, genes encoding proteins that potentially confer metal resistance were found in 10 phages. These are: (i, ii) tellurium resistance proteins TerB (phage vB_PamS_Pami1) and TerC (vB_PspS_PD44), (iii, iv) arsenite resistance protein ArsB (vB_PcoS_PD6 and vB_PsaS_PD20), (v, vi) zinc/cadmium/lead-transporting ATPase ZntA (vB_PhoS_PD13 and vB_PspS_PD34), (vii) multidrug efflux system AcrABCR (vB_PsaS_PD23), (viii) lead/cadmium/zinc/mercury transporter, copper transporting ATPase and a multi-copper oxidase (vB_PspS_PD33), (ix) cobalt transporter CorA (vB_PyeS_PD47) and (x) zinc transporter ZitB (vB_PyeS_PD49) (Supplementary Table S4). Heavy metal-rich regions are ubiquitous all over the planet and this is a consequence of natural processes (e.g. bioweathering of metal-containing minerals) and anthropogenic activities (e.g. burning of fossil fuels)39. Paracoccus spp. are frequently found in metal-rich environments (including metal mines and contaminated soils) and therefore acquisition of metal resistance genes may be beneficial for these bacteria40,41,42. Acquired (with a phage) resistance genes may modify bacterial host reaction to toxic elements and therefore enhance its overall fitness under detrimental, environmental conditions and, in a consequence, facilitates production of the virus progeny.
It was also shown that, in addition to two previously described phages, eight of Paracoccus prophages carried TA modules. The type of TA modules varied: two were RelBE-like, two HicAB-like, two VapBC-like, one ParDE-like and one HigBA-like (Supplementary Table S4).
Comparative genomics of Paracoccus (pro)phages
In this study, 5 novel active lysogenic Paracoccus phages and 53 prophages were identified. These, together with six previously characterized prophages of P. aminophilus JCM 7686 (of which vB_PamS_Pami6 was shown to be an active lysogenic virus) and two lytic phages (vB_PmaS_IMPE1 and vB_PmaS_Shpa), constitute the current virome of the genus Paraccocus, which consists of 66 (pro)phages in total. This provided the opportunity for comprehensive genomic studies to reveal the common and unique features of these (pro)phages.
A first step in the comparison of Paracoccus (pro)phages was whole genome all-against-all BLASTn searches and their visualization with Circoletto (Fig. 3). This analysis revealed that at the nucleotide level (with a threshold e-value of <1e-100), 5 (pro)phages (lytic phages vB_PmaS_IMEP1 and vB_PmaS_Shpa (Shpa), and prophages vB_PsaS_PD22, vB_PspP_PD41 and vB_PyeS_PD51) were unique, 9 other prophages were nearly (if not) identical to at least one other, while the rest showed only local (limited to mostly short genomic regions) identities (of at least 73%).

Comparison of Paracoccus (pro)phage genomes. Whole-genome similarity analysis was performed using Circoletto with e-100 as the threshold. The ribbon colours reflect the percentage identity of particular genomic regions. The bars within the first ring represent subsequent phages. The next ring, comprised of histograms, shows the frequency of hits in certain regions of the analyzed genomes. The outer-most ring reflects the (pro)phage classification: orange – Siphoviridae, green – Myoviridae and violet – Podoviridae. Outer-most, gray curves indicate polylysogenic host strains: 1 – P. aminophilus JCM 7686, 2 – P. aminovorans HPD-2, 3 – P. contaminans RKI, 4 - P. denitrificans PD1222, 5 – P. sanguinis 39524, 6 – P. sanguinis 4681, 7 – P. sanguinis 5503, 8 – P. sanguinis DSM 29303, 9 – Paracoccus sp. BM15, 10 – Paracoccus sp. CBA4604, 11 – Paracoccus sp. S4493, 12 – Paracoccus sp. SCN 68–21, 13 – P. yeei ATCC BAA-599, 14 – P. yeei TT13.
The first group of identical prophages consists of members of the Podoviridae, vB_PdeP_PD8, vB_PdeP_PD9 and vB_PdeP_PD12, sharing 99–100% sequence identity, all of which were identified in closely related P. denitrificans strains. Another group is composed of two prophages, vB_PcoS_PD6 and vB_PsaS_PD20, sharing 99% sequence identity. Interestingly, both of these prophages carry an auxiliary gene encoding the arsenite efflux pump ArsB integrated in the opposite orientation to the surrounding structural genes. It is noteworthy that these prophages are present in strains isolated on different continents, from different environments and 10 years apart. Another two pairs of identical prophages are the Mu-like viruses vB_PsaS_PD18 and vB_PsaS_PD27, and vB_PsaS_PD19 and vB_PsaS_PD28, respectively. These were identified within the genomes of human blood-borne P. sanguinis strains 39524 and DSM 29303.
Comparison of the Paracoccus (pro)phages was continued by constructing protein-based similarity networks (Fig. 4A). In total, the 66 analyzed (pro)phages encode 3,891 putative proteins, of which 2,062 are similar to at least one other protein. Comparative analysis of whole proteomes showed that the Paracoccus (pro)phages can be grouped into three major clusters and three orphan nodes (representing: vB_PmaS_IMEP1, vB_PsaS_PD22 and vB_PyeS_PD53). The densest (i.e. the most similar to one another) cluster is composed of a set of viruses of the Siphoviridae, while the most numerous cluster (31 elements) is more relaxed, reflecting a lower number of reciprocally similar proteins of phages that comprise this group (Fig. 4A). It is important to mention that these clusters are linked via a common protein – a truncated IS3 family transposase encoded by vB_PsaS_PD25 and vB_PamS_Pami4 (Fig. 4A). The relaxed cluster contains Podoviridae (pro)phages on the peripheries, while the core is built by representatives of Siphoviridae and Myoviridae. Interestingly, vB_PkoS_Pkon1 constitutes an internal linker within this cluster because it encodes proteins (e.g. endolysin Pkon1_p73 exhibiting similarity to appropriate proteins of vB_PdeS_PD10, vB_PdeS_PD11 and vB_PthS_Pthi1) whose homologues are present in proteomes of Paracoccus phages classified to the Siphoviridae and Podoviridae. The third cluster of similar phages consists of all five Mu-like Siphoviridae viruses identified within the opportunistic human pathogens P. sanguinis 39542 and DSM 29303.

Protein-based similarity network of Paracoccus (pro)phages. The general clustering of (pro)phages based on their summarized proteomes (A), integrases (B), large terminase subunits (C) and major capsid proteins (D). Nodes represent a single (pro)phage, while edges correspond to the summarized quantity of reciprocally similar proteins. Orphan nodes are made transparent for better visibility. On (A) the size of the node corresponds to the number of prophages with which they share proteins. The nodes represent the following (pro)phages (Paracoccus strain): Pben1 – vB_PbeS_Pben1 (P. bengalensis); Pkon1 – vB_PkoS_Pkon1 (P. kondratieve); Psul1 – vB_PsuS_Psul1 (P. sulfuroxidans); Pthi1 – vB_PthS_Pthi1 (P. thiocyanatus); Pyei1 – vB_PyeM_Pyei1 (P. yeei CCUG 32053); IMEP1 – vB_PmaS_IMEP1 (P. marcusii); Shpa – vB_PmaS_Shpa (P. marinus); Pami1-Pami6 – vB_PamS_Pami1-vB_PamS_Pami6 (P. aminophilus); 1 – vB_PamS_PD1 (P. aminovorans DSM 8537); 2–3 – vB_PamP_PD2-vB_PamS_PD3 (P. aminovorans HPD-2); 4–7 – vB_PcoS_PD4-vB_PcoS_PD7 (P. contaminans); 8 – vB_PdeP_PD8 (P. denitrificans DSM 413); 9 – vB_PdeP_PD9 (P. denitrificans DSM 415); 10 – vB_PdeS_PD10 (P. denitrificans ISTOD1); 11–12 – vB_PdeS_PD11-vB_PdeP_PD12 (P. denitrificans PD1222); 13 – vB_PhoS_PD13 (P. homiensis); 14 – vB_PpaP_PD14 (P. pantotrophus DSM 1403); 15 – vB_PpaP_PD15 (P. pantotrophus J46); 16 – vB_PsaS_PD16 (P. saliphilus); 17 – vB_PsaS_PD17 (P. sanguinis 10990); 18–19 – vB_PsaS_PD18-vB_PsaS_PD19 (P. sanguinis 39524); 20–21 – vB_PsaS_PD20-vB_PsaS_PD21 (P. sanguinis 4681); 22–26 – vB_PsaS_PD22-vB_PsaS_PD26 (P. sanguinis 5503); 27–29 – vB_PsaS_PD27-vB_PsaS_PD29 (P. sanguinis DSM 29303); 30 – vB_PseS_PD30 (P. sediminis); 31 – vB_PsoS_PD31 (P. solventivorans); 32–33 – vB_PspS_PD32-vB_PspS_PD33 (Paracoccus sp. BM15); 34–36 – vB_PspM_PD34-vB_PspS_PD36 (Paracoccus sp. CBA4604); 37 – vB_PspS_PD37 (Paracoccus sp. J39); 38 – vB_PspS_PD38 (Paracoccus sp. N5); 39–40 – vB_PspS_PD39-vB_PspS_PD40 (Paracoccus sp. S4493); 41–44 – vB_PspP_PD41-vB_PspS_PD44 (Paracoccus sp. SCN 68–21); 45 – vB_PspS_PD45 (P. sphaerophysae); 46 – vB_PveS_PD46 (P. versutus); 47–49 – vB_PyeS_PD47-vB_PyeS_PD49 (P. yeei ATCC BAA-599); 50–53 – vB_PyeM_PD50-vB_PyeS_PD53 (P. yeei TT13).
Several polylysogenic host strains were identified in this study and we checked the reciprocal similarity of their phage proteomes. Within P. aminophilus JCM 7686 and Paracoccus sp. BM15, all identified prophages (six and two, respectively) show similarity of their encoded proteins; they share between five and 30 highly similar proteins. It is also worth mentioning that P. aminophilus JCM 7686 contains the highest number of prophages11. Some prophages of the other polylysogenic host strains also exhibit similarities, including (i) three (out of four) prophages of P. contaminans RKI (vB_PcoS_PD5-PD7) that share between 14 and 29 common proteins, (ii) two (out of three) prophages of Paracoccus sp. CBA4604 (vB_PspS_PD34 and vB_PspS_PD35), sharing 19 proteins, (iii) two (out of three) prophages of P. yeei ATCC BAA-599 (vB_PyeS_PD47 and vB_PyeS_PD49), sharing 11 proteins and (iv) two (out of four) prophages of P. yeei TT13 (vB_PyeS_PD52 and vB_PyeS_PD53), sharing 16 proteins. In contrast, of the five prophages of P. sanguinis 5503, only vB_PsaS_PD23 shares a single protein with vB_PsaS_PD24 and vB_PsaS_PD25. Similarly, amongst four prophages of Paracoccus sp. SCN 68-21 only vB_PspS_42 and vB_PspS_43 encode a single similar protein.
Detailed analysis of the (pro)phage integration module sequences showed that they group into 10 clusters and 30 unique nodes (Fig. 4B). The largest cluster is composed of 11 (out of 15) serine recombinases for which integration sites were not identified. The four remaining serine recombinases (of vB_PamS_Pami6, vB_PamS_PD1, vB_PsoS_PD31 and vB_PsaS_PD35) form single nodes. It is worth emphasizing that the specific integration site was predicted (tRNAMet gene11) for only one phage encoding serine recombinase – the active lysogenic phage vB_PamS_Pami6 (Supplementary Fig. S1). This analysis revealed that 10 other (pro)phages also integrated within the tRNAMet gene, but these viruses all encode tyrosine recombinases (Supplementary Fig. S1). Another group is composed of Podoviridae (pro)phages (i.e. vB_PdeP_PD8, vB_PdeP_PD9, vB_PdeP_DP12, vB_PamP_PD2, vB_PspP_PD41), integrated into tRNAThr genes with various anticodons (Supplementary Fig. S1). Interestingly, one podovirus, vB_PpaP_PD15, encodes a tyrosine recombinase highly similar (92.7%) to that of siphovirus vB_PkoS_Pkon1 and both have integrated into OmpR family transcriptional regulator genes. Another two clusters group phages encoding Mu-like transposases (Supplementary Fig. S1).
The other two networks of large terminase subunits and major capsid protein sequences show a higher level of conservation among these proteins than in the case of recombinases (Fig. 4C,D). In both cases, 12 clusters grouping between two and 15 (pro)phages were found. Moreover, there is significant congruency between these networks, which is especially visible amongst Mu-like prophages, myoviruses and podoviruses (Fig. 4C,D). This finding, together with our previous observations for Sinorhizobium (pro)phages, indicates that terminase large subunits and major capsid proteins as markers representing congruent clustering are the most convenient tools for phylogenetic analyses of alphaproteobacterial viruses31.
Diversity of Paracoccus (pro)phages in comparison to the general diversity of bacteriophages
Paracoccus (pro)phages were subjected to comparative analyses with all bacteriophage genomes deposited in the NCBI Viruses database by constructing a complex protein similarity network composed of 6,126 nodes and 330,592 edges (Fig. 5). Interestingly, Paracoccus phages created separate clusters (Fig. 5).

Protein-based similarity network analysis of Paracoccus (pro)phages and other bacteriophages retrieved from the NCBI Viruses database. (A) Overall similarity network of known bacterial phages. Nodes are coloured based on the taxonomy of the phage host (at phylum level, except Proteobacteria where classes are considered). Paracoccus (pro)phages are distinguished within the network. The host taxonomy is based on manually-curated qualifiers in the source section and organism name of the virus GenBank files. (B) Magnified image of Alphaproteobacteria (pro)phage network. The colour scheme is based on the host genus classification of the phages. The topology of the clustering of Paracoccus phages is the same as the one presented on Fig. 4, where (pro)phages were coloured based on their classification to Sipho-, Myo- and Podoviridae families.
We focused on detailed analysis of the relationship between phages infecting bacteria of the Alphaproteobacteria class. In the analyzed network, 156 Alphaproteobacteria phages, infecting hosts from 38 different genera were included. Our analysis revealed that these viruses are highly diverse and do not show similarity to other phages (Fig. 5A). They created 24 clusters (between two and 35 nodes) and 40 orphan nodes. The two most numerous clusters are composed of 35 Brucella phages and 13 Caulobacter phages. Interestingly, there are only five multi-host clusters grouping phages that infect Alphaproteobacteria representing various genera. The largest of such clusters groups nine related phages infecting Reugeria (five phages), Dinoroseobacter (two) and Sulfitobacter (two) (Fig. 5B).
The network analysis (Fig. 5) showed the distinction of Alphaproteobacteria phages from other bacteriophages and hence, searching for potential links between these phages and other viruses, we have used the IMG/VR database resources43. Amongst over 700,000 viral contigs present within the IMG/VR database, less than 1% (4,915) encoded at least a single protein similar to those of Alphaproteobacteria phages. From these viral contigs only 212 with completeness parameter over 75% were overlaid onto the global network (Supplementary Fig. S2). As a result, it was shown, that vB_PkoS_Pkon1 has been connected with Gammaproteobacteria phages (via pkon1_p50, hypothetical protein with the P63C domain), while vB_PmaS_Shpa, vB_PsaS_PD19, vB_PsaS_PD26 and vB_PsaS_PD28 have been linked with other Alphaproteobacteria phages (via single-stranded DNA-binding protein and large terminase subunit protein). Interestingly, newly added contigs retrieved from the IMG/VR database extended many other clusters and linked Alphaprotoebacteria phages with viruses infecting Terrabacteria and Gammaproteobacteria (Supplementary Fig. S2). However, despite several new links/connections, this extended network analysis still indicates that Alphaproteobacteria (and particularly Paracoccus) phages create separate groups.
It is important to mention that since the number of alphaproteobacterial virus genomes currently available for comparison is low (especially compared to the number of other phage genomes in the NCBI database), all performed analyses still have some limitations. Therefore, network analyses should be repeated once the database has been enriched in the future.
Conclusion
In this study, five novel active temperate phages and 53 prophages of Paracoccus spp. (Alphaproteobacteria) were identified and analyzed together with six previously identified prophages of P. aminophilus JCM 7686 and two lytic phages (vB_PmaS_IMEP1 and vB_PmaS_Shpa). Four of the newly discovered active phages represent the Siphoviridae family, while vB_PyeM_Pyei1 is the first active Myoviridae phage infecting Paracoccus spp. Moreover, amongst the identified prophages, the first Podoviridae viruses infecting Paracoccus spp. were distinguished. Several auxiliary metabolic genes were found within the genomes of the identified Paracoccus (pro)phages. These genes encode proteins that potentially confer metal resistance. This may be highly beneficial to bacterial hosts, as many Paracoccus spp. have been isolated from various contaminated environments. Amongst other genes found within analysed (pro)phages, these encoding DNA methyltransferases are very common. It was shown that, 58 of identified methylases were classified as m6A/m4C DNA MTases and 30 as m5C DNA MTases. In similarity network analysis, these MTases formed highly conserved clusters, possibly grouping enzymes with common specificities. Interestingly, 57 genes encoding MTases were localized in a common region, i.e. the ParB-Tls locus. This location was also shared by a large group of genes encoding MTases fused with ParB-like proteins, that may be involved in directing the DNA-modification apparatus during packaging. Finally, it was shown that Paracoccus (pro)phages form a separate group of viruses, that is not only distinct from other phages of Alphaproteobacteria, but also from all other bacterial viruses.
Methods
Bacterial strains, plasmids and culture conditions
The following strains were used in this study: E. coli DH5α44, P. alcaliphilus JCM 736445, P. aminophilus JCM 76863, P. aminovorans JCM 76853, P. alkenifer DSM 115932, P. bengalensis DSM 170994, P. ferroxidans NCCB 130006646, P. haeundaensis LGM P-219035, P. halophilus JCM 14014T6, P. homiensis DSM 1786247, P. kondratievae NCIMB 13773T48, P. pantotrophus DSM 1107249, P. seriniphilus DSM 148277, P. solventivorans DSM 115922, P. sulfuroxidans JCM 1401350, P. thiocyanatus JCM 2075651, P. versutus UW1R52 and UW225 (Rifr-derivative of a wild-type strain)53, and P. yeei CCUG 3205354. All strains were grown in lysogeny broth (LB) medium at 37 °C (E. coli) and 30 °C (Paracoccus spp.). Liquid cultures were incubated with shaking. When required, media were supplemented with kanamycin (50 μg ml−1) and rifampin (50 μg ml−1). Plasmid pABW3 was used for the stability testing52.
Standard molecular biology procedures
Standard DNA manipulation methods were performed as described by Sambrook and Russell (2001)55. Transformation of E. coli strains and triparental mating of P. versutus were performed according to previously described methods52,56. The test for the presence of cohesive ends of the phage genome was performed as previously described57, using various restriction enzymes (Thermo Fisher Scientific, Waltham, MA, USA).
Cloning of the toxin-antitoxin systems
Toxin-antitoxin (TA) systems of vB_PbeS_Pben1 and vB_PkoS_Pkon1 were PCR amplified using Phusion High-Fidelity DNA Polymerase (Thermo Fisher Scientific) with appropriate primer pairs, i.e. TABamHf 5′-GTTGTTGGATCCATGATCTCGGCATCAGCAG-3′ and TAEcoRr 5′-GGTGGTGAATTCAACACATTGCAGCAATGCTC-3′ for the TA module of vB_PbeS_Pben1, and PKTABamHI 5′-TGCAGGATCCAATACCGCATCCGTTCG-3′ and PKTAEcoRI 5′-AGCTGAATTCCATGGCCGCCTCAATCC-3′ for the TA module of vB_PkoS_Pkon1 (introduced restriction sites are underlined). The following PCR program was applied using a Mastercycler (Eppendorf, Hamburg, Germany) to amplify the desired products: initial denaturation at 95 °C for 3 min followed by 35 cycles of denaturation at 98 °C for 20 s, annealing at 64 °C for 1 min, extension at 72 °C for 1 min/kb and then a final extension at 72 °C for 1 min/kb. The obtained PCR amplicons were analyzed by agarose gel electrophoresis and purified using a Gel Out kit (A&A Biotechnology, Gdynia, Poland). The DNA fragments were then digested with EcoRI and BamHI and cloned in the vector pABW3 cleaved with the same restriction endonucleases. The resulting plasmid constructs were named pABW3-TA_PBE and pABW3-TA_PKO, respectively.
Mitomycin induction of prophages and purification of phage particles for DNA preparation and transmission electron microscopy
Paracoccus cultures were grown to an OD600 of 0.4, then mitomycin C (Sigma-Aldrich, St. Louis, MO, United States) was added to 500 µg ml−1 and incubation was continued for 6 h. The cells were then pelleted by centrifugation and phage particles in the supernatant precipitated using PEG/NaCl55. Bacteriophage particles were collected by centrifugation and resuspended in SM buffer55. Phage DNA was isolated by phenol-chloroform extraction and isopropanol precipitation55, and analyzed by 0.7% agarose gel electrophoresis. For transmission electron microscopy (TEM) analysis, phage particles were purified on a 1 ml Convective Interaction Media (CIM®) anion-exchange monolith column (BIA Separations, Ajdovscina, Slovenia) using an ÄKTApurifier system (GE Healthcare, Little Chalfont, UK) running UNICORN™ software, according to a recently published protocol58. Briefly, phages from the initial purification were loaded on the column at a flow rate of 2 ml/min. Impurities were then washed out by increasing the proportion of elution buffer (20 mM Tris-HCl, pH 7.5; 2 M NaCl) in the mobile phase from 0 to 10%, at a flow rate of 4 ml/min. Elution of the phage particles was achieved by increasing the proportion of elution buffer to 35%.
Transmission electron microscopy (TEM)
For TEM analysis, 10 μl samples of purified phage were adsorbed onto carbon-coated grids (Sigma-Aldrich) for 3 min, stained with 1.5% uranyl acetate (Sigma-Aldrich) and examined using a Tecnai Spirit BioTWIN transmission electron microscope (FEI Company, Hillsboro, OR, USA). Images were collected using iTEM software (FEI Company). The visualization of phages was performed at the Laboratory of Electron Microscopy, Faculty of Biology, University of Gdansk, Gdansk, Poland.
Determination of phage host range by spot testing
To determine bacterial susceptibility to phage-mediated lysis, 17 Paracoccus strains (P. alcaliphilus JCM 7364, P. aminophilus JCM 7686, P. aminovorans JCM 7685, P. alkenifer DSM 11593, P. bengalensis DSM 17099, P. ferroxidans NCCB 1300066, P. haeudaensis LGM P-21903, P. halophilus JCM 14014T, P. homiensis DSM 17862, P. kondratievae NCIMB 13773T, P. pantotrophus DSM 11072, P. seriniphilus DSM 14827, P. solventivorans DSM 11592, P. sulfuroxidans JCM 14013, P. thiocyanatus JCM 20756, P. versutus UW1R and P. yeei CCUG 32053) were grown in liquid LB medium and plated onto LB agar plates. A drop of each phage suspension was spotted onto the bacterial lawns and the plates were incubated at 30 °C. The plates were examined for evidence of bacterial lysis after 72 h.
Plasmid stability testing
The stability of plasmids in P. versutus cells was tested as described previously25. Briefly, P. versutus UW225 containing the introduced plasmids (pABW3-TA_PKO, pABW3-TA_PBE or pABW3 as a control) were grown overnight at 30 °C in LB medium supplemented with kanamycin. Stationary phase cultures were then diluted in fresh medium without added antibiotic and cultivated for approximately 10, 20 or 30 generations. Samples were diluted and plated onto solid medium lacking kanamycin. One hundred colonies from each plate were then tested for the presence of the selection marker by replica plating. The percentage of kanamycin resistant colonies was used as a measure of the retention of the different plasmids. All plasmid stability assays were performed in triplicate.
DNA sequencing and phage genome assembly
The complete nucleotide sequences of Paracoccus phages were determined in the DNA Sequencing and Oligonucleotide Synthesis Laboratory (oligo.pl) at the Institute of Biochemistry and Biophysics, Polish Academy of Sciences. The phage genomes were sequenced using an Illumina MiSeq instrument in paired-end mode with a v3 chemistry kit. The obtained sequence reads were filtered for quality with cutAdapt v1.15 trimming bases on 3′ ends (with quality lower than Q20) and removing reads containing Ns or shorter than 50 bp59. Processed reads were afterwards assembled using Newbler v3.0 software (Roche, Basel, Switzerland) with default settings. Final gap closure was performed by capillary sequencing of PCR amplicons using an ABI3730xl DNA Analyser (Applied Biosystems, Waltham, MA, USA).
Prophage detection and classification
On June 25th 2018, 9 complete and 55 draft Paracoccus species genomes were retrieved from the National Center for Biotechnology Information (NCBI) genome browser. The sequences of these genomes together with all complete plasmids of Paracoccus spp., were screened for the presence of prophages using PhiSpy33 and the results were verified by manual inspection. Assessment of the prophage genome completeness was based on the presence of modules responsible for phage integration, lysis/lysogeny switch, DNA packaging, head-tail assembly and lysis.
Taxonomy assignment of all prophages was conducted using the VIRFAM service, which also allowed more precise identification of certain structural proteins34.
Genome annotation
The identified prophage sequences, as well as those of the mitomycin C-induced Paracoccus phages, were manually annotated using Clone Manager (Sci-Ed8) and Artemis software60. Annotation was based on homology searches performed using BLAST programs, including domain searches with CD-Search61. Putative tRNA genes were identified with the tRNAScan-SE 2.0 and ARAGORN programs62,63. Methyltransferase classification was performed using the REBASE database64 and manual inspection. For the identification of transmembrane proteins, i.e. holins, TMHMM65 and TMPRED (https://embnet.vital-it.ch/software/TMPRED_form.html) were used. The annotation of the identified heavy metal resistance genes was assisted by searches against the BacMet and PRIAM databases66,67.
Comparative genomics
Phage genome comparisons were performed with the Circoletto tool, using an e-value of 1e-100 as the threshold68. The construction of similarity networks was based on all-against-all BLASTp comparisons of three sets of proteomes: (i) those derived from 66 Paracoccus (pro)phages, (ii) those of the Paracoccus (pro)phages combined with all 6,253 viruses infecting Bacteria available in the NCBI genome browser (as of August 3rd 2018), and (iii) the previous two datasets extended with the part of the IMG/VR database version 3 (as of July 1st 2018)43. From the set of bacteriophages deposited in the NCBI database, 191 not encoding any proteins based on their annotations were excluded from the analyses. For the construction of the network only the IMG/VR viral contigs encoding at least a single protein similar to proteins encoded by known Alphaproteobacteria phages were used. From 4,915 resulting viral contigs (out of 760,453), these duplicating the nodes and with parameter of the genome completeness below 75% were excluded from further analysis. The following thresholds were used during the BLASTp searches: e-value 1e-10 (to avoid losing small, i.e. <100 aa, proteins from the analysis), query coverage of HSP of at least 75% and sequence identity of 80%. Within the obtained networks, each node represents a single (pro)phage (or IMG/VR-retrieved viral contig) and each edge corresponds to a common reciprocated similarity of at least one protein encoded by two connected (pro)phages or viral contigs. The thickness of the edge reflects the number of common proteins between two analyzed (pro)phages. These networks were created using self-written Python scripts and visualized in Gephi69 using ForceAtlas 2 layout70.
Nucleotide sequence accession numbers
The nucleotide sequences of the vB_PbeS_Pben1, vB_PkoS_Pkon1, vB_PsuS_Psul1, vB_PthS_Pthi1 and vBPyeM_Pyei1 phages have been deposited in the GenBank (NCBI) database with the accession numbers MK291441, MK291442, MK291443, MK291444 and MK291445, respectively.
References
Baker, S. C. et al. Molecular genetics of the genus Paracoccus: metabolically versatile bacteria with bioenergetic flexibility. Microbiol. Mol. Biol. Rev. 62, 1046–1078 (1998).
Lipski, A., Reichert, K., Reuter, B., Sproer, C. & Altendorf, K. Identification of bacterial isolates from biofilters as Paracoccus alkenifer sp. nov. and Paracoccus solventivorans with emended description of Paracoccus solventivorans. Int. J. Syst. Bacteriol. 48, 529–536 (1998).
Urakami, T., Araki, H., Oyanagi, H., Suzuki, K. & Komagata, K. Paracoccus aminophilus sp. nov. and Paracoccus aminovorans sp. nov., which utilize N,N-dimethylformamide. Int. J. Syst. Bacteriol. 40, 287–291 (1990).
Ghosh, W., Mandal, S. & Roy, P. Paracoccus bengalensis sp. nov., a novel sulfur-oxidizing chemolithoautotroph from the rhizospheric soil of an Indian tropical leguminous plant. Syst. Appl. Microbiol. 29, 396–403 (2006).
Lee, J. H., Kim, Y. S., Choi, T. J., Lee, W. J. & Kim, Y. T. Paracoccus haeundaensis sp. nov., a Gram-negative, halophilic, astaxanthin-producing bacterium. Int. J. Syst. Evol. Microbiol. 54, 1699–1702 (2004).
Liu, Z.-P. et al. Paracoccus halophilus sp. nov., isolated from marine sediment of the South China Sea, China, and emended description of genus Paracoccus Davis 1969. Int. J. Syst. Evol. Microbiol. 58, 257–261 (2008).
Pukall, R. et al. Paracoccus seriniphilus sp. nov., an L-serine-dehydratase-producing coccus isolated from the marine bryozoan Bugula plumosa. Int. J. Syst. Evol. Microbiol. 53, 443–447 (2003).
Lasek, R. et al. Genome structure of the opportunistic pathogen Paracoccus yeei (Alphaproteobacteria) and identification of putative virulence factors. Front. Microbiol. 9, 2553 (2018).
Uemoto, H. & Saiki, H. Nitrogen removal reactor using packed gel envelopes containing Nitrosomonas europaea and Paracoccus denitrificans. Biotechnol. Bioeng. 67, 80–86 (2000).
Zhang, J. et al. Biodegradation of chloroacetamide herbicides by Paracoccus sp. FLY-8 in vitro. J. Agric. Food Chem. 59, 4614–4621 (2011).
Dziewit, L. et al. Architecture and functions of a multipartite genome of the methylotrophic bacterium Paracoccus aminophilus JCM 7686, containing primary and secondary chromids. BMC Genomics 15, 124 (2014).
Czarnecki, J. et al. Lifestyle-determining extrachromosomal replicon pAMV1 and its contribution to the carbon metabolism of the methylotrophic bacterium Paracoccus aminovorans JCM 7685. Environ. Microbiol. 19, 4536–4550 (2017).
Aurass, P. et al. Genome sequence of Paracoccus contaminans LMG 29738T, isolated from a water microcosm. Genome Announc. 5, e00487–17 (2017).
Lim, J. Y. et al. Complete genome sequence of Paracoccus yeei TT13, isolated from human skin. Genome Announc. 6, e01514–17 (2018).
Wu, Z.-G. et al. Paracoccus zhejiangensis sp. nov., isolated from activated sludge in wastewater-treatment system. Antonie Van Leeuwenhoek 104, 123–128 (2013).
Xu, Y., Zhang, R. & Jiao, N. Complete genome sequence of Paracoccus marcusii phage vB_PmaS-R3 isolated from the South China Sea. Stand. Genomic Sci. 10, 94 (2015).
van Zyl, L. J., Nemavhulani, S., Cass, J., Cowan, D. A. & Trindade, M. Three novel bacteriophages isolated from the East African Rift Valley soda lakes. Virol. J. 13, 204 (2016).
Casjens, S. R. & Gilcrease, E. B. Determining DNA packaging strategy by analysis of the termini of the chromosomes in tailed-bacteriophage virions. Methods Mol. Biol. 502, 91–111 (2009).
Canchaya, C., Proux, C., Fournous, G., Bruttin, A. & Brüssow, H. Prophage genomics. Microbiol. Mol. Biol. Rev. 67, 238–276 (2003).
Groth, A. C. & Calos, M. P. Phage integrases: biology and applications. J. Mol. Biol. 335, 667–678 (2004).
Schubert, R. A., Dodd, I. B., Egan, J. B. & Shearwin, K. E. Cro’s role in the CI Cro bistable switch is critical for λ’s transition from lysogeny to lytic development. Genes Dev. 21, 2461–2472 (2007).
Magnuson, R. D. Hypothetical functions of toxin-antitoxin systems. J. Bacteriol. 189, 6089–6092 (2007).
Jørgensen, M. G., Pandey, D. P., Jaskolska, M. & Gerdes, K. HicA of Escherichia coli defines a novel family of translation-independent mRNA interferases in bacteria and archaea. J. Bacteriol. 191, 1191–1199 (2009).
Pedersen, K. et al. The bacterial toxin RelE displays codon-specific cleavage of mRNAs in the ribosomal A site. Cell 112, 131–140 (2003).
Dziewit, L., Jazurek, M., Drewniak, L., Baj, J. & Bartosik, D. The SXT conjugative element and linear prophage N15 encode toxin-antitoxin-stabilizing systems homologous to the tad-ata module of the Paracoccus aminophilus plasmid pAMI2. J. Bacteriol. 189, 1983–1997 (2007).
Oliveira, H., São-José, C. & Azeredo, J. Phage-derived peptidoglycan degrading enzymes: challenges and future prospects for in vivo therapy. Viruses 10, E292 (2018).
Young, R. Bacteriophage holins: deadly diversity. J. Mol. Microbiol. Biotechnol. 4, 21–36 (2002).
Murphy, J., Mahony, J., Ainsworth, S., Nauta, A. & van Sinderen, D. Bacteriophage orphan DNA methyltransferases: insights from their bacterial origin, function, and occurrence. Appl. Environ. Microbiol. 79, 7547–7555 (2013).
Dempsey, R. M. et al. Sau42I, a BcgI-like restriction-modification system encoded by the Staphylococcus aureus quadruple-converting phage Phi42. Microbiology 151, 1301–1311 (2005).
Dziewit, L., Oscik, K., Bartosik, D. & Radlinska, M. Molecular characterization of a novel temperate Sinorhizobium bacteriophage, ΦLM21, encoding DNA methyltransferase with CcrM like specificity. J. Virol. 88, 13111–13124 (2014).
Decewicz, P., Radlinska, M. & Dziewit, L. Characterization of Sinorhizobium sp. LM21 prophages and virus-encoded DNA methyltransferases in the light of comparative genomic analyses of the sinorhizobial virome. Viruses 9, 161 (2017).
Johnson, T. J., Wannemeuhler, Y. M., Scaccianoce, J. A., Johnson, S. J. & Nolan, L. K. Complete DNA sequence, comparative genomics, and prevalence of an IncHI2 plasmid occurring among extraintestinal pathogenic Escherichia coli isolates. Antimicrob. Agents Chemother. 50, 3929–33 (2006).
Akhter, S., Aziz, R. K. & Edwards, R. A. PhiSpy: a novel algorithm for finding prophages in bacterial genomes that combines similarity- and composition-based strategies. Nucleic Acids Res. 40, e126 (2012).
Lopes, A., Tavares, P., Petit, M. A., Guerois, R. & Zinn-Justin, S. Automated classification of tailed bacteriophages according to their neck organization. BMC Genomics 15, 1027 (2014).
Williams, K. P. Integration sites for genetic elements in prokaryotic tRNA and tmRNA genes: sublocation preference of integrase subfamilies. Nucleic Acids Res. 30, 866–875 (2002).
Cheng, H., Shen, N., Pei, J. & Grishin, N. V. Double-stranded DNA bacteriophage prohead protease is homologous to herpesvirus protease. Protein Sci. 13, 2260–2269 (2004).
Iyer, L. M., Zhang, D., Burroughs, A. M. & Aravind, L. Computational identification of novel biochemical systems involved in oxidation, glycosylation and other complex modifications of bases in DNA. Nucleic Acids Res. 41, 7635–7655 (2013).
Iida, S. et al. DNA restriction-modification genes of phage P1 and plasmid p15B. Structure and in vitro transcription. J. Mol. Biol. 165, 1–18 (1983).
Adriano, D. C. Trace elements in terrestrial environments: biogeochemistry, bioavailability, and risks of metals. (Springer, 2001).
Shi, Z. et al. Correlation models between environmental factors and bacterial resistance to antimony and copper. PLoS One 8, e78533 (2013).
Drewniak, L., Styczek, A., Majder-Lopatka, M. & Sklodowska, A. Bacteria, hypertolerant to arsenic in the rocks of an ancient gold mine, and their potential role in dissemination of arsenic pollution. Environ. Pollut. 156, 1069–1074 (2008).
Zhang, J. et al. Anaerobic arsenite oxidation by an autotrophic arsenite-oxidizing bacterium from an arsenic-contaminated paddy soil. Environ. Sci. Technol. 49, 5956–5964 (2015).
Paez-Espino, D. et al. IMG/VR v.2.0: an integrated data management and analysis system for cultivated and environmental viral genomes. Nucleic Acids Res. 47, D678–D686 (2019).
Hanahan, D. Studies on transformation of Escherichia coli with plasmids. J. Mol. Biol. 166, 557–580 (1983).
Urakami, T., Tamaoka, J., Suzuki, K. & Komagata, K. Paracoccus alcaliphilus sp. nov., an alkaliphilic and facultatively methylotrophic bacterium. Int. J. Syst. Bacteriol. 39, 116–121 (1989).
Kumaraswamy, R., Sjollema, K., Kuenen, G., van Loosdrecht, M. & Muyzer, G. Nitrate-dependent [Fe(II)EDTA]2- oxidation by Paracoccus ferrooxidans sp. nov., isolated from a denitrifying bioreactor. Syst. Appl. Microbiol. 29, 276–286 (2006).
Kim, B. Y. et al. Paracoccus homiensis sp. nov., isolated from a sea-sand sample. Int. J. Syst. Evol. Microbiol. 56, 2387–2390 (2006).
Doronina, N. V. & Trotsenko, Y. A. A novel plant-associated thermotolerant alkalophilic methylotroph of the genus Paracoccus. Microbiology 69, 593–598 (2000).
Kelly, D. P., Euzeby, J. P., Goodhew, C. F. & Wood, A. P. Redefining Paracoccus denitrificans and Paracoccus pantotrophus and the case for a reassessment of the strains held by international culture collections. Int. J. Syst. Evol. Microbiol. 56, 2495–2500 (2006).
Liu, X. Y., Wang, B. J., Jiang, C. Y. & Liu, S. J. Paracoccus sulfuroxidans sp. nov., a sulfur oxidizer from activated sludge. Int J Syst Evol Microbiol 56, 2693–2695 (2006).
Katayama, Y., Hiraishi, A. & Kuraishi, H. Paracoccus thiocyanatus sp. nov., a new species of thiocyanate-utilizing facultative chemolithotroph, and transfer of Thiobacillus versutus to the genus Paracoccus as Paracoccus versutus comb. nov. with emendation of the genus. Microbiology 141, 1469–1477 (1995).
Bartosik, D., Szymanik, M. & Wysocka, E. Identification of the partitioning site within the repABC-type replicon of the composite Paracoccus versutus plasmid pTAV1. J. Bacteriol. 183, 6234–6243 (2001).
Bartosik, D., Baj, J., Plasota, M., Piechucka, E. & Wlodarczyk, M. Analysis of Thiobacillus versutus pTAV1 plasmid functions. Acta Microbiol. Pol. 39, 5–11 (1993).
Daneshvar, M. I. et al. Paracoccus yeeii sp. nov. (formerly CDC group EO-2), a novel bacterial species associated with human infection. J. Clin. Microbiol. 41, 1289–1294 (2003).
Sambrook, J. & Russell, D. W. Molecular cloning: A laboratory manual. (Cold Spring Harbor Laboratory Press, 2001).
Kushner, S. R. In Genetic engineering (eds Boyer, H. B. & Nicosia, S.) 17–23 (Elsevier/North-Holland, 1978).
Forsman, P. & Alatossava, T. Genetic variation of Lactobacillus delbrueckii subsp. lactis bacteriophages isolated from cheese processing plants in Finland. Appl. Environ. Microbiol. 57, 1805–1812 (1991).
Vandenheuvel, D., Rombouts, S. & Adriaenssens, E. M. Purification of bacteriophages using anion-exchange chromatography. Methods Mol. Biol. 1681, 59–69 (2018).
Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 17, 10 (2011).
Carver, T., Harris, S. R., Berriman, M., Parkhill, J. & McQuillan, J. A. Artemis: an integrated platform for visualization and analysis of high-throughput sequence-based experimental data. Bioinformatics 28, 464–469 (2012).
Marchler-Bauer, A. et al. CDD: a Conserved Domain Database for the functional annotation of proteins. Nucleic Acids Res. 39, D225–229 (2011).
Lowe, T. M. & Chan, P. P. tRNAscan-SE On-line: integrating search and context for analysis of transfer RNA genes. Nucleic. Acids Res. 44, W54–W57 (2016).
Laslett, D. & Canback, B. ARAGORN, a program to detect tRNA genes and tmRNA genes in nucleotide sequences. Nucleic Acids Res. 32, 11–16 (2004).
Roberts, R. J., Vincze, T., Posfai, J. & Macelis, D. REBASE - a database for DNA restriction and modification: enzymes, genes and genomes. Nucleic Acids Res. 43, D298–D299 (2015).
Chen, Y., Yu, P., Luo, J. & Jiang, Y. Secreted protein prediction system combining CJ-SPHMM, TMHMM, and PSORT. Mamm. Genome 14, 859–865 (2003).
Pal, C., Bengtsson-Palme, J., Rensing, C., Kristiansson, E. & Larsson, D. G. BacMet: antibacterial biocide and metal resistance genes database. Nucleic Acids Res. 42, D737–D743 (2014).
Claudel-Renard, C., Chevalet, C., Faraut, T. & Kahn, D. Enzyme-specific profiles for genome annotation: PRIAM. Nucleic Acids Res. 31, 6633–6639 (2003).
Darzentas, N. Circoletto: visualizing sequence similarity with Circos. Bioinformatics 26, 2620–2621 (2010).
Bastian, M., Heymann, S. & Jacomy, M. Gephi: an open source software for exploring and manipulating networks. Third International AAAI Conference on Weblogs and Social Media (2009).
Jacomy, M., Venturini, T., Heymann, S. & Bastian, M. ForceAtlas2, a continuous graph layout algorithm for handy network visualization designed for the Gephi software. PLoS One 9, e98679 (2014).
Acknowledgements
This research was funded by the Ministry of Science and Higher Education, Poland (program “Iuventus Plus” conducted in 2015–2016; Grant No. IP2014 009073) and partially by the National Science Centre, Poland, on the basis of the Decision Number DEC-2013/09/B/NZ1/00133. Library construction and genome assembly were carried out at the DNA Sequencing and Oligonucleotide Synthesis Laboratory of the IBB Polish Academy of Science using the CePT infrastructure financed by the European Union – the European Regional Development Fund [Innovative economy 2007–13, Agreement POIG.02.02.00-14-024/08-00]. We thank Jan Gawor for his technical assistance in DNA sequencing and Magdalena Narajczyk for her technical assistance in TEM analysis.
Author information
Authors and Affiliations
Contributions
P.D., L.D., D.B. and M.R. conceived and designed the experiments; P.D., P.G., P.K. and M.R. conducted the experiments; P.D. and M.R. performed bioinformatic analyses; P.D., L.D. and M.R. analyzed the results; L.D., D.B. and M.R. contributed reagents, materials, analysis tools; P.D., L.D. P.G. and M.R. wrote the paper. P.D., L.D. and D.B. revised and modified the manuscript to its final version. All authors reviewed and approved the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Decewicz, P., Dziewit, L., Golec, P. et al. Characterization of the virome of Paracoccus spp. (Alphaproteobacteria) by combined in silico and in vivo approaches. Sci Rep 9, 7899 (2019). https://doi.org/10.1038/s41598-019-44460-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-44460-4
This article is cited by
-
Genomic profiling and characteristics of a C1 degrading heterotrophic fresh-water bacterium Paracoccus sp. strain DMF
Archives of Microbiology (2024)
-
Functional characterization of the ABC transporters and transposable elements of an uncultured Paracoccus sp. recovered from a hydrocarbon-polluted soil metagenome
Folia Microbiologica (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
