
Abstract
Resistance to key first-line drugs is a major hurdle to achieve the global end tuberculosis (TB) targets. A prodrug, pyrazinamide (PZA) is the only drug, effective in latent TB, recommended in drug resistance and susceptible Mycobacterium tuberculosis (MTB) isolates. The prodrug conversion into active form, pyrazinoic acid (POA), required the activity of pncA gene encoded pyrazinamidase (PZase). Although pncA mutations have been commonly associated with PZA resistance but a small number of resistance cases have been associated with mutationss in RpsA protein. Here in this study a total of 69 PZA resistance isolates have been sequenced for pncA mutations. However, samples that were found PZA resistant but pncA wild type (pncAWT), have been sequenced for rpsA and panD genes mutation. We repeated a drug susceptibility testing according to the WHO guidelines on 18 pncAWT MTB isolates. The rpsA and panD genes were sequenced. Out of total 69 PZA resistant isolates, 51 harbored 36 mutations in pncA gene (GeneBank Accession No. MH46111) while, fifteen different mutations including seven novel, were detected in the fourth S1 domain of RpsA known as C-terminal (MtRpsACTD) end. We did not detect any mutations in panD gene. Among the rpsA mutations, we investigated the molecular mechanism of resistance behind mutations, D342N, D343N, A344P, and I351F, present in the MtRpsACTD through molecular dynamic simulations (MD). WT showed a good drug binding affinity as compared to mutants (MTs), D342N, D343N, A344P, and I351F. Binding pocket volume, stability, and fluctuations have been altered whereas the total energy, protein folding, and geometric shape analysis further explored a significant variation between WT and MTs. In conclusion, mutations in MtRpsACTD might be involved to alter the RpsA activity, resulting in drug resistance. Such molecular mechanism behind resistance may provide a better insight into the resistance mechanism to achieve the global TB control targets.
Similar content being viewed by others


Introduction
According to the World Health Organization report 2018, 23% of the world’s population (1.7 billion), are infected with latent TB1. Among the anti-tuberculosis agents, pyrazinamide (PZA) is the only drug that kills MTB in the latent state which has successfully reduced the time span of TB therapy from 9 to 6 months2,3,4. PZA is a prodrug that depends on MTB encoded, pyrazinamidase (PZase) activity for conversion into pyrazinoic acid (POA), an active form of PZA that targets the trans-translation5,6 by interfering with ribosomal protein S1 (RpsA), where POA has been disrupting the transfer messenger RNA complex formation with RpsA (RpsA-tmRNA)3,7,8,9.
In MTB, the RpsA protein has four S1 domains (36–105, 123–188, 209–277, and 294–363)10. Residues from 292–363, forming fourth S1 domain which is also known as C-terminus domain of RpsA (MtRpsACTD), is highly conserved among MTB strains and fully capable with POA binding. Amino acids at positions, F307, F310, H322, D352, and Arg357 are present in RNA binding sites of fourth S1 domain11. PZA resistance emerges due to mutations at MtRpsACTD of mycobacterial species, causing conformational changes in the POA binding site3. Residues, Lys303, Phe307, Phe310, and Arg357, also known as tmRNA binding site, have been identified, involving in the interactions with two POA molecules3,12. In Mycobacterium smegmatis, Alanine deletion at the C-terminal (RpsAΔA438) results in the lack of binding with RpsA. MtRpsACTD is the POA binding site where tmRNA bind each other to form a complex for initiation of translation12. Protein structure may have drastic effects due to amino acid substitution, altering the protein structure and function, especially in the active site or binding pockets13,14,15. Mutation may also produces effects at a long-range position16. Exploring the mechanism of such changes behind a mutation, has been investigated for better understanding of a particular phenomenon. However, these are time-consuming and very expensive investigations when addressed by experimental procedure alone.
Molecular dynamic (MD) simulations is a method of choice that have been applied widely in exploring the mechanisms of conformational changes in protein, especially in drug resistance mechanisms caused by mutations. MD simulation studies of ligand-protein interactions are widely applied approach to explain the mechanisms of drug resistance due to mutations especially in target protein, which is one of the major causes behind resistance. In vivo experimental research, the crystal structure is analyzed for drug resistance while in comparison with experimental approach, MD simulation has a particular advantage to explain the mechanisms of drug resistance at molecular level14,17. Further, the structural dynamics of protein complexes and other residues level information can be accessed through MD which have been considered difficult by experimental procedures18,19,20,21.
In our recent studies, we identified different mutations in pncA gene22 (GeneBank Accession No. MH46111) and RpsA23, whose molecular mechanism of resistance have been investigated through MD simulation24,25,26. Here, we analyzed the effect of our novel mutations, D342N, D343N, A344P, and I351F, on RpsA activity which have been detected in the conserved region in our previous study23 among PZA-resistance isolates. We have investigated the possible changes in the RpsA dynamics, that results due to mutations in MtRpsACTD which may provide useful information behind the drug resistance.
Results
The repeating DST results demonstrated that the isolates are resistant to PZA. Further, the resistant samples were also analyzed manually where growth was occurred against the critical concentration of PZA. In the previous study, out of total 69 PZA resistant isolates, 51 harbored 36 mutations in pncA gene (GeneBank Accession No. MH46111). The remaining 18 isolates were pncAWT PZA resistance isolates. Out of 18 PZA resistants but pncAWT isolates, 11 (61%) samples have fifteen non-synonymous mutations, while seven isolates were RpsAWT (Table 1). We did not detect any mutation in panD genes. Mutations, S324F, E325K, G341R, D342N, D343N, A344P, and I351F, were present in the conserved region (292–363) of the rpsA gene (Table 1). Mechanism of PZA resistance behind mutations S324F, E325K, and G341R in rpsA have been already investigated in our previous study. Here, we analyzed the mechanism of resistance behind mutation, Asp342Asn, Asp343Asn, Ala344Pro, and Ile351Phe, which have been involved in the conformational changes that might be associated with RpsA activity.
Binding pocket and shape complementarity
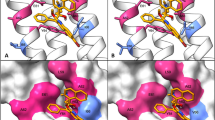
Any diviation from pocket volume may effect the interaction with drug. Compared with WT (4638.493 Å), MTs Asp342Asn (4622.668 Å), Asp343Asn (4623.892 Å), Ala344Pro (4618.064 Å), and Ile351Phe (4623.253 Å), have been altered the binding pocket volume. Shape complementarity score of WT was also found higher (3862) than the MT (Table 2). Further, the RMSD of superimposed MT and WT has a significance difference showing the effect of mutation effect on protein structure (Fig. 1).

Superimposition of WT-RpsA (A) and mutant (B–J) in apo state and complex (Com) state. RpsA and POA binding residues are 303, 307, 310, and 357. Superimposition of mutant D342N, D343N, A344P, and I351F and WT-RpsA before and after simulation. The difference in RMSDs have been shown with each superimposed structure.
Proteins-ligand interactions
For a strong binding affinity, hydrogen and hydrophobic are essential interactions between ligand and receptor. Overall residues, R357, F309, G319, L320, A287, I358, D352, D350, S359, L353, F307, F310, E318, and K303 have been involved in the interactions. WT formed more interactions than MTs (Fig. 2).

Wild type and mutant’s interactions with POA. (A) MTB RpsA WT and MT secondary structure. Mutation site has been pointed, where the conversion of loop into helix and sheet into coil, encircled in D342N, D343N, A344P, and I351F. (B) WT formed two H-bonds with two POA molecules. MT had fewer hydrogen as compared to WT.
Protein and drug trajectory
RMSD is a measure of structure stability during simulation period. During the period of simulation, substantial higher deviation specify that a molecule may be unstable. In apo-state, WT RpsA attained RMSD value between 1.6 Å and 4.0 Å at 70 ns and 15–20 ns repectively. However, the RMSD is seemed to be consistent at 3 Å from 25 to 100 ns. In comparison with WT, MTs, D342N and D343N attained RMSD between 1.5 Å, 3.5 Å and 1.3 Å and 4.3 Å respectively. The RMSD of D343N is seemed to be inconsistent after 75 ns. MT A344P and I351F attained RMSDs between 1.5 Å, 3.5 Å.and 1.0 Å, 4 Å respectively. However, the RMSDs of mutant I351F and A344P are seemed to be unstable as it still in rise at 100 ns.
In complex with POA, the RMSD of WT RpsA is seemed to be consistent (1.5 Å–3.2 Å) throughout simulations. The MTs, D342N (1.1 Å–3.5 Å), D343N (1.0 Å–3.5 Å), A344P (1.2 Å–3.4 Å), and I351F (1.2 Å–3.3 Å) exhibited variations in RMSDs during the simulation period (Fig. 3). The MT, D343N and I351F are more deviated at 1000 ns, while, MTs D342N and D343N have been in rise at 100 s.

RMSD of wild type and mutants RpsA. Apo state (A–D) Complex with PZA (E–H). RpsA(WT); ribosomal protein S1 (RpsA) WT (black) retained a constant RMSD from 25 ns to 100 ns. RMSD of D342N seems to be inconsistent as it rises at 100 ns. In apo state, D343N exhibited a continuous rise in RMSD from 0 ns to 78 ns. However, a little fall have been shown at 95 ns–100 ns. While in complex state, the RMSD is little stable but seems, still rising at 83 ns–100 ns. RMSD of MT A344P is rising at 80 ns–100 ns in apo state, however, it seems stable in complex state. MT I351F attained a little higher RMSD in both, apo and complex state. However, it seems highly unstable in apo state, rising continuously 20 ns–100 ns.
In complex with a drug, RpsA WT exhibited lower flexibility as compared to MTs (Fig. 4). D342N, D343N, and A344P exhibited a higher flexibility at residues lcation 330, 390 and 293, 330, 355, 395–400 in both states. Residues flexibility in MT I351F is higher that WT in apo state.

RMSF of wild type and mutant MtRpsACTD. In apo state, residues at locations, 390–410 are more flexible in MT. Residues in D342N exhibited a higher flexibility at lcation 330 and 390. MT, D343N attaind a higher flexibility at position 293, 330, 355, 395–400 in both states. In A344P, the flexibility is seeming almost similar to WT throughout the simulation period. Residues flexibility in MT I351F is higher that WT.
Folding dynamics (Radius of gyration (Rg))
The MT exhibited Rg value between 17 and 19 whereas the WT attained the Rg values between 17.4 and 18.3. In both the states, MTs, D342N, D343N, A344P and I351F demonstrated a degree of variation in folding during the simulation period, showing unstable folding than WT. Variations in Rg with time show unstable folding while a stable Rg value indicate compactness in proteins folding. The Rg which measured the degree of compactness and folding is plotted against time. A stable folded protein maintain a steady value of Rg whereas in case of misfolding, the Rg will show variation over time (Fig. 5).

Radius of gyration and B-Factor of wild type and mutant RpsA. A constant Rg value is a measure of correct folding. MT RpsA, D342N, D343N, A344P, and I351F remained unstable throughout simulation period.
Essential dynamics
The mutant’s structures covered more area in both, apo and complex states and is more scattered, showing its variation in dynamics of motions (uncorrelated). PCs of WT and MT have been shown (Fig. 6). MTs, D342N, D343N, A344P, and I351F exhibited a scattered type of motion than WT. In apo state, a cluster type of motion was observed in WT, covering a small area on PC1 and PC2 between −50 and 50, −100 and 100 respectively. MTs RpsA, D342N, D343N, A344P and I351F exhibited a more scattered type of motion on PC1 (between −100 and 50, −125 and 100,–125 and 100, −100 and 125) and PC2 (−80 and 80, −60 and 90, −60 and 90, −100 and 60).

PCA of wild type and mutant RpsA (apo and complex state). MTs RpsA, D342N, D343N, A344P, and I351F scattered over a large area, showing a dispersed type of motion on first two components, PC1 and PC2. These variations in dynamics of WT and MT are more evident in the complex state with the drug. The PC2 motions of MT are significantly more scatered and disperse type, especially I351F.
In complex state WT is more dispersed on PC2 (−150 and 100) than mutants D342N, D343N, A344P and I351F (−50 and 50, −75 and 75, −100 and 50, −125 and 125). However, the dynamics of MTs is more scattered (−80 and 70, −70 and 120, −100 and 70, −100 and 100) than WT (−70 and 80).
Free energy landscape and Gibbs free energy
Gibbs free energy (GFE) is a measure of work of a closed system when exchanging heat with the surroundings. The differences in GFE values may have importance in the stability calculation. A protein with native structure has the minimum GFE. WT exhibited a significant difference in GFE than MT, depicted in (Fig. 7). The color (red) in both states of the MT is more unstable compared to the MT.

Free energy Landscape (FEL) of the wild type and mutants, D342N, D343N, A344P and I351F. High energy state is represented by red color in the plot. WT has a significant GFE variations in both states (apo and complex) as compared to MT, as indicated by the color of the plot. The color of WT demonstrated more stability compared to MT.
The ΔGbindof WT (−6.88 kcal/mol) is very low in comparison with ΔGbind of D342N (−3.408 kcal/mol), D343N (−3.605 kcal/mol), A344P (−4.620 kcal/mol), and I351F (−3.937 kcal/mol) (Table 2), showing a very low binding affinity and stability. The results of Gibbs free energy and ΔGbind is showing that these mutations might be involved in no or low binding affinity with POA, causing resistance.
Distance matrix
The average distance of WT RpsA and drug is almost constant during the simulation period as compared to MTs, where a high degree of fluctuation (Fig. 8) representing the variation in distance between drug and protein during the whole simulation period. The MT, D343N and I351F, exhibited a high average distance between target and drug, signifiying the effect of mutations on proteins binding affinity. The total energy of WT RpsA (−76000 kJ/mol) was significantly lower than MTs (74500kj/mol, 73000kj/mol, 71500kj/mol, and 70500kj/mol) throughout simulation as shown in Fig. 9.

Distance matrix of wild type and mutant RpsA with drug. The average distance of WT and POA is constant. MT, D342N, D343N, A344P, and I351F exhibited variations in distances throughout simulation period. D343N and I351 exhibited much higher distance.

Total estimated energy of wild type and mutant RpsA. Total energy measured for WT was significantly higher than MT D342N, D343N, A344P and I351F.
Discussion
Drug resistance is the main obstacle towards the WHO end TB 2030. Investigating the prevalence of PZA resistance in TB high burden countries23,27 and insight into drug resistance mechanisms is vital for better supervision of global TB control. PZA is the only drug that kills persister MTB under latent state. According to the previous investigations, the emergence of PZA resistance is associated with mutations in pncA, but not in all cases, where resistance may be emerged due to mutations in the target RpsA23,28,29,30. In our previous study23, we reported novel mutations in conserved region called MtRpsACTD (Table 1). However, the molecular mechanism behind resistance was still unknown. Here, we explored the inside mechanism of resistance due to RpsA mutations which have been associated in-vivo PZA resistance. MD simulations provide the interaction mechanisms at the molecular level17,31,32 along with structural and dynamical information, which is difficult to be determined by experimental procedures. Protein’s structure and function are maintained by conserved region residues where mutations have been reported, leading to conformational changes or a loss of function33,34,35. To explore these mechanistic effects behind the PZA resistance, we analyzed multiple characteristics of protein affected by conserved region mutations in MtRpsACTD.
A higher RMSD and RMSF values of MT, D342N, D343N, A344P, and I351F signifies the effect of point mutations when compared with WT, affecting the activity of RpsA to interact with the POA. Stability and flexibility are essential properties maintaining the activity of biomolecules36. Mutations in drug target often affect these properties, making them a vulnerable target to interact with drugs. Increases in residue flexibility have a significant effect on activity. The earlier reports32,37,38,39 have also found the effect of mutations in a specific location, resulting in the loss of activity. Thus RMSD and RMSF may be as a measure of effect behind mutations in targets, resulting in drug resistance or disease where stability is a fundamental property, affecting the function, activity, and regulation of biomolecules. Changes in stability and flexibility of targets proteins may be the loss of thermodynamic stability and protein folding40. Destabilization in folding and thermodynamic stability may affect the total energy of biomolecules (Fig. 9).
Further, proteins folding stability may also be measured by Radius of gyration (Rg) which is a degree of compactness, commonly measured in MD simulation by a ratio of the accessible surface area to the surface area of the ideal sphere of the same volume, plotted against time. A stable Rg value signifies the proteins correct folding (Fig. 5), while any deviation is regarded as folding instability41,42,43. In comparison with MTs, a more stable Rg attanied by WT is an indication of correct folding during simulations (Fig. 5).
Oftenly, mutations may occurr far from the active site, causing some drastic changes in binding pocket volume. These distant sites may have a good communication with enzyme function in signal transmissions from one functional site to far site through a series of pathways during biological function44,45. Any change to the binding pocket may lead to the loss of interaction with inhibitors. Resistance to the drug may be developed due to change in the binding pocket38,46,47. The binding affinity of RpsA to POA may also be lost due to alteration in pocket size. In the binding pocket, the primary interactions accounted with ligand are, hydrogen bonds, van der Waals and electrostatic forces36. The three-dimensional structure of a protein has hydrogen and hydrophobic interactions playing a vital role in interactions. Weak interactions have fewer hydrogen and hydrophobic interactions. WT protein has more and MT, D342N, D343N, A344P, and I351F, has significantly fewer interactions indicating the strength of protein folding andbinding stability. Hydrogen bonds support the core, which is comprised of α-helices and β-sheets48,49,50. In spite of mutations, a low level of resistance may be developed due to efflux or influx51. Fore more better understanding of drug resistance, the role of the efflux needs to be specified.
We analyzed the consequences of our novel mutations D342N, D343N, A344P, and I351F on RpsA dynamics by comparing them with WT in PZA-resistant pncAWT MTB strains. These mutations have been involved, changing the RpsA activity by alltering the total energy, flexibility, folding and stability, thereby affecting the interactions with POA. In comparison with WT, interaction of POA and RpsA seemed to be altered due to variations in the binding pocket of MTs D342N, D343N, A344P, and I351F. The overall investigations supports the hypothesis that mutaions in the conserved region of the rspA gene might be involved in PZA-resistance. To the best of our knowledge, we presented a first comprehensive investigation of such kind where multiple characteristics have been investigated for better insight into mutations affecting RpsA activity and caused PZA resistance.
Materials and Methods
Samples collection
Provincial Tuberculosis Reference Laboratory (PTRL) is the Central laboratory of Khyber Pakhtunkhwa (KPK) province of Pakistan. We sequenced 69 PZA resistance isolates for pncA gene mutations, out of which 51 has 36 different mutations in pncA gene22 while 18 isolates were detected as pncAWT. We collected all the 18 isolates, previously detected as resistant to PZA (PZA-R) but pncAWT. To screen mutation in rpsA and panD genes, all these samples were grown on 7H9 media in the mycobacterium growth indicator tube (MGIT) 960 system for confirmation as MTB52. Approximately 100 µl of the sample of the positive tube was added to TBc ID device that indicates a positive MTB by the emergence of pink to red at the test and control location, confirming the antigen, MPT64 in the sample53,54. All the positive MTB tubes were subjected for repetition of drug susceptibility testing (DST).
PZA DST
MTB Drug susceptibility testing (DST) is now routinely performed through automated BACTEC MGIT 960 system55. The samples were repeated for PZA DST along with positive (ATCC 25618/H37Rv) and negative controls (Mycobacterium bovis). A sample was marked as PZA-resistance when growth occurred at 100 μg/ml of PZA critical concentration55.
Interpretation ofsci drug susceptibility testing
The test completion is interpreted when the growth unit (GU) value of control (GC) attained as 400 or more. An inventory report is printed as “S” for sensitive isolate and “R” for resistance. The growth unit (GU) value was less than 100 for sensitive isolates and greater than 100 for resistants55,56,57.
rpsA and panD gene sequencing
DNA was isolated from the PZA resistance samples using the sonication method58,59. Forward and reverse primers, F-5′CGGAGCAACCCAACAATA-3′ and R-5′ GTGGACAGCAACGACTTC-3′)60 and F-5′TCAACGGTTCCGGTCGGCTGCT-3′, R-5′TATCCGCCACTGCTGCACGACCTT-3′61 were used to amplify rpsA and panD genes. Each 50-µl reaction consist of molecular grade water (34.8 µl), genomic DNA (4 µl), DNTs (0.1 µl), MgCl2 (3 µl), polymerase chain reaction (PCR) buffer (5 µl), Taq (0.8 µl) (New England Biolabs, UK)), forward and reverse primers (1 µl each). The PCR conditions were adjusted as, 94 °C (5 minutes), as denaturation step of 30 cycles (30 seconds), 30 seconds (56 °C), and for 1 minute (72 °C); 72 °C for 5 minutes as an extension step. The amplified rpsA (PCR product) was sent for sequencing (6 Applied Biosystems 3730xl, Macrogen, Korea).
rpsA sequencing data analysis
The rpsA and panD sequence data was loaded into Mutation Surveyor V5.0.162 compared with rpsA (Rv1630) RefSeq (NC_000962.3) gene.
Crystal structure retrieval
A crystal structure of MTB RpsA3 (PDB ID 4NNI) was retrieved from Brookhaven Raster Display (BRAD) protein data bank (PDB)63. Mutant structures of RpsA were generated by inducing mutations at D342N, D343N, A344P, and I351F residues using PYMOL64. The POA structure was retrieved from PubChem database (PubChem CID: 1047)65 and energy minimized.
Molecular docking
Structures were loaded into Chimera66,67, selenomethionines were changed into methionine and hydrogen atoms were corrected. Shape complementarity measuring score of drug and protein structures was performed using PatchDock server68, which is a kind of geometric matching, where drug and protein features are compared for the docking purpose. Molecular complexes have been found with good interactions are oftenly exhibited good shape complementarity. Mutations often cause conformational changes, affecting the interactions with a drug69,70. Pocket volumes of WT and mutant D342N, D343N, A344P, and I351F were compared through CastP server71.
Molecular dynamics (MD) simulation
Molecular dynamics (MD) simulations provide plenteous dynamical structural and energetic information about the target protein and drug interactions. The essence of receptor and drug interaction can be analyzed accurately as MD simulation provides useful information in understanding the structure-function relationship, guiding the drug discovery and designing practice47. MD simulation was performed on all the structures using AMBER while topological parameters were generated using the antechamber tool (. frcmod). A transferable intermolecular potential with 3 points (TIP3P) water box was used to solvate the proteins and ligand coordinates with a distance of 8.0 Å72. Sodium and chloride ions were added to neutralize the systems, as these ions possess high electrostatic potential in the replacement of water molecules. The steepest descent algorithm was used to minimize the energy.
Constant volume and temperature (NVT) ensemble and pressure and temperature (NPT) ensemble was equilibrated and set out at 1-bar pressure for 100 ps. Temperature and pressure were maintained constant using Berendsen thermostat73 and Parrinello–Rahman barostat74 methods respectively. Linear constraint solver (LINCS) algorithm75 was used for bond length rectification and CPPTRAJ was used for the post-dynamics simulation analysis such as RMSD, RMSF, radius of gyration (Rg), and total energy. Further, a free energy landscape calculation methodology, implemented in Gromacs was used to calculate the energy changes and metastable states during the course of simulation.
Essential dynamics
To obtained internal motion of the system, Principal Component Analysis (PCA) of trajectory was performed on the mass-weighted cartesian coordinates, where rotation and its translation were removed. Low modes are recognize by long dynamics in proteins76,77. Further, PCA reduces the motion in a trajectory78,79,80 where A set of variables z1, z2…, zp, transformed, called principal components (PCs) are generated. Free Energy Landscape (FEL)81,82 is applied to calculate the energies of macromolecule conformations sets. The first two PCs (PC1 vs. PC2) of PCA computed, give the trajectories on the two principal components of motion83. The amplitude of fluctuations in proteins were captured through PCA and the first two components (PC1 and PC2) were plotted to analyze their fluctuation boundries84,85.
The lowest energy stable state is represented by the free energy landscape (FEL). The stable state with minimal energy on a plot is shown by deep valleys while boundaries between the deep valleys represent the intermediate conformations86. FEL was plotted, using g_sham module while PC1 and PC2 were used to analyze the FEL using the equation:
Here, PC1 and PC2 are the reaction coordinates, KB represents the Boltzmann constant and P (PC1, PC2) is the first two principal components probability distribution of the system.
The Gibbs and total binding free energy
Gibbs free energy (G), is a single value, combining enthalpy and entropy while the change in free energy (ΔG) is the sum of the enthalpy plus the product of entropy and the temperature of the system.
The G is minimized to the chemical equilibrium state of the system and it is a thermodynamic potential at constant pressure and temperature, where a reduction is a vital state for processes. The G (Sugita & Kitao, 1998) was plotted against WT. The binding free energy was measured from 500 snapshots at the end of 10-ns trajectory. The free energy of each component was estimated using the following equation:
∆Gbind represents the total binding free energy The free energy of each component was estimated as:
Gbond, represents bonded, Gele is electrostatic, and GvdW is van der Waals interactions while G-pol and Gnpol represent polar and nonpolar to the total solvated free energies.
References
WHO, 2018. WHO|Global tuberculosis report 2018. WHO Available at, http://www.who.int/tb/publications/global_report/en/. (Accessed: 5th October 2018).
Mitchison, D. A. The action of antituberculosis drugs in short-course chemotherapy. Tubercle 66, 219–225 (1985).
Yang, J. et al. Structural basis for targeting the ribosomal protein S1 of Mycobacterium tuberculosis by pyrazinamide. Mol. Microbiol. 95, 791–803 (2015).
Yadon, A. N. et al. A comprehensive characterization of PncA polymorphisms that confer resistance to pyrazinamide. Nat. Commun. 8 (2017).
Lu, P. et al. Pyrazinoic Acid Decreases the Proton Motive Force, Respiratory ATP Synthesis Activity, and Cellular ATP Levels. Antimicrob. Agents Chemother. 55, 5354–5357 (2011).
Ying, Z., Wade, M. M., Scorpio, A., Zhang, H. & Sun, Z. Mode of action of pyrazinamide: disruption of Mycobacterium tuberculosis membrane transport and energetics by pyrazinoic acid. J. Antimicrob. Chemother. 52, 790–795 (2003).
Sørensen, M. A., Fricke, J. & Pedersen, S. Ribosomal protein S1 is required for translation of most, if not all, natural mRNAs in Escherichia coli in vivo 11 Edited by D. Draper. J. Mol. Biol. 280, 561–569 (1998).
Simons, S. O., Mulder, A., van Ingen, J., Boeree, M. J. & van Soolingen, D. Role of rpsA gene sequencing in diagnosis of pyrazinamide resistance. J. Clin. Microbiol. 51, 382–382 (2013).
Tan, Y. et al. Role of pncA and rpsA gene sequencing in detection of pyrazinamide resistance in Mycobacterium tuberculosis isolates from southern China. J. Clin. Microbiol. 52, 291–297 (2014).
Salah, P. et al. Probing the relationship between Gram-negative and Gram-positive S1 proteins by sequence analysis. Nucleic Acids Res. 37, 5578–5588 (2009).
Bycroft, M., Hubbard, T. J., Proctor, M., Freund, S. M. & Murzin, A. G. The solution structure of the S1 RNA binding domain: a member of an ancient nucleic acid-binding fold. Cell 88, 235–242 (1997).
Shi, W. et al. Pyrazinamide inhibits trans-translation in Mycobacterium tuberculosis. Science 333, 1630–1632 (2011).
Worth, C. L., Gong, S. & Blundell, T. L. Structural and functional constraints in the evolution of protein families. Nat. Rev. Mol. Cell Biol. 10, 709–720 (2009).
Ganesan, P. & Ramalingam, R. Investigation of structural stability and functionality of homodimeric gramicidin towards peptide-based drug: a molecular simulation approach. J. Cell. Biochem, https://doi.org/10.1002/jcb.27765 (2018).
Bartlett, G. J., Borkakoti, N. & Thornton, J. M. Catalysing new reactions during evolution: economy of residues and mechanism. J. Mol. Biol. 331, 829–860 (2003).
Kosloff, M. & Kolodny, R. Sequence-similar, structure-dissimilar protein pairs in the PDB. Proteins 71, 891–902 (2008).
Liu, H. & Yao, X. Molecular basis of the interaction for an essential subunit PA-PB1 in influenza virus RNA polymerase: insights from molecular dynamics simulation and free energy calculation. Mol. Pharm. 7, 75–85 (2010).
Xue, W., Pan, D., Yang, Y., Liu, H. & Yao, X. Molecular modeling study on the resistance mechanism of HCV NS3/4A serine protease mutants R155K, A156V and D168A to TMC435. Antiviral Res. 93, 126–137 (2012).
Hou, T., McLaughlin, W. A. & Wang, W. Evaluating the potency of HIV-1 protease drugs to combat resistance. Proteins 71, 1163–1174 (2008).
Ding, B., Li, N. & Wang, W. Characterizing binding of small molecules. II. Evaluating the potency of small molecules to combat resistance based on docking structures. J. Chem. Inf. Model. 53, 1213–1222 (2013).
Xue, W., Liu, H. & Yao, X. Molecular mechanism of HIV-1 integrase-vDNA interactions and strand transfer inhibitor action: a molecular modeling perspective. J. Comput. Chem. 33, 527–536 (2012).
khan, M. T. et al. Pyrazinamide resistance and mutations in pncA among isolates of Mycobacterium tuberculosis from Khyber Pakhtunkhwa, Pakistan. BMC Infect. Dis. 19, 116 (2019).
Khan, M. T. et al. Pyrazinamide-resistant mycobacterium tuberculosis isolates from Khyber Pakhtunkhwa and rpsA mutations. J. Biol. Regul. Homeost. Agents 32, 705–709 (2018).
Khan, M. T., Rehaman, A. U., Junaid, M., Malik, S. I. & Wei, D.-Q. Insight into novel clinical mutants of RpsA-S324F, E325K, and G341R of Mycobacterium tuberculosis associated with pyrazinamide resistance. Comput. Struct. Biotechnol. J, https://doi.org/10.1016/j.csbj.2018.09.004 (2018).
Junaid, M., Khan, M. T., Malik, S. I. & Wei, D.-Q. Insights into the mechanisms of pyrazinamide resistance of three pyrazinamidase mutants N11K, P69T and D126N. J. Chem. Inf. Model, https://doi.org/10.1021/acs.jcim.8b00525 (2018).
Khan, M. T. et al. Pyrazinamide resistance and mutations L19R, R140H, and E144K in Pyrazinamidase of Mycobacterium tuberculosis. J. Cell. Biochem, https://doi.org/10.1002/jcb.27989 (2018).
Khan, M. T. et al. Prevalence of Pyrazinamide Resistance in Khyber Pakhtunkhwa, Pakistan. Microb. Drug Resist. Larchmt. N, https://doi.org/10.1089/mdr.2017.0234 (2018).
Akhmetova, A. et al. Mutations in the pncA and rpsA genes among 77 Mycobacterium tuberculosis isolates in Kazakhstan. Int. J. Tuberc. Lung Dis. 19, 179–184 (2015).
Shi, W. et al. Aspartate decarboxylase (PanD) as a new target of pyrazinamide in Mycobacterium tuberculosis. Emerg. Microbes Infect. 3, e58 (2014).
Nusrath Unissa, A. & Hanna, L. E. Molecular mechanisms of action, resistance, detection to the first-line anti tuberculosis drugs: Rifampicin and pyrazinamide in the post whole genome sequencing era. Tuberculosis 105, 96–107 (2017).
Liu, X. et al. Molecular dynamics simulations and novel drug discovery. Expert Opin. Drug Discov. 13, 23–37 (2018).
He, M., Li, W., Zheng, Q. & Zhang, H. A molecular dynamics investigation into the mechanisms of alectinib resistance of three ALK mutants. J. Cell. Biochem. 119, 5332–5342 (2018).
Miller, M. P. & Kumar, S. Understanding human disease mutations through the use of interspecific genetic variation. Hum. Mol. Genet. 10, 2319–2328 (2001).
Suemori, A. Conserved and non-conserved residues and their role in the structure and function of p-hydroxybenzoate hydroxylase. Protein Eng. Des. Sel. PEDS 26, 479–488 (2013).
Frieden, C. ApoE: The role of conserved residues in defining function. Protein Sci. 24, 138–144 (2015).
Nagasundaram, N. et al. Analysing the Effect of Mutation on Protein Function and Discovering Potential Inhibitors of CDK4: Molecular Modelling and Dynamics Studies. Plos One 10 (2015).
Aggarwal, M. et al. Role of pnc A gene mutations W68R and W68G in pyrazinamide resistance. J. Cell. Biochem. 13, 593 (2017).
Vats, C. et al. Mechanistic analysis elucidating the relationship between Lys96 mutation in Mycobacterium tuberculosis pyrazinamidase enzyme and pyrazinamide susceptibility. BMC Genomics 1 16 SRC-GoogleScholar (2015).
Doss, C. G. P. et al. Screening of mutations affecting protein stability and dynamics of FGFR1—A simulation analysis. Appl. Transl. Genomics 1, 37–43 (2012).
Chen, J. & Shen, B. Computational Analysis of Amino Acid Mutation: A Proteome Wide Perspective, https://doi.org/10.2174/157016409789973734 (2009).
Lobanov, M. Y., Bogatyreva, N. S. & Galzitskaya, O. V. Radius of gyration as an indicator of protein structure compactness. Mol. Biol. 42, 623–628 (2008).
Smilgies, D.-M. & Folta-Stogniew, E. Molecular weight–gyration radius relation of globular proteins: a comparison of light scattering, small-angle X-ray scattering and structure-based data. J. Appl. Crystallogr. 48, 1604–1606 (2015).
Yoon, J.-H., Nam, J.-S., Kim, K.-J. & Ro, Y.-T. Characterization of pncA mutations in pyrazinamide-resistant Mycobacterium tuberculosis isolates from Korea and analysis of the correlation between the mutations and pyrazinamidase activity. World J. Microbiol. Biotechnol. 30, 2821–2828 (2014).
Tyukhtenko, S. et al. Effects of Distal Mutations on the Structure, Dynamics and Catalysis of Human Monoacylglycerol Lipase. Sci. Rep. 8, 1719 (2018).
Lee, J. & Goodey, N. M. Catalytic contributions from remote regions of enzyme structure. Chem. Rev. 111, 7595–7624 (2011).
Swier, L. J. Y. M. et al. Insight into the complete substrate-binding pocket of ThiT by chemical and genetic mutations. MedChemComm 8, 1121–1130 (2017).
Coleman, R. G. & Sharp, K. A. Protein Pockets: Inventory, Shape, and Comparison. J. Chem. Inf. Model. 50, 589–603 (2010).
Gerlt, J. A., Kreevoy, M. M., Cleland, W. & Frey, P. A. Understanding enzymic catalysis: the importance of short, strong hydrogen bonds. Chem. Biol. 4, 259–267 (1997).
Hubbard, R. E. & Haider, M. K. Hydrogen Bonds in Proteins: Role and Strength. in eLS, https://doi.org/10.1002/9780470015902.a0003011.pub2 (American Cancer Society, 2010).
Pace, C. N. et al. Contribution of hydrogen bonds to protein stability. Protein Sci. Publ. Protein Soc. 23, 652–661 (2014).
Zhang, Y., Zhang, J., Cui, P., Zhang, W. & Zhang, Y. Identification of novel efflux proteins Rv0191, Rv3756c, Rv3008 and Rv1667c involved in pyrazinamide resistance in Mycobacterium tuberculosis. Antimicrob. Agents Chemother. AAC, 00940–17, https://doi.org/10.1128/AAC.00940-17 (2017).
Pardini, M. et al. Usefulness of the BACTEC MGIT 960 system for isolation of mycobacterium tuberculosis from sputa subjected to long-term storage. J. Clin. Microbiol. 45, 575–576 (2007).
Said, H. M., Ismail, N., Osman, A., Velsman, C. & Hoosen, A. A. Evaluation of TBc Identification Immunochromatographic Assay for Rapid Identification of Mycobacterium tuberculosis Complex in Samples from Broth Cultures∇. J. Clin. Microbiol. 49, 1939–1942 (2011).
Machado, D. et al. Assessment of the BD MGIT TBc Identification Test for the Detection of Mycobacterium tuberculosis Complex in a Network of Mycobacteriology Laboratories. BioMed Research International, https://doi.org/10.1155/2014/398108 (2014).
Aono, A., Hirano, K., Hamasaki, S. & Abe, C. Evaluation of BACTEC MGIT 960 PZA medium for susceptibility testing of Mycobacterium tuberculosis to pyrazinamide (PZA): compared with the results of pyrazinamidase assay and Kyokuto PZA test. Diagn. Microbiol. Infect. Dis. 44, 347–352 (2002).
Demers, A.-M. et al. Direct Susceptibility Testing of Mycobacterium tuberculosis for Pyrazinamide by Use of the Bactec MGIT 960 System. J. Clin. Microbiol. 54, 1276–1281 (2016).
Mustazzolu, A., Iacobino, A., Giannoni, F., Piersimoni, C. & Fattorini, L. Improved Bactec MGIT 960 Pyrazinamide Test Decreases Detection of False Mycobacterium tuberculosis Pyrazinamide Resistance. J. Clin. Microbiol. 55, 3552–3553 (2017).
Buck, G. E., O’Hara, L. C. & Summersgill, J. T. Rapid, simple method for treating clinical specimens containing Mycobacterium tuberculosis to remove DNA for polymerase chain reaction. J. Clin. Microbiol. 30, 1331–1334 (1992).
Kirschner, P. et al. Genotypic identification of mycobacteria by nucleic acid sequence determination: report of a 2-year experience in a clinical laboratory. J. Clin. Microbiol. 31, 2882–2889 (1993).
Xia, Q. et al. Phenotypic and Genotypic Characterization of Pyrazinamide Resistance among Multidrug-Resistant Mycobacterium tuberculosis Isolates in Zhejiang, China. Antimicrob. Agents Chemother. 59, 1690–1695 (2015).
Zhang, S. et al. Mutations in panD encoding aspartate decarboxylase are associated with pyrazinamide resistance in Mycobacterium tuberculosis. Emerg. Microbes Infect. 2, e34 (2013).
Dong, C. & Yu, B. Mutation surveyor: an in silico tool for sequencing analysis. Methods Mol. Biol. Clifton NJ 760, 223–237 (2011).
Berman, H. M. et al. The Protein Data Bank. Nucleic Acids Res. 28, 235–242 (2000).
L DeLano, W. The PyMOL Molecular Graphics System (2002) DeLano Scientific, Palo Alto, CA, USA, http://www.pymol.org. (2002).
Kim, S. et al. PubChem Substance and Compound databases. Nucleic Acids Res. 44, D1202–D1213 (2016).
Pettersen, E. F. et al. UCSF Chimera–a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 (2004).
Rodríguez-Guerra Pedregal, J. & Maréchal, J.-D. PyChimera: use UCSF Chimera modules in any Python 2.7 project. Bioinformatics 34, 1784–1785 (2018).
Schneidman-Duhovny, D., Inbar, Y., Nussinov, R. & Wolfson, H. J. PatchDock and SymmDock: servers for rigid and symmetric docking. Nucleic Acids Res. 33, W363–W367 (2005).
Norel, R., Petrey, D., Wolfson, H. J. & Nussinov, R. Examination of shape complementarity in docking of unbound proteins. In Proteins: Structure, Function, and Genetics 36 307–317 (1999).
Zhang, Q., Sanner, M. & Olson, A. J. Shape Complementarity of Protein-Protein Complexes at Multiple Resolutions. Proteins 75, 453–467 (2009).
Binkowski, T. A., Naghibzadeh, S. & Liang, J. CASTp: Computed Atlas of Surface Topography of proteins. Nucleic Acids Res. 31, 3352–3355 (2003).
Wu, Y., Tepper, H. & Voth, G. A. Flexible simple point-charge water model with improved liquid-state properties. Phys 024503 124 SRC-GoogleScholar (2006).
Berendsen, H. J. C., van der Spoel, D. & van Drunen, R. GROMACS: A message-passing parallel molecular dynamics implementation. Comput. Phys. Commun. 91, 43–56 (1995).
Parrinello, M. & Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 52, 7182–7190 (1981).
Hess, B., Bekker, H., Berendsen, H. J. C. & Fraaije, J. G. E. M. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 18, 1463–1472 (1997).
Balsera, M. A., Wriggers, W., Oono, Y. & Schulten, K. Principal Component Analysis and Long Time Protein Dynamics. J. Phys. Chem. 100, 2567–2572 (1996).
Sittel, F., Jain, A. & Stock, G. Principal component analysis of molecular dynamics: On the use of Cartesian vs. internal coordinates. J. Chem. Phys. 141, 014111 (2014).
Ernst, M., Sittel, F. & Stock, G. Contact- and distance-based principal component analysis of protein dynamics. J. Chem. Phys. 143, 244114 (2015).
Kume, A. et al. Exploring high-affinity binding properties of octamer peptides by principal component analysis of tetramer peptides. J. Biosci. Bioeng. 123, 230–238 (2017).
Ouaray, Z., ElSawy, K. M., Lane, D. P., Essex, J. W. & Verma, C. Reactivation of mutant p53: Constraints on mechanism highlighted by principal component analysis of the DNA binding domain. Proteins 84, 1443–1461 (2016).
Iida, S. et al. Variation of free-energy landscape of the p53 C-terminal domain induced by acetylation: Enhanced conformational sampling. J. Comput. Chem. 37, 2687–2700 (2016).
Tripathi, S., Srivastava, G. & Sharma, A. Molecular dynamics simulation and free energy landscape methods in probing L215H, L217R and L225M βI-tubulin mutations causing paclitaxel resistance in cancer cells. Biochem. Biophys. Res. Commun. 476, 273–279 (2016).
David, C. C. & Jacobs, D. J. Principal Component Analysis: A Method for Determining the Essential Dynamics of Proteins. Methods Mol. Biol. Clifton NJ 1084, 193–226 (2014).
Amadei, A., Linssen, A. B. & Berendsen, H. J. Essential dynamics of proteins. Proteins 17, 412–425 (1993).
Amadei, A. et al. Essential dynamics of pr oteins. Proteins Struct Funct Bioinf Sel Ction Vitro Mutants Pyrazinamideresistant Mycobacterium Tuberc. Anti 17 SRC-GoogleScholar, 412–425 (2002).
Hoang, T. X., Trovato, A., Seno, F., Banavar, J. R. & Maritan, A. Geometry and symmetry presculpt the free-energy landscape of proteins. Proc. Natl. Acad. Sci. 101, 7960–7964 (2004).
Acknowledgements
We are thankful for technical support by project director Dr Maqsood, PTRL KPK, Dr Anwar Sheed and Dr Sajid Ali, Microbiologist at PTRL. The works was supported by the grants from the Key Research Area Grant 2016YFA0501703 of the Ministry of Science and Technology of China, the National Natural Science Foundation of China (Contract no. 61832019, 61503244), the State Key Lab on Microbial Metabolism, and Joint Research Funds for Medical and Engineering & Scientific Research at Shanghai Jiao Tong University (YG2017ZD14). The simulations in this work were supported by the Center for High-Performance Computing, Shanghai Jiao Tong University. This study was also supported by Higher Education Commission Islamabad, Pakistan under IRSIP No: 1–8/HEC/HRD/2017/8392.
Author information
Authors and Affiliations
Contributions
Muhammad Tahir Khan performed experiments, analyzed and wrote manuscript. Abbas Khan performed MD simulation and analysis. Ashfaq Ur Rehman prepared the figures. Khalid Akhtar, revised the discussion, conclusion, and the plot. Yanjie Wang, analyzed the data, revised the manuscript. Shaukat Iqbal Malik, designed the research, revised the manuscript and experimental data. Dong-Qing Wei, designed the research, revised the manuscript and supervised the simulations and analysis.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Khan, M.T., Khan, A., Rehman, A.U. et al. Structural and free energy landscape of novel mutations in ribosomal protein S1 (rpsA) associated with pyrazinamide resistance. Sci Rep 9, 7482 (2019). https://doi.org/10.1038/s41598-019-44013-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-44013-9
This article is cited by
-
Theophylline as a quorum sensing and biofilm inhibitor in Pseudomonas aeruginosa and Chromobacterium violaceum
International Microbiology (2024)
-
In silico protein engineering shows that novel mutations affecting NAD+ binding sites may improve phosphite dehydrogenase stability and activity
Scientific Reports (2023)
-
Mutational analysis of catalytic site domain of CCHFV L RNA segment
Journal of Molecular Modeling (2023)
-
Comparative structural study of selective and non-selective NSAIDs against the enzyme cyclooxygenase-2 through real-time molecular dynamics linked to post-dynamics MM-GBSA and e-pharmacophores mapping
Journal of Molecular Modeling (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
