
Abstract
Melanoma is the most lethal cutaneous cancer. New drugs have recently appeared; however, not all patients obtain a benefit of these new drugs. For this reason, it is still necessary to characterize melanoma at molecular level. The aim of this study was to explore the molecular differences between melanoma tumor subtypes, based on BRAF and NRAS mutational status. Fourteen formalin-fixed, paraffin-embedded melanoma samples were analyzed using a high-throughput proteomics approach, combined with probabilistic graphical models and Flux Balance Analysis, to characterize these differences. Proteomics analyses showed differences in expression of proteins related with fatty acid metabolism, melanogenesis and extracellular space between BRAF mutated and BRAF non-mutated melanoma tumors. Additionally, probabilistic graphical models showed differences between melanoma subgroups at biological processes such as melanogenesis or metabolism. On the other hand, Flux Balance Analysis predicts a higher tumor growth rate in BRAF mutated melanoma samples. In conclusion, differential biological processes between melanomas showing a specific mutational status can be detected using combined proteomics and computational approaches.
Similar content being viewed by others


Introduction
Melanoma is the most lethal cutaneous cancer, with over 11,000–15,000 estimated deaths in the United States and Europe every year1,2. Better understanding of the molecular biology of this tumor has allowed the development of new effective drugs for the treatment of advanced disease, both in the fields of targeted therapies and immunotherapy3. However, as not all patients obtain a benefit from new drugs, further insight into the biology of melanoma is needed.
Gene signatures, genomic hybridization, whole-exome genome sequencing, microRNA analysis and other techniques have widely addressed the genomic landscape of melanoma, contributing to significant advances4,5. Given the heterogeneity of melanoma and the complex interaction of this tumor with the immune system, the need for combination of biomarkers assays has been recently proposed to properly analyze the disease6.
Proteins determine cell phenotype, so proteomics analyses offer the possibility to measure the biological effects caused by genomic abnormalities7. The most adapted technique for massive quantification of proteins is mass spectrometry. The recent technological advances in the field allow the identification and quantification of thousands of proteins per sample. Therefore, proteomics offers complementary information to that provided by standard pathology and genomics. We recently demonstrated the feasibility of high-throughput label-free quantitative proteomics to analyze breast cancer from paraffin-embedded samples8. In the present study we sought to determine whether high-throughput proteomics combined with computational approaches, such as probabilistic graphical models and Flux Balance Analysis, are useful tools to explore functional differences between groups of melanoma tumors.
Results
Patients and samples
Primary melanoma samples coming from 14 patients with advanced disease were included. Samples were split into three groups according to mutational status: BRAF-mutant (n = 3), NRAS-mutant (n = 5) or double negative (n = 6). BRAF and NRAS mutations had been previously determined in local laboratories with standard polymerase chain reaction-based tests.
Mass-spectrometry analysis
FFPE melanoma tumor samples were analysed by mass-spectrometry (Supplementary Table S1). 4,006 protein groups were identified, of which 1,606 present detectable measurements in at least 75% of the samples and at least two unique peptides. Label-free quantification data from these 1,606 proteins were employed for consequent analyses.
Differential protein expression patterns between subtypes
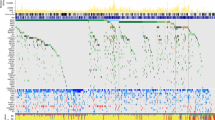
A Significance Analysis of Microarrays (SAM) was done to find differences among samples at the protein level. Seventeen proteins were found differentially expressed between BRAF mutated and BRAF wild type tumors, all of them underexpressed in BRAF-mutated tumors (Fig. 1, Supplementary Table S2). These proteins are mainly related with fatty acid metabolism.

Differential proteins obtained by Significance Analysis of Microarrays between BRAF positive and negative tumors. 17 proteins were found differentially expressed between BRAF positive and BRAF negative tumors (green = underexpressed, red = overexpressed; BRAF-0 = BRAF negative, BRAF-1 = BRAF positive, NRAS-0 = NRAS negative, NRAS-1 = NRAS positive).
In addition, delta values between BRAF-mutated and BRAF-wild type, and NRAS-mutated and NRAS-wild type tumors were calculated (Supplementary Table S3). Delta values higher than 1.5 or lower than −1.5 were used to perform gene ontology analyses as well. Proteins related with keratinization, epidermis development and cytoskeleton were underexpressed, whereas proteins involved in melanogenesis and extracellular space were overexpressed in BRAF-mutant as compared with BRAF-wild type samples. SAM and delta analyses did not find significant differences between NRAS-mutant and NRAS-wild type tumors.
Probabilistic graphical model and functional node activity measurements
A probabilistic graphical model (PGM) was assembled using proteomics data with any a priori information. The resulting network was handled to build a functional structure, as described in previous works9,10,11. The network was split into thirteen branches, and gene ontology analyses were performed to establish functional structure. Finally, twelve principal functions were assigned to different branches (from now on, functional nodes) and there was a branch to which no main function could be assigned (Fig. 2, Supplementary Table S4).

Probabilistic graphical model built using protein expression data from melanoma tumors which showed a functional structure. The network was divided into thirteen functional nodes and one branch without any function (yellow). Proteins are represented by squares.
Functional node activity measurements were calculated for each functional node using proteins related with the main assigned function and a comparison between BRAF-mutant, NRAS-mutant and double-negative groups was performed. Although the limited number of samples did not allow seeing significant differences, some trends in functional activities were found. For instance, NRAS-mutant had a lower melanosome functional node activity than BRAF-mutant or double negative tumors. On the other hand, BRAF-mutant tumors had a higher metabolism functional node activity than NRAS-mutant or double negative (Fig. 3).

Activity measurements calculated for each network functional node according to biomarkers features. Boxplots comparing functional node activities between BRAF (n = 3), NRAS (n = 5) and double negative (n = 6) melanoma tumors. ***p < 0.0001, **p < 0.001, *p < 0.05.
Flux balance analysis
Flux Balance Analysis is a computational approach to assess biochemical networks through the calculation of the flow of metabolites through this network. FBA can be used to calculate the growth rate of an organism or a tumor or the rate of generation of a metabolite. Our model suggest that BRAF mutated tumors may have a higher tumor growth rate than the two other subtypes (Fig. 4, Supplementary Table S5).

FBA predicted tumor growth rates. FBA predicted a higher growth rate for BRAF mutated tumors. The medium line represents the mean and the whiskers are the standard deviation (BRAF pos = BRAF positive, NRAS pos = NRAS positive, DOUBLE neg = double negative).
Discussion
In this study, proteomics coupled with probabilistic graphical models and flux balance analysis were used to describe differences between melanoma biomarker subgroups in melanoma samples.
Proteomics analyses from FFPE samples are complicated because fixation process provokes chemical alterations. Actually, the number of works in this field in melanoma is scarce. Rezaul et al. analysed by MS one microdissected FFPE melanoma sample and identify 935 proteins, confirming the presence of some of them by immunohistochemistry12. A previous work comparing two different FFPE samples of melanocytic nevus and advanced melanoma identified two proteins differentially expressed between those type of tumors13. Moreover, Byrum et al. compared primary melanoma, advanced melanoma and benign nevi and identified 171 varying proteins between these diseases14. In this work, we demonstrated that it is also possible, using proteomics and FFPE samples, to see more subtle differences such as differential proteins between mutational-status groups.
Mass-spectrometry workflow allowed the detection of 1,606 proteins with detectable expression in at least 75% of the samples and two unique peptides. Differences in fatty acid metabolism, cytoskeleton or keratinization were observed between BRAF–mutant and BRAF-wild type tumors. Also, it seems that differences in functions such as melanogenesis or metabolism may be existed between subgroups.
SAM and gene ontology analysis found 17 proteins differentially expressed between BRAF-mutant and the two other subgroups (NRAS-mutant and double-negative), all of them were underexpressed in BRAF-mutated tumors. These proteins are mainly involved in fatty acid metabolism: acyl-Co A dehydrogenases P11310 (acyl-CoA dehydrogenase medium chain, ACADM), P42765 (acetyl-CoA acyltransferase 2, ACAA2) and P49748 (acyl-CoA dehydrogenase very long chain, ACADVL). It has been previously described that melanoma cells reduce fatty acids from glutamine through tricarboxylic acid cycle15. On the other hand, some of the proteins underexpressed in BRAF-mutated tumors have antiproliferative functions. For instance, Q9Y3Z3 (histidine/aspartate (HD)- domain containing protein 1, SAMHD1) is implicated in regulation of DNA replication and damage repair and it is proposed to have antiproliferative and tumor suppressive functions in many cancers16. Q96CX2 (potassium channel tetradimerization domain containing 12, KCTD12) inhibits proliferation in uveal melanoma cells17. O14745 (SLC9A3R1) is involved in suppressing breast cancer cells proliferation18. Other proteins of those 17 were previously related with melanoma or melanogenesis processes. For example, P31040 (succinate dehydrogenase complex flavoprotein subunit A,SDHA), which encodes a major catalytic subunit of succinate-ubiquinone reductase, a complex of the mitochondria chain, it was previously related with melanogenesis process19. Another protein differentially expressed is P00488 (coagulation factor XIII, F13A1) which it was previously associated with chemotherapy response in melanoma tumors20. P08133 (annexin A6, ANXA6) acts as a tumor suppressor in skin cancer and it is involved in in the conversion of melanocytes to malignant melanomas21. Lastly, it was previously described that metastatic melanoma tumors have a decreased expression of signal transducer and activator of transcription P42224 (signal transducer and activator of transcription 1, STAT1) and it could be one of the mechanism by which melanoma can evade immune detection22. Finally, P04899 (G protein subunit alpha i2, GNAI2) contributes to melanoma cell growth23. O60749 (Sorting nexin 2, SNX2) is involved in membrane trafficking of growth factor receptors including epidermal growth factor receptor and c-Met24. P05107 (integrin subunit beta 2, ITGB2) participates in cell adhesion as well as cell-surface mediated signalling and it is correlated with survival in other cancers such as renal or colorectal tumors25,26. As far we know, P14317 (hematopoietic cell-specific Lyn substrate, HCLS1), P63027 (vesicle-associated membrane protein 2, VAMP2), P16402 (histone cluster 1H1 family member d, HIST1H1D) and Q9H0W9 (chromosome 11 open reading frame 54, C11orf54) were not previously related with melanoma or other cancers.
Differential analyses did not show differences between NRAS-mutant and NRAS-wild type tumors, which are attributable to the small sample size. The present study was limited in this regard because it was designed just as a proof of principle that high-throughput proteomics can be used to study clinical samples of melanoma. Future studies with larger sample size will be needed to establish significant differences among subtypes. Interestingly, it seems that delta analyses and SAM provide complementary information about different protein expression patterns, because differential proteins provided by these two analyses were different and they were also related to different biological processes.
Besides that, a PGM was used to generate a network based on protein expression data. It is remarkable that, despite the low number of samples, the PGM clearly showed a functional structure. This type of analysis previously demonstrated its utility to characterize other tumor types such as bladder carcinoma or breast cancer and may complement the information provided by genomics10,11. Although there are no significant differences in functional node activities between groups, the PGM provides some biological processes as candidates to be deregulated among subtypes. Despite the fact that there are a node without an assigned function, it is remarkable that this node contains proteins related with lipoprotein metabolism and melanine synthesis, both previously related with melanoma progression27,28. What is more, despite the reduced number of samples, the high growth rate in BRAF-mutant tumors suggested by FBA, although it is not statistically significant, agrees with previous observations, as BRAF-mutated tumors are more proliferative29.
Our study demonstrates that proteomics and computational methods can be applied to the study of formalin-fixed, paraffin-embedded melanoma samples, suggesting that melanoma subgroups, defined by mutational status, present molecular differences. Despite the reduced number of samples analyzed, the probabilistic graphical model showed a functional structure and allowed characterizing differences at biological processes regarding melanoma mutational status. Additionally, flux balance analysis was capable to predict differences at tumor growth rate between these groups. In conclusion, this proof-of-principle work demonstrate the usefulness of proteomics and computational approaches in the molecular characterization of melanoma and suggest some proteins and biological processes that could be used as therapeutic targets after proper validation in larger cohorts.
Methods
Samples
Fourteen melanoma cancer patients were included in the study. FFPE samples were retrieved from Biobanks in IdiPAZ, Hospital Universitario Gregorio Marañón and Hospital Universitario Ramón y Cajal, all integrated in the Spanish Hospital Biobank Network (RetBioH; http://www.redbiobancos.es/). Patients provided informed consent. All experiments were performed in accordance with relevant guidelines and regulations. The histopathological features of each sample were reviewed by an experienced pathologist to confirm diagnosis and tumor content. Eligible samples had to include at least 50% of tumor cells. Approval from the Ethical Committees of Hospital Universitario La Paz was obtained for the conduct of the study.
Mass-spectrometry analysis, protein identification and quantification
Proteins were isolated from FFPE samples as previously described30. Peptides were desalted using C18 stage tips, dried and resolubilized with 3% acetonitrile, 0.1% formic acid. Samples were analyzed on a QExactive mass spectrometer coupled to a nano EasyLC 1000 (Thermo Fisher Scientific) as previously described31 Briefly, 3 μL of each sample were loaded. Samples were acquired in a randomized order. Elution was performed at a flow rate of 300 nL/min. Mass spectrometer was operated in data-dependent mode (DDA), acquiring a full-scan MS spectra followed by HCD (higher-energy collision dissociation) fragmentation on the twelve most intense signals per cycle. The samples were acquired using internal lock mass calibration on m/z 371.1010 and 445.1200.
The acquired raw MS data were processed by MaxQuant (version 1.5.2.8), protein identification was done using the integrated Andromeda search engine as previously described31. Briefly, spectra were searched against a forward Swiss Prot-human database, concatenated to a reversed decoyed fasta database (NCBI taxonomy ID9606, release date 2014-05-06). Carbamidomethylation of cysteine, methionine oxidation and N-terminal protein acetylation were set as modifications. Enzyme specificity was set to trypsin/P. The maximum false discovery rate (FDR) was set to 0.01 for peptides and 0.05 for proteins. Intensity, defined as the sum of the precursor intensities of all identified peptides for the respective protein group, was used for protein abundance calculation. All the mass spectrometry raw data files acquired in this study can be downloaded from Chorus (http://chorusproject.org) under the project name “Melanoma proteomics in FFPE samples”.
Following MS workflow, identified protein groups were filtered by the presence of at least two unique peptides. Also, detectable expression in at least 75% of the total of samples was required for a protein to be included in subsequent analyses. Additionally, batch effects were removed using limma package32 and R v 3.2.533.
Protein differential expression analyses
Significance Analysis of Microarrays (SAM) was performed using MeV to find significant differences in protein expression among samples34. SAM consists on a t-test corrected by permutations over the number of samples. The significance is determined using a delta parameter, based on the false discovery rate (FDR)35. In this analysis, the threshold was a delta = 1.6, equivalent to a FDR = 0.61 (90th percentile). Protein expression patterns were also compared calculating delta expression values by subtracting the protein expression value for each biomarker status against the expression value for the rest of the tumor samples. Proteins showing a change in their delta expression higher than 1.5 or lower than −1.5 were selected.
Probabilistic graphical model and activity measurements
A PGM was constructed using R v 3.2.533 and grapHD package36. The correlation coefficient was selected as the associative method9,10,11. PGM are undirected acyclic graphs based on obtaining the spanning tree that maximizes the likelihood and then favoring the graph with the simplest structure preserving the decomposability of tree and minimizing the Bayesian Information Criterion (BIC) as well37. Protein expression data was used to build the network with any a priori information. This type of networks reflects relationships between expression patterns, i.e. proteins associated in the network by an edge have related expression patterns. The network was divided into several branches or functional nodes. Network functional structure was explored by Gene Ontology analysis. A major biological function was assigned to each branch. Activity measurements were then calculated by the mean expression of all the proteins in the functional node related to the assigned function. Gene Ontology Analyses were performed in DAVID webtool. “Homo sapiens” was set as background and only Biocarta, GOTERM-FAT and KEGG categories were selected.
Flux balance analysis
Flux Balance Analysis (FBA) was performed using COBRA Toolbox38 and whole metabolism human reconstruction Recon 239 both available for MATLAB. As an objective function, biomass reaction supplied by the Recon 2 was used as representative of tumor growth rate. Recon 2 is a human metabolic model formed by 7,440 reactions and 5,063 metabolites grouped in 101 metabolic pathways. These models could incorporate gene or protein expression data in order to make accurate predictions by solving Gene-Protein-Reaction (GPR) rules which contains the association between proteins and reactions. Then, proteomics expression data from the 1,606 proteins previously identified was incorporated into the model as described in previous works10. Briefly, GPR rules, which associate genes and proteins with the enzymes involved in each metabolic reaction, were estimated using the sum for “ORs” expressions and minimum for “ANDs” expressions. Then, E-flux algorithm40 was used to normalize the GPR values dividing by the maximum value in each tumor and incorporate protein expression data into the model. Predicted values of biomass were compared between BRAF, NRAS and double negative tumors.
Statistical analyses
GraphPad Prism v6 was used for statistical analyses, whereas Cytoscape was used for network analysis.
Data Availability
All data employed in this study are provided as supplementary files.
References
Ferlay, J. et al. Cancer incidence and mortality patterns in Europe: estimates for 40 countries in 2012. Eur J Cancer 49, 1374–1403, https://doi.org/10.1016/j.ejca.2012.12.027 (2013).
Ferlay, J. et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer 136, E359–386, https://doi.org/10.1002/ijc.29210 (2015).
Dummer, R., Keilholz, U. & Committee, E. G. appendix 2: Cutaneous melanoma (2): eUpdate published online September 2016, (http://www.esmo.org/Guidelines/Melanoma). Ann Oncol 27, v136–v137, https://doi.org/10.1093/annonc/mdw432 (2016).
Lin, W. M. & Fisher, D. E. Signaling and Immune Regulation in Melanoma Development and Responses to Therapy. Annu Rev Pathol 12, 75–102, https://doi.org/10.1146/annurev-pathol-052016-100208 (2017).
Bauer, J. The Molecular Revolution in Cutaneous Biology: Era of Cytogenetics and Copy Number Analysis. J Invest Dermatol 137, e57–e59, https://doi.org/10.1016/j.jid.2016.11.043 (2017).
Blank, C. U., Haanen, J. B., Ribas, A. & Schumacher, T. N. CANCER IMMUNOLOGY. The “cancer immunogram”. Science 352, 658–660, https://doi.org/10.1126/science.aaf2834 (2016).
Ellis, M. J. et al. Connecting genomic alterations to cancer biology with proteomics: the NCI Clinical Proteomic Tumor Analysis Consortium. Cancer Discov 3, 1108–1112, https://doi.org/10.1158/2159-8290.CD-13-0219 (2013).
Gámez-Pozo, A. et al. Prediction of adjuvant chemotherapy response in triple negative breast cancer with discovery and targeted proteomics. PLoS One 12, e0178296, https://doi.org/10.1371/journal.pone.0178296 (2017).
Gámez-Pozo, A. et al. Combined Label-Free Quantitative Proteomics and microRNA Expression Analysis of Breast Cancer Unravel Molecular Differences with Clinical Implications. Can Res 75, 2243–2253 (2015).
Gámez-Pozo, A. et al. Functional proteomics outlines the complexity of breast cancer molecular subtypes. Scientific Reports 7, 10100, https://doi.org/10.1038/s41598-017-10493-w (2017).
de Velasco, G. et al. Urothelial cancer proteomics provides both prognostic and functional information. Sci Rep 7, 15819, https://doi.org/10.1038/s41598-017-15920-6 (2017).
Rezaul, K., Murphy, M., Lundgren, D. H., Wilson, L. & Han, D. K. Combined mass spectrometry- and immunohistochemistry-based approach to determine protein expression in archival melanoma–proof of principle. Pigment Cell Melanoma Res 23, 849–852, https://doi.org/10.1111/j.1755-148X.2010.00774.x (2010).
Byrum, S. et al. A quantitative proteomic analysis of FFPE melanoma. J Cutan Pathol 38, 933–936, https://doi.org/10.1111/j.1600-0560.2011.01761.x (2011).
Byrum, S. D. et al. Quantitative Proteomics Identifies Activation of Hallmark Pathways of Cancer in Patient Melanoma. J Proteomics Bioinform 6, 43–50, https://doi.org/10.4172/jpb.1000260 (2013).
Scott, D. A. et al. Comparative metabolic flux profiling of melanoma cell lines: beyond the Warburg effect. J Biol Chem 286, 42626–42634, https://doi.org/10.1074/jbc.M111.282046 (2011).
Kohnken, R., Kodigepalli, K. M. & Wu, L. Regulation of deoxynucleotide metabolism in cancer: novel mechanisms and therapeutic implications. Mol Cancer 14, 176, https://doi.org/10.1186/s12943-015-0446-6 (2015).
Luo, L. et al. Lentiviral-mediated overexpression of KCTD12 inhibits the proliferation of human uveal melanoma OCM-1 cells. Oncol Rep 37, 871–878, https://doi.org/10.3892/or.2016.5325 (2017).
Liu, H. et al. SLC9A3R1 stimulates autophagy via BECN1 stabilization in breast cancer cells. Autophagy 11, 2323–2334, https://doi.org/10.1080/15548627.2015.1074372 (2015).
Boulton, S. J. & Birch-Machin, M. A. Impact of hyperpigmentation on superoxide flux and melanoma cell metabolism at mitochondrial complex II. FASEB J 29, 346–353, https://doi.org/10.1096/fj.14-261982 (2015).
Azimi, A. et al. Proteomics analysis of melanoma metastases: association between S100A13 expression and chemotherapy resistance. Br J Cancer 110, 2489–2495, https://doi.org/10.1038/bjc.2014.169 (2014).
Qi, H. et al. Role of annexin A6 in cancer. Oncol Lett 10, 1947–1952, https://doi.org/10.3892/ol.2015.3498 (2015).
Osborn, J. L. & Greer, S. F. Metastatic melanoma cells evade immune detection by silencing STAT1. Int J Mol Sci 16, 4343–4361, https://doi.org/10.3390/ijms16024343 (2015).
Hermouet, S., Aznavoorian, S. & Spiegel, A. M. In vitro and in vivo growth inhibition of murine melanoma K-1735 cell by a dominant negative mutant alpha subunit of the Gi2 protein. Cell Signal 8, 159–166 (1996).
Ogi, S. et al. Sorting nexin 2-mediated membrane trafficking of c-Met contributes to sensitivity of molecular-targeted drugs. Cancer Sci 104, 573–583, https://doi.org/10.1111/cas.12117 (2013).
Boguslawska, J. et al. Expression of Genes Involved in Cellular Adhesion and Extracellular Matrix Remodeling Correlates with Poor Survival of Patients with Renal Cancer. J Urol 195, 1892–1902, https://doi.org/10.1016/j.juro.2015.11.050 (2016).
Cavalieri, D. et al. Analysis of gene expression profiles reveals novel correlations with the clinical course of colorectal cancer. Oncol Res 16, 535–548 (2007).
Pencheva, N. et al. Convergent multi-miRNA targeting of ApoE drives LRP1/LRP8-dependent melanoma metastasis and angiogenesis. Cell 151, 1068–1082, https://doi.org/10.1016/j.cell.2012.10.028 (2012).
Rodriguez-Leyva, I. et al. The Presence of Alpha-Synuclein in Skin from Melanoma and Patients with Parkinson’s Disease. Mov Disord Clin Pract 4, 724–732, https://doi.org/10.1002/mdc3.12494 (2017).
Wellbrock, C. et al. Oncogenic BRAF regulates melanoma proliferation through the lineage specific factor MITF. PLoS One 3, e2734, https://doi.org/10.1371/journal.pone.0002734 (2008).
Gámez-Pozo, A. et al. Shotgun proteomics of archival triple-negative breast cancer samples. Proteomics Clin Appl 7, 283–291, https://doi.org/10.1002/prca.201200048 (2013).
Trilla-Fuertes, L. et al. Molecular characterization of breast cancer cell response to metabolic drugs. Oncotarget 9, 9645–9660, https://doi.org/10.18632/oncotarget.24047 (2018).
Ritchie, M. et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acid. Research 43, e47 (2015).
R Foundation for Statistical Computing (2013).
Saeed, A. I. et al. TM4: a free, open-source system for microarray data management and analysis. Biotechniques 34, 374–378 (2003).
Tusher, V. G., Tibshirani, R. & Chu, G. Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci USA 98, 5116–5121, https://doi.org/10.1073/pnas.091062498 (2001).
Abreu, G., Edwards, D. & Labouriau, R. igh-Dimensional Graphical Model Search with the gRapHD R Package. Journal of Statistical Software 37, 1–18 (2010).
Lauritzen, S. Graphical Models. Oxford,UK.: Oxford University Press (1996).
Schellenberger, J. et al. Quantitative prediction of cellular metabolism with constraint-based models: the COBRA Toolbox v2.0. Nature Protocols 6, 1290–1307 (2011).
Thiele, I. et al. A community-driven global reconstruction of human metabolism. Nat Biotechnol 31, 419–425, https://doi.org/10.1038/nbt.2488 (2013).
Colijn, C. et al. nterpreting expression data with metabolic flux models: Predicting Mycobacterium tuberculosis mycolic acid production. PLOS Comput Bio, Vol. 5 (2009).
Acknowledgements
We want to particularly acknowledge the patients in this study for their participation and to IdiPAZ, as well as participating Biobanks. LT-F is supported by the Spanish Economy and Competitiveness Ministry (DI-15-07614). GP-V is supported by Conserjería de Educación, Juventud y Deporte of Comunidad de Madrid (IND2017/BMD7783). This work was supported by Instituto de Salud Carlos III, Spanish Economy and Competitiveness Ministry, Spain and co-funded by the FEDER program, “Una forma de hacer Europa” (PI15/01310). The funders had no role in the study design, data collection and analysis, decision to publish or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
R.L.-V. contributed the protein extraction. C.F. contributed the mass spectrometry data. A.G.-P., L.T.-F., G.P.-V., A.Z.-M. and M.F.-G. contributed the probabilistic graphical models. M.D.-A. contributed the GPR rule method. V.P.-B., I.M.-R., A.S. and E.E. contributed the clinical data and the analyses related. A.G.-P. and L.T.-F. contributed in the design of the study and the statistical and gene ontology analyses. L.T.-F. drafted the manuscript. L.T.-F. contributed the FBA analyses. J.A.F.V., A.G.-P. and E.E. conceived of the study, and participated in its design and interpretation. J.A.F.V. and E.E. coordinated the research.
Corresponding authors
Ethics declarations
Competing Interests
J.A.F.V., E.E. and A.G.-P. are shareholders in Biomedica Molecular Medicine S.L. A.G.-P., L.T.-F. and G.P.-V. are employees of Biomedica Molecular Medicine S.L. The other authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Trilla-Fuertes, L., Gámez-Pozo, A., Prado-Vázquez, G. et al. Melanoma proteomics suggests functional differences related to mutational status. Sci Rep 9, 7217 (2019). https://doi.org/10.1038/s41598-019-43512-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-43512-z
This article is cited by
-
BRAF activation by metabolic stress promotes glycolysis sensitizing NRASQ61-mutated melanomas to targeted therapy
Nature Communications (2022)
-
Proteogenomic analysis of melanoma brain metastases from distinct anatomical sites identifies pathways of metastatic progression
Acta Neuropathologica Communications (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
