
Abstract
Ferula sinkiangesis is a valuable medicinal plant that has become endangered. Improving the soil habitat of Ferula sinkiangesis can alleviate plant damage. Fungi play an important role in the soil, but current information on the fungal community structure in the habitat of Ferula sinkiangesis and the relationship between soil fungi and abiotic factors remains unclear. In this study, we analyzed the relative abundance of fungal species in the rhizosphere of Ferula sinkiangesis. Spearman correlation analysis showed that the abiotic factor total potassium (TK) significantly explained the alpha diversity of the fungal community. At altitude, available phosphorus (AP), nitrate nitrogen (NN) and TK were significantly associated with the fungal species. In addition, a two-way ANOVA showed that soil depth had no significant effects on the alpha diversity of rhizosphere and non-rhizosphere fungi. Interestingly, linear discriminant effect size (LEfSe) analysis indicated that different biomarkers were present at varying soil depths. These findings may be related to the growth and medicinal properties of Ferula Sinkiangensis.
Similar content being viewed by others
Introduction
More than 150 species of ferula exist globally, mainly in Southern Europe, North Africa Central Asia and their adjacent ares1. Approximately 26 species and a single variant are present in China2. Ferula Sinkiangensis is an early spring ephemeral plant of the Umbelliferae family. It begins to pump stems at the end of March and blooms the end of April. Full bloom occurs in mid-May3. At this time, the roots and stems of Ferula Sinkiangensis contain large amounts of milky white resin. The resin is termed ferulic and has high medicinal value including anticancer4,5, anti-influenza6,7 antibacterial8, and antioxidant activity9. Ferula sinkiangensis is the major species of the genus ferula2 and was included in the Pharmacopoeia of the People’s Republic of China in 197710. In recent years, Ferula sinkiangensis has approached extinction due to natural and human factors5. Considering its important medicinal value and endangered status, the protection of Ferula sinkiangensis is extremely important.
As the foundation of plant life, soil provides the nutrients for plant growth. Hiltner first coined the term “rhizosphere area” in 1904 to describe the soil affected by rhizosphere sediment exudates, mucus, and exfoliated cells11,12. These soils contain energy that can be supplied to microorganisms12,13,14. Diverse microbial communities concentrate in these soils. At the same time, microbes enriched in the roots of the plants contribute to the nutrient absorption and plant growth15,16, increasing the stress resistance16, and ecological adaptability of the plants. Both benefit from this interaction and participate in the complex rhizosphere ecosystem. In the rhizosphere niche, it is believed that the carbon sources released by plant roots enters the soil food chain through bacterial channels17,18. However, studies tracking the fate of root carbon using stable isotope detection (SIP) have shown that rhizosphere microorganisms add recently fixed plant 13 C to fungi to much higher levels than bacteria. This applies not only to mycorrhizal fungi but also to saprophytic fungi19,20. In addition, bacteria bind to mycorrhizal fungi and the mycelia of saprophytic fungi, passively dependent on the compounds secreted by the mycelia) and actively (obtaining the energy present in the mycelia) to obtain energy from the fungi19,21. It is noteworthy that because the cell walls of fungi vary in composition22,23, different bacteria may be selected from different bacterial communities. We may thus have underestimated the role of fungi in the rhizosphere niche of plants, which may explain the lack of studies on Ferula sinkiangensis rhizosphere fungi. We believe that this is not conducive to performing research on the rhizosphere ecological mechanisms of Ferula sinkiangensis to reveal the cause of extinction. Thus, this study explores the role of fungi in the rhizosphere community structure and provides diversity information on Ferula sinkiangensis.
Understanding the relationship between the structural diversity of the Ferula sinkiangensis fungal community and other factors is also considered. Biological factors, (plant species, developmental stages, physiological conditions) and abiotic factors (soil chemistry and planting systems) affect the structure and diversity of rhizosphere microbial communities24,25,26,27,28. Previous studies have shown that changes in water, nutrient availability, and salinity, regulate plant photosynthetic rates and growth, through altering the composition of rhizosphere bacterial communities through changes in the exudation patterns29,30. Marques and coworkers showed that both the growth and genotype of sweet potato affects the structure of the microbial community in the rhizosphere31. Ndour and colleagues indicated that the genetic characteristics of pearl millet determined the diversity of rhizosphere bacteria32 Studies in natural ecosystems have found that increasing nitrogen (N) reduces the microbial biomass33,34,35,36. Compost and biochar alter mycorrhization and tomato root exudation37. pH also affects the growth of soil microbial communities38,39. Root traits and microbial community interactions can be regulated by phosphorus availability and acquisition40. Rhizospheric microorganisms may be indirectly or directly affected by these factors, but in most cases can simultaneously exist. Therefore, in a complex habitat, understanding the key factors that drive and maintain microbial diversity can predict the response strategies of the ecosystem to future environmental changes. In this study, our key objective was to understand the fungal community structure and diversity by artificially changing factors that are convenient for human operation. On this basis, we aimed to understand the relationship between fungal communities, diversity, and other factors, so as to indirectly protect and increase plant productivity.
The rapid development of high-throughput sequencing technology has facilitated the diversity and structural analysis of rhizosphere bacterial communities41,42. Recording the diversity and richness of the rhizosphere bacterial community, permits an assessment of its contribution to the rhizosphere core, functional bacteria, and flora that changes with geological isolation and soil depth43. We used Illumina HiSeq sequencing and multivariate analysis to assess the diversity and structure of the rhizosphere bacterial community in Ferula sinkiangensis. To our knowledge, this is the first study that analyzes the diversity and structure of the rhizosphere fungi community in Ferula sinkiangensis through high-throughput sequencing.
Materials and Methods
Site description and experimental design
During the Ferula sinkiangensis growing season (April to May), we collected samples of Ferula sinkiangensis (Good growth, no pests and diseases) rhizosphere soil and non-rhizosphere soil from three different locations in Yining city, Xinjiang, China (Table S1). Rhizosphere and non-rhizosphere soil samples were collected using the Riley and Barber’s shaking method44,45. In the three different geographical conditions, nine ferula were randomly selected and the entire roots were excavated from the soil profile (Amongst the nine Ferula sinkiangesis, every three were divided into a group as a repeating unit). Soil samples were collected at a root depth of 0–5 cm, 5–15 cm and 15–40 cm. Samples were marked using a mixed marker system. An initial set of letters were used to indicate the site number, rhizosphere, and non-rhizosphere (E, R and S represent the rhizosphere areas at sites 1, 2 and 3, respectively), (NE, NR and NS represent the non-rhizosphere areas of sites 1, 2 and 3, respectively). The second number indicated the depth (1, 2 and 3 represent the 0–5, 5–15, and 15–40 cm depths, respectively) and the third number indicates the number of repetitions. For example, E2.3 represents the third replicate soil sample at a depth of 5–15 cm in the Ferula sinkiangesis root of the first site.
DNA extraction, amplification, and sequencing of the ITS rRNA gene
We extracted DNA from each sample using the Centrifugal Soil Genomic DNA Extraction Kit. DNA concentration and purity were monitored on a 1% agarose gel. The DNA was diluted to 1 ng/μL using sterile water. We used the Internal Transcribed Spacer (ITS) 36, technique to analyze nucleic acid sequences. ITS1 is located between 18 S and 5.8 S of the eukaryotic ribosomal rDNA sequence. Because it does not need to be added to mature ribosomes, ITS1 can withstand additional mutations during evolution. Its evolution rate is 10 times that of 18SrDNA, which is moderately conserved. The area can be used to study the classification order below. The ITS rRNA gene was amplified in the ITS1–5F region using ITS5–1737 Forward (GGAAGTAAAAGTCGTAACAAGG),andITS2-2043Reverse(GCTGCGTTCTTCATCGATGC) primers. All PCR reactions were performed using Phusion® High-Fidelity PCR Master Mix (New England Biolabs). The X1 (containing SYB green) buffer was mixed with an equal volume of PCR product and electrophoresed on a 2% agarose gels. Samples with bright master bands between 400–450 bp were selected for further experiments. PCR products were mixed in equal density ratios. PCR products were purified using Qiagen Gel Extraction Kits (Qiagen, Germany). Sequencing libraries were generated using TruSeq DNA PCR-Free Sample Preparation Kits (Illumina, USA) following the manufacturer’s recommendations. Index codes were added. The library quality was assessed on Qubit 2.0 Fluorometer (ThermoScientific) and an Agilent Bioanalyzer 2100 system. Finally, the library was a sequenced on an Illumina HiSeq46,47. 2500 platform to generate 250 bp paired-end reads.
Soil physicochemical properties
The content of organics was determined by external heating with potassium bichromate. Total nitrogen content was determined using the perchlorate-sulfuric acid digestion method, and fox 1035 automatic nitrogen determination apparatus. The content of total phosphorus was determined by acid soluble molybdenum anti-colorimetry and agilent CARY60 UV spectrophotometer. The total kalium content was determined using the acid dissolution - atomic absorption method using the Thermo Scientific Series Atomic Absorption Spectrometer. Nitrate nitrogen and ammonium nitrogen content was determined through the 0.01 M calcium chloride extraction method using a BRAN + LUEBBE flow analyzer. Available phosphorus was determined through extraction with sodium bicarbonate and molybdenum inverse colorimetry. pH measurements were performed using a mettler tolido FiveEasy Plus pH meter.
Data analysis
Cutadapt8 software was used to filter and control data. Low -quality areas were removed and reads that conformed to the length of the target fragment were retained. According to the barcode, sample data were extracted from the reads obtained. Barcode and primer sequences were cut off and the Cut adapt parameter (−q) was set for quality control to obtain raw reads. The original sequencing data possessed a proportion of interference data to allow the results of information analysis to be accurate and reliable. We performed splicing and filtering of the raw datasets to obtain valid data. OUT clustering and species classification analysis were then performed based on valid data.
Alpha Diversity was used to analyze the complexity of species diversity for a sample through six indices48, including observed-species, Chao1, Shannon, Simpson, ACE, and Good-coverage. All indices in the samples were calculated with QIIME (Version 1.7.0) and displayed with R software (Version 2.15.3). Chao1 and Abundance Coverage based Estimators (ACE) were selected to identify community richness. Shannon and Simpson indexes were used to identify community diversity. Good coverage was used to characterize the sequencing depth. Observed species were used to directly reflect the rationality of data sequencing, and indirectly reflected the species richness in the sample.
Beta diversity analysis was used to evaluate differences in sample species complexity, Beta diversity on both weighted and unweighted unifrac were calculated using QIIME software (Version 1.7.0). Cluster analysis was preceded by principal component analysis (PCA) which was applied to reduce the dimensions of the original variables using the FactoMineR package and ggplot2 package in R software (Version 2.15.3). Principal Coordinate Analysis (PCoA) was performed to obtain principal coordinates and to visualize complex and multidimensional data. A distance matrix of weighted or unweighted unifrac amongst the samples was obtained prior to being transformed to a new set of orthogonal axes, by which the maximum variation factor was demonstrated by the first principal coordinates, and the second maximum was obtained by the second principal coordinates. PCoA analysis was displayed through the WGCNA package, Stat packages and ggplot2 package in R software (Version 2.15.3). Unweighted Pair-group Methods with Arithmetic Mean (UPGMA) clustering was performed using QIIME software (version 1.7.0) as a hierarchical clustering method to interpret the distance matrix using average linkages. Linear discriminant analysis (LDA) effect sizes (LEfSe) was used for significant differences between statistical groups49. At the same time, CCA/RDA/dbRDA analysis and correlation analysis of the diversity index and environmental factors could be combined with environmental factors, and environmental impact factors that significantly affected community changes between the groups.
Results
Rhizospheric and non-rhizospheric soil areas share or have specific fungal operational taxonomic units, and the fungal communities are not evenly distributed
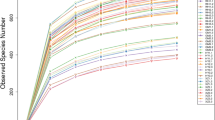
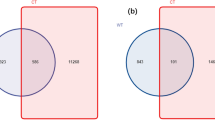
Petal maps revealed 224 core OTUs in the soil samples from different locations and depths in the rhizosphere soil region (Fig. 1a), whilst 129 core OTUs samples from different locations and depths were obtained in the non-rhizosphere soil (Fig. 1b). Interestingly, whether in the rhizosphere or non-rhizosphere, the number of unique OTUs for each soil sample differed. Specifically, the number of OTUs in each sample of the rhizosphere region were significantly higher than those of the non-rhizosphere region, and the number of OTUs in many of the soil samples in the non-rhizosphere soil region were extremely low. Rarefaction curves were constructed and founded on the levels of data extracted using the Observed species index and the corresponding number of species (Supplementary Fig. S1). The curve in the Rarefaction diagram tends to be flat, and more data is produced for new OTUs, indicating that the levels of sequencing are reasonable. Good coverage ranged from 99.4 to 99.8% (Supplementary Table S1) and the depth of measurements aligned with all requirements. The Rank Abundance Curve (Supplementary Fig. S2) showed a rapid decline, indicating that only a few dominant fungi account for a large proportion of the total fungal population.

Operational Taxonomic Units (OTU) based petal maps. Ferula sinkiangesis rhizospheric (a) and non-rhizospheric (b) petal maps. Description: Each petal in the diagram represents a (Group) sample. Different colors represent different samples (Groups). Middle core numbers represent the number of OTUs common to all samples, and the number on the petal represents the number of OTUs unique to this sample (Group). E, R, and S represent the rhizosphere of Ferula sinkiangesis 1, 2, and 3 plots, respectively. NE, NR, and NS represent the non-rhizosphere of Ferula sinkiangesis 1, 2, and 3 plots, respectively. Numbers indicate the root depth (1, 2 and 3 indicate depths of 0–5 cm, 5–15 cm and 15–35 cm, respectively).
Soil TK can Significantly explain the alpha diversity of fungal communities
Spearman correlation analysis showed that the abiotic factor TK had a significant and strongly positive correlation with the Shannon and Simpson indexes (Shannon index, R2 = 0.59, simpson index, R2 = 0.57, p < 0.01, Table 1). Conversely, other abiotic Factors (pH, TOC, TN, TP, NN, AN, AP, TS and Altitude) did not exhibit significant correlation with Shannon and Simpson indexes (Table 1). These diversity indices reflect the diversity and uniformity of species communities; the greater the index, the higher the species diversity, the more uniform the distribution. Therefore, TK is accountable for the diversity of fungal communities. simultaneously, the stepwise multiple linear regression model shows that TK is the only important influencing factor among all the abiotic factors listed in the article, affecting the alpha diversity of the rhizosphere community (Shannon index: y = 0.3245 × −1.8459, p < 0.01; simpson index: y = 0.0071 × +0.6231, p < 0.01, Fig. 2).

Stepwise multiple linear regression model. Description: The best model for the diversity of the rhizosphere fungi community of Ferula sinkiangesis was explained. Multiple nonlinear regressions were performed using the stepwise method. The abiotic factors were used as independent variables, and the Shannon or Chao 1 index were used as dependent variables.
Dominant fungal phyla and their relationship with abiotic factors
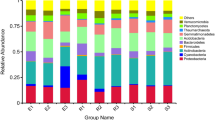
Analysis of dominant bacteria in all soil samples found that Ascomycota (42.87%), Basidiomycota (3.21%), Mortierellomycota (2.66%), Chytridiomycota (0.21%), Glomeromycota (0.05%), Rozellomycota (0.04%), Olpidiomycota (0.01%), Zoopagomycota (0.01%), Mucoromycota (0.01%) and Blastocladiomycota (0.01%) are fungi with higher phyla levels. Amongst the ten phyla, Ascomycota, Mortierellomycota and Basidiomycota were relatively richer than other phyla, with Ascomycota having the highest abundance (Fig. 3). Step by step, we found that there is a significant correlation between dominant fungal phyla and abiotic factors (Table 2). As showed in Table 2, pH, TK, NN, AP and TS were markedly correlation with fungal phyla. For example, pH markedly positively correlated with the relative abundances of Basidiomycota and Rozellomycota. TK showed markedly positively relationships with Basidiomycota and Mucoromycota. NN and AP showed obviously positively relationships with Basidiomycota. And TS have important relationships with Mucoromycota (Table 2). Contrary, pH markedly negative correlated with the relative abundances of Mortierellomycota and Olpidiomycota (Table 2). Meanwhile, Kickxellomycota, Zoopagomycota,Glomeromycota, Chytridiomycota and Ascomycota have no significant correlation with various factors. No obviously relationships was found between the relative abundance of fungi and TOC, TN, TP, AN and altitude.

UPGMA distance sample cluster tree. Description: The UPGMA cluster analysis was performed with the Weighted Unifrac distance matrix, and the clustering results were integrated with the species relative abundance of each sample at the Phylum level. The left side is the UPGMA clustering tree structure, and the right side is the species relative abundance distribution of each sample at the phylum level.
Dominant fungal genus and their relationship with abiotic factors
OTUs and species classification analysis found that in the rhizosphere and non-rhizosphere soils of Ferula sinkiangesis, Polythrincium, Mortierella, Striatibotrys, Pyrenochaeta, Schizophyllum, Gibberella, Alternaria, Myrothecium, Arthrocladium and Graphium were relatively more enriched than other genera. Among the top thirty genera, Polythrincium, Mortierell and Striatibotrys were more highly enriched than the other genera, with Polythrincium having the highest abundance (Fig. 4). Gradually, Spearman correlation analysis showed a significant correlation between abiotic factors and fungal communities. We screened four abiotic factors associated with the majority of fungal communities, including altitude, AP, NN, and TK. Amongst them, altitude showed a significant positive correlation with Ascotremella, Fusarium, Graphium, Knufia, Paraphaeosphaeria, Phlyctochytrium, Pyrenochaeta, Sarocladium, Stachybotrys and Striatibotrys, and a significant negative correlation with Corticium, Myrothecium and Plectosphaerella (Fig. 5a). AP showed a significant positive correlation with Ascotremella, Gibberella, Graphium, Knufia, Paraphaeosphaeria, Phlyctochytrium, Pyrenochaeta, Sarocladium and Scutellinia, and a significant negative correlation with Scutellinia (Fig. 5b). NN showed a significant positive correlation with Scutellinia, Thelebolus, Stachybotrys, Myrothecium and Striatibotrys. Plectosphaerella, and a significant negative correlation with Aspergillus and Fusarium (Fig. 5c). TK showed a significant positive correlation with Phlyctochytrium, Ascotremella, Cladosporium, Knufia, Fusarium, Preussia, Paraphaeosphaeria and Pyrenochaeta (Fig. 5d). The relationship between all abiotic factors and fungal genus (Supplementary Fig. S3).

Histogram of relative abundance of rhizosphere microorganisms in Ferula sinkiangesis. Description: Relative abundance of the top thirty genera in the rhizosphere and non-rhizosphere of Ferula sinkiangesis. Sample names and relative abundance are shown. Others indicate the sum of the relative abundance of all genera except the thirty genera of the Figure.

Spearman correlation analysis was used to evaluate the relationship between abiotic factors ((a–d) respectively represent Altitude, AP, NN, and TK) and fungal communities (genera). Description: p < 0.5 represents a significant correlation, (*) p < 0.01 is very significant (**), Positive correlation (+), Negative correlation (−). Abbreviations: phenyl group (PH),Total Organic Carbon (TOC), Total nitrogen (TN), Total phosphorus (TP), Total kalium (TK), Nitrate nitrogen (NN), Ammoniacal nitrogen (AN), Available phosphorus (AP), Total salt (TS).
Different biomarkers are present in the rhizosphere and non-rhizosphere at different soil depth and sites
LDA effect size (LEfSe) analysis revealed different biomarkers in the rhizosphere at different soil depths from the same site. For example, in the rhizosphere of the Ferula sinkiangesis in the third site, enrichment of didymosphaeriaceae, Ceratobasidiaceae, Plecosporales and Cantharellales were significant in 0–5 cm soil, enrichment of eurotiomycetes and eurotiales were significant in deep soil (5 cm–15 cm) and enrichment of cucurbitariacea and graphiaceae were significant at a depth of 15–40 cm (Fig. 6a). Biomarkers of the rhizospheric area differed according to location at a depth of 0–5 cm. For example, at a soil depth of 0–5 cm, the accumulation of pleosporaceae in site 1 was significant. In site 2 Glomerellales, Stachybotryaceae, Hypocreales and Sordariomycetes were significant. In site 3, Didymosphaeriaceae, Pleosporales, Ceratobasidiaceae and Cantharellales were 144 abundant (Fig. 6a). At the same location, the enrichment of species in rhizosphere and non-rhizosphere soils at a depth of 0–5 cm also differed. For example, at a soil depth of 0–5 cm at site 1, Cladosporiaceae and Hypocrees enrichment in the non-rhizosphere soil was significant, and Pleosporacea enrichment in the rhizosphere soil was significant (Fig. 6b).

LDA Effect Size (LEfSe) analysis between groups. Biomarkers with statistical differences in different regions and depths (a) and Biomarker with statistical difference at 0–5 cm depths (b). Description: In the evolutionary branch diagram, the circle radiating from inside to outside represents the classification level from the gate to the genus (or species). Each small circle at a different classification level represents a classification at that level, and the diameter of the small circle is proportionate to the relative abundance. Coloring: species with no significant differences are uniformly colored yellow. Red nodes indicate the microbial group playing an important role and green nodes indicates an important role. Microbial Populations, if one group in the figure is missing this indicates that no significant differences are observed in the group. The name of the species represented by letters is shown in the legend (right side).
Discussion
Microorganisms participate in soil food webs as decomposers50,51. In this study, we found that the dominant fungi in the root soil of Ferula Sinkiangensis were Ascomycota, Basidiomycota, Chytridiomycota, Mortierellomycota, Glomeromycota, Rozellomycota, Zoopagomycota, Olpidiomycota, Mucoromycota and Blastocladiomycota. Amongst them, Ascomycota, Basidiomycota, and Chytridiomycota had absolute quantitative advantages. These conclusions are consistent with the results of the dominant position of ascomycetes observed by Chen and colleagues in the grassland soil fungal community, and the dominant results of the ascomycetes identified by Hugoni and colleagues in the soil fungal community of Gramineae28,52. Ascomycota is considered a key player in the decomposition of soil organic matter53,54 and largely dominates the active fungal community through its involvement in root exudation assimilation and SOM degradation28. In addition, Ascomycetes, Blastocladiomycota, Mortierellomycota, Olpidiomycota and Chytridiomycota can be saprophytic on the residual limbs of plants and animals and decompose their remains55,56,57,58,59. Glomeromycota can form arbuscular mycorrhiza with plants and are able to absorb nutrients directly60. Rozellomycota and Zoopagomycotan gain nutrients from invading plants61. This indicates that fungi in the rhizosphere niche perform a variety of tasks. This is supported by studies from Ballhausen and coworkers. A saprophytic rhizosphere term defines the area affected by saprophytic mycelium. Its contribution to the food web is underestimated, particularly from the view of saprophytic fungi19. This consequently underestimates the status of fungi in the ecological cycle.
Linear Discriminant Analysis effect size (LEfSe) analysis showed significant differences between rhizosphere and non-rhizosphere biomarkers in different regions at a depth of 0–5 cm (Fig. 6b). We believe that these differences are due to natural environments in the regions, the trend effect of different environmental factors on the microorganisms62,63 and explains why microorganisms in the topsoil are more susceptible to the environment64,65,66. We observed different markers at varying depths (Fig. 6a), which were roughly divided into three layers. (1) the surface layer of plant litter accumulation; (2) the bottom layer of root exfoliation accumulation; and (3) the middle layer. Due to nutritional trends, the fungi that feed on cellulose and lignin accumulate on the surface and bottom layer, as the surface layer is sensitive to various environmental factors to which the fungi can adapt. The secretion of sugars and simple compounds permits bacteria to be enriched to high levels, at which point more competitive fungi become enriched. In the middle layer, it is difficult to decompose lignin, so lignin-feeding fungi become enriched, resulting in different biomarkers at each soil depth. However, the root system environment is more complex and requires experimental evidence. In addition, this study indicates that no significant differences between the rhizosphere and non-rhizosphere below 0–5 cm exist. We suspect that this is due to the influence of volatile components in the roots of Ferula sinkiangesis, The volatile substances of Ferula sinkiangesis are related, permitting a new layer of experimental design.
Dominant soil microbes (genus) have specific relationships to abiotic factors. In addition to altitude, TK, AP and NN have significant interrelationships with the majority of dominant microbes and other non-biological factors identified in this study. Some correlation with microbes also exists. For example, pH negatively correlates with mortierella and Arthrocladium, but positively correlates with Gibberella and Alternaria. There is also a significant positive correlation between TPand Alternaria, Paraphaeosphaeria, Knufia and Coniochaeta. A significant positive correlation between AN and Solicoccozyma and Papulaspora also occurred, whilst a significant negative correlation with Ascotremella, Knufia, Paraphaeosphaeria and Pyrenochaeta was observed. There was a significant positive correlation between TN and Pyrenochaeta and Knufia and between TS and Wardomycopsis No correlation between TOC and the dominant microbial genus occurred (Supplementary Fig. 3). Understanding this inter-relationship between abiotic factors and microorganisms is of great significance to reveal themechanistic interactions between plants and microorganisms, and to understand the role of microorganisms in this niche.
A significant correlation exists between abiotic factors and soil microbial community diversity and abundance. Yao qin and colleagues demonstrated that soil pH and TK have significant effects on the microbial community structure, and artificially increasing biochars can influence the properties of the soil67. In this study, we found significant positive correlation between TK and fungal biodiversity, and so identified that K fertilizers in the habitat soil of Ferula Sinkiangensiscan affect the fungal community, indirectly optimizing the soil structure. The purpose of this is to improve the growth environment of the ferula. The investment in TK is not unlimited, and a threshold is likely to be reached and various factors are likely to interact. The correct allocation of the investment requires further experiments to explore the most reasonable input.
In summary, this study provides information on the rhizosphere fungal community and the diversity of Ferula Sinkiangensis. Moreover, different biomarkers were identified at varying depths, which may be related to the medicinal components of Ferula Sinkiangensis. We also identified a link between environmental factors and fungal community diversity, in addition to fungal genus and gates. We hope that by mastering these relationships, we can artificially intervene and regulate the diversity of fungal communities to improve soil structure and ferlity. This information is useful for the protection and commercial cultivation of Ferula Sinkiangensis.
References
Xu, E. & Zhang, H. Spatially-explicit sensitivity analysis for land suitability evaluation. Applied Geography 45, 1–9 (2013).
Xing, F. & Qin, X. A supplement to the flora reipublicae popularis sinicae (volume 51, violaceae). Journal of South China Agricultural University 25, 120–121 (2004).
Fang, L. I., Zhao, W. B., Cheng, Y. H. & Wang, Z. H. A Study on Biological Characters of Wild Ferula sinkiangensis K. M. SHENin. Xinjiang. Lishizhen Medicine & Materia Medica Research (2008).
Alkhatib, R. et al. Activity of elaeochytrin A from Ferula elaeochytris on leukemia cell lines. Phytochemistry 69, 2979–2983 (2009).
Sun, L., Shi, S. B., Zhu, J., Xiao-Jin, L. I. & University, X. A. Traditional Use and Modern Research Review of Ferula in China. Modern Chinese Medicine (2013).
Lee, C. L. et al. Influenza A (H(1)N(1)) Antiviral and Cytotoxic Agents from Ferula assa-foetida. Journal of Natural Products 72, 1568–1572 (2009).
ELRAZEK et al. Terpenoid coumarins of the genus Ferula. Cheminform 34 (2003).
Matejić, J. S. et al. Total phenolic content, flavonoid concentration, antioxidant and antimicrobial activity of methanol extracts from three Seseli L. taxa. Central European Journal of Biology 7, 1116–1122 (2012).
Kartal, N. et al. Investigation of the antioxidant properties of Ferula orientalis L. using a suitable extraction procedure. Food Chemistry 99, 584–589 (2007).
Qian, Z. Z., Dan, Y., Liu, Y. Z. & Peng, Y. Pharmacopoeia of the People’s Republic of China (2010 Edition): A Milestone in Development of China’s Healthcare. 02, 157–160 (2010).
Hiltner, L. Uber neuere Erfahrungen und Probleme auf dem Gebeit der Bodenbakteriologie und unter besonder Berucksichtigung der Grundungung und Brache (1904).
Bais, H. P., Weir, T. L., Perry, L. G., Gilroy, S. & Vivanco, J. M. The role of root exudates in rhizosphere interactions with plants and other organisms. Annual Review of Plant Biology 57, 233–266 (2006).
Hütsch, B. W., Augustin, J. & Merbach, W. Plant rhizodeposition — an important source for carbon turnover in soils. Journal of Plant Nutrition and Soil Science=Zeitschrift fuer Pflanzenernaehrung und Bodenkunde 165, 397–407 (2015).
Haichar, F. E. Z., Santaella, C., Heulin, T. & Achouak, W. Root exudates mediated interactions belowground. Soil Biology &. Biochemistry 77, 69–80 (2014).
Avis, T. J. & Antoun Tweddell, R. J. Multifaceted beneficial effects of rhizosphere microorganisms on plant health and productivity. Soil Biology & Biochemistry 40, 1733–1740 (2008).
Gaskins, M. H., Albrecht, S. L. & Hubbell, D. H. Rhizosphere bacteria and their use to increase plant productivity: A review. Agriculture Ecosystems &. Environment 12, 99–116 (1985).
Cocking, E. C. Endophytic colonization of plant roots by nitrogen-fixing bacteria. Plant & Soil 252, 169–175 (2003).
Aravena, J. E., Berli, M., Ghezzehei, T. A. & Tyler, S. W. Effects of Root-Induced Compaction on Rhizosphere Hydraulic Properties - X-ray Microtomography Imaging and Numerical Simulations. Environmental Science & Technology 45, 425–431 (2011).
Ballhausen, M. B. & Boer, W. D. The sapro-rhizosphere: Carbon flow from saprotrophic fungi into fungus-feeding bacteria. Soil Biology &. Biochemistry 102, 14–17 (2016).
Butler, J. L., Williams, M. A., Bottomley, P. J. & Myrold, D. D. Microbial Community Dynamics Associated with Rhizosphere Carbon Flow. Appl Environ Microbiol 69, 6793–6800 (2003).
Rudnick, M. B., Veen, J. A. V. & Boer, W. D. Baiting of rhizosphere bacteria with hyphae of common soil fungi reveals a diverse group of potentially mycophagous secondary consumers. Soil Biology &. Biochemistry 88, 73–82 (2015).
Bartnickigarcia, S. Cell Wall Chemistry, Morphogenesis, and Taxonomy of Fungi. Annual Review of Microbiology 22, 87–108 (1968).
Qiu, X. et al. Changes in the composition of intestinal fungi and their role in mice with dextran sulfate sodium-induced colitis. Scientific Reports 5, 10416 (2015).
Lauber, C. L., Strickland, M. S., Bradford, M. A. & Fierer, N. The influence of soil properties on the structure of bacterial and fungal communities across land-use types. Soil Biology &. Biochemistry 40, 2407–2415 (2008).
Berg, G. & Smalla, K. Plant species and soil type cooperatively shape the structure and function of microbial communities in the rhizosphere. Fems Microbiology Ecology 68, 1–13 (2010).
Guyonnet, J. P., Cantarel, A. A. M., Simon, L. & Haichar, F. E. Z. Root exudation rate as functional trait involved in plant nutrient‐use strategy classification. Ecology & Evolution 8, 8573–8581 (2018).
Tu, S., Sun, J., Guo, Z. & Feng, G. On relationship between root exudates and plant nutrition in rhizosphere. Soil & Environmentalences 9, 64–67 (2000).
Hugoni, M., Luis, P., Guyonnet, J. & Haichar, F. E. Z. Plant host habitat and root exudates shape fungal diversity. Mycorrhiza 28, 451–463 (2018).
Kavamura, V. N. et al. Water Regime Influences Bulk Soil and Rhizosphere of Cereus jamacaru Bacterial Communities in the Brazilian Caatinga Biome. Plos One 8, e73606 (2013).
Howard, A. G., Comber, S. D. W., Kifle, D., Antai, E. E. & Purdie, D. A. Arsenic Speciation and Seasonal Changes in Nutrient Availability and Micro-plankton Abundance in Southampton Water, U.K. Estuarine Coastal & Shelf Science 40, 435–450 (1995).
Marques, J. M. et al. Plant age and genotype affect the bacterial community composition in the tuber rhizosphere of field-grown sweet potato plants. Fems Microbiology Ecology 88, 424–435 (2014).
Ndour, P. M. S. et al. Pearl Millet Genetic Traits Shape Rhizobacterial Diversity and Modulate Rhizosphere Aggregation. Frontiers in Plant Science 8, 1288- (2017).
Copeland, S. M., Bruna, E. M., Silva, L. V. B., Mack, M. C. & Vasconcelos, H. L. Short-term effects of elevated precipitation and nitrogen on soil fertility and plant growth in a Neotropical savanna. Ecosphere 3, art31 (2012).
Pinggera, J., Geisseler, D., Merbach, I., Joergensen, R. G. & Ludwig, B. Effect of substrate quality on the N uptake routes of soil microorganisms in an incubation experiment. European Journal of Soil Biology 69, 17–23 (2015).
Yevdokimov, I. V., Gattinger, A., Buegger, F., Schloter, M. & Munch, J. C. Changa in the structure and activity of a soil microbial community caused by inorganic nitrogen fertilization. Microbiology 81, 743–749 (2012).
Jones, R., Chambers, J. C., Johnson, D. W., Blank, R. R. & Board, D. I. Effect of repeated burning on plant and soil carbon and nitrogen in cheatgrass (Bromus tectorum) dominated ecosystems. Plant & Soil 386, 47–64 (2015).
Akhter, A., Hageahmed, K., Soja, G. & Steinkellner, S. Compost and biochar alter mycorrhization, tomato root exudation, and development of Fusarium oxysporum f. sp. lycopersici. Frontiers in Plant Science 6, 529 (2015).
Schnittler, M. & Stephenson, S. L. Myxomycete biodiversity in four different forest types in Costa Rica. Mycologia 92, 626–637 (2000).
Fontúrbel, M. T. et al. Effects of an experimental fire and post-fire stabilization treatments on soil microbial communities. Geoderma 191, 51–60 (2012).
Hunter, P. J., Teakle, G. R. & Bending, G. D. Root traits and microbial community interactions in relation to phosphorus availability and acquisition, with particular reference to Brassica. Frontiers in Plant Science 5, 27 (2014).
Lu, L. et al. Nitrification of archaeal ammonia oxidizers in acid soils is supported by hydrolysis of urea. Isme Journal 6, 1978–1984 (2012).
DONN et al. Rhizosphere microbial communities associated with Rhizoctonia damage at the field and disease patch scale. Applied Soil Ecology 78, 37–47 (2014).
Cardinale, M., Grube, M., Erlacher, A., Quehenberger, J. & Berg, G. Bacterial networks and co-occurrence relationships in the lettuce root microbiota. Environmental Microbiology 17, 239–252 (2015).
Riley, D. & Barber, S. A. Bicarbonate Accumulation and pH Changes at the Soybean (Glycine max (L.) Merr.) Root-Soil Interface 1. Soil Science Society of America Journal 33, 905–908 (1969).
Riley, D. & Barber, S. A. Salt accumulation at the soybean (Glycine max (L.) Merr) root-soil interface. Soil Science Society of America Journal 34, 154–155 (1970).
Degnan, P. H. & Ochman, H. Illumina-based analysis of microbial community diversity. Isme Journal 6, 183 (2012).
Caporaso, J. G. et al. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. Isme Journal Multidisciplinary Journal of Microbial Ecology 6, 1621–1624 (2012).
Li, B., Zhang, X., Guo, F., Wu, W. & Zhang, T. Characterization of tetracycline resistant bacterial community in saline activated sludge using batch stress incubation with high-throughput sequencing analysis. Water Research 47, 4207–4216 (2013).
Rebollar, E. A. et al. Using “Omics” and Integrated Multi-Omics Approaches to Guide Probiotic Selection to Mitigate Chytridiomycosis and Other Emerging Infectious Diseases. Frontiers in Microbiology 7, 68 (2016).
Morrison, E. et al. Mycorrhizal inoculation increases genes associated with nitrification and improved nutrient retention in soil. Biology & Fertility of Soils 53, 1–5 (2017).
Philippot, L., Raaijmakers, J. M., Lemanceau, P. & Wh, V. D. P. Going back to the roots: the microbial ecology of the rhizosphere. Nature Reviews Microbiology 11, 789–799 (2013).
Chen, L. et al. Responses of soil microeukaryotic communities to short-term fumigation-incubation revealed by MiSeq amplicon sequencing. Frontiers in Microbiology 6, 1149 (2015).
Osono, T. & Takeda, H. Organic chemical and nutrient dynamics in decomposing beech leaf litter in relation to fungal ingrowth and succession during 3-year decomposition processes in a cool temperate deciduous forest in Japan. Ecological Research 16, 649–670 (2010).
Voříšková, J. & Baldrian, P. Fungal community on decomposing leaf litter undergoes rapid successional changes. Isme Journal 7, 477–486 (2013).
Ekman, S. & Jørgensen, P. M. Towards a molecular phylogeny for the lichen family Pannariaceae (Lecanorales, Ascomycota). Canadian Journal of Botany 80, 625–634 (2002).
Lücking, R., Hodkinson, B. P. & Leavitt, S. D. The 2016 classification of lichenized fungi in the Ascomycota and Basidiomycota – Approaching one thousand genera. Bryologist 119, 361–416 (2016).
Wilfried, T. et al. Conserving the functional and phylogenetic trees of life of European tetrapods. Philosophical Transactions of the Royal Society B Biological Sciences 370, 20140005–20140005 (2015).
Blackwell, M. The fungi: 1, 2, 3… 5.1 million species? American Journal of Botany 98, 426–438 (2011).
Longcore, J. E., Pessier, A. P. & Nichols, D. K. in Mycologia. 219.
Haug, I. et al. Graffenrieda emarginata (Melastomataceae) forms mycorrhizas with Glomeromycota and with a member of the Hymenoscyphus ericae aggregate in the organic soil of a neotropical mountain rain forest. Revue Canadienne De Botanique (2004).
Cai, L. et al. The evolution of species concepts and species recognition criteria in plant pathogenic fungi. Fungal Diversity 50, 121–133 (2011).
Arora, D. K. & S., G. Effect of different environmental conditions on bacterial chemotaxis toward fungal spores. Can.j.microbiol 39, 922–931 (1993).
Chung, L. et al. Bacteroides fragilis Toxin Coordinates a Pro-carcinogenic Inflammatory Cascade via Targeting of Colonic Epithelial Cells. Cell Host & Microbe 23, 421 (2018).
Bone, T. L. & Balkwill, D. L. Morphological and cultural comparison of microorganisms in surface soil and subsurface sediments at a pristine study site in Oklahoma. Microbial Ecology 16, 49–64 (1988).
Vargas, D. Y. et al. Single-molecule imaging of transcriptionally coupled and uncoupled splicing. Cell 147, 1054–1065 (2011).
Wang, M. et al. Effect of Collembola on mineralization of litter and soil organic matter. Biology & Fertility of Soils 53, 563–571 (2017).
Yao, Q. et al. Three years of biochar amendment alters soil physiochemical properties and fungal community composition in a black soil of northeast China. Soil Biology &. Biochemistry 110, 56–67 (2017).
Acknowledgements
In this study, we would like to thank professor LZ for his guidance and all the authors for their joint efforts. This study was supported by the Chinese National Basic Research Program (2014CB954203) and the National Natural Science Foundation of China (31360139, 41561010, 31560177).
Author information
Authors and Affiliations
Contributions
T.Z. is the first author of this article, L.Z. are the corresponding authors, T.Z. and L.Z. designed this study and conducted data analysis, T.Z., Z.K.W., X.H.Lv. and Y.L. conducted the research, T.Z. wrote the manuscript and had primary responsibility for the final content. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhang, T., Wang, Z., Lv, X. et al. High-throughput sequencing reveals the diversity and community structure of rhizosphere fungi of Ferula Sinkiangensis at different soil depths. Sci Rep 9, 6558 (2019). https://doi.org/10.1038/s41598-019-43110-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-43110-z
This article is cited by
-
Similarities and differences in the rhizosphere biota among different ephemeral desert plants in Gurbantünggüt Desert
Environmental Sciences Europe (2023)
-
Asafoetida exerts neuroprotective effect on oxidative stress induced apoptosis through PI3K/Akt/GSK3β/Nrf2/HO-1 pathway
Chinese Medicine (2022)
-
Study on Physicochemical Properties of High-Density Sludge Sediment in Copper Mine After Phytoremediation
Journal of Soil Science and Plant Nutrition (2021)
-
High-throughput sequencing analysis of the rhizosphere arbuscular mycorrhizal fungi (AMF) community composition associated with Ferula sinkiangensis
BMC Microbiology (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
