
Abstract
The 37 currently recognized Bemisia tabaci cryptic species are economically important species and contain both primary and secondary endosymbionts, but their diversity has never been mapped systematically across the group. To achieve this, PacBio sequencing of full-length bacterial 16S rRNA gene amplicons was carried out on 21 globally collected species in the B. tabaci complex, and two samples from B. afer were used here as outgroups. The microbial diversity was first explored across the major lineages of the whole group and 15 new putative bacterial sequences were observed. Extensive comparison of our results with previous endosymbiont diversity surveys which used PCR or multiplex 454 pyrosequencing platforms showed that the bacterial diversity was underestimated. To validate these new putative bacteria, one of them (Halomonas) was first confirmed to be present in MED B. tabaci using Hiseq2500 and FISH technologies. These results confirmed PacBio is a reliable and informative venue to reveal the bacterial diversity of insects. In addition, many new secondary endosymbiotic strains of Rickettsia and Arsenophonus were found, increasing the known diversity in these groups. For the previously described primary endosymbionts, one Portiera Operational Taxonomic Units (OTU) was shared by all B. tabaci species. The congruence of the B. tabaci-host and Portiera phylogenetic trees provides strong support for the hypothesis that primary endosymbionts co-speciated with their hosts. Likewise, a comparison of bacterial alpha diversities, Principal Coordinate Analysis, indistinct endosymbiotic communities harbored by different species and the co-divergence analyses suggest a lack of association between overall microbial diversity with cryptic species, further indicate that the secondary endosymbiont-mediated speciation is unlikely to have occurred in the B. tabaci species group.
Similar content being viewed by others


Introduction
Insects’ bacterial endosymbionts play a critical role in supplementing the diet of their hosts, particularly for those on nutritionally simple diets such as cellulose or plant-phloem sap1. Endosymbionts are classified into primary (P-endosymbionts) and secondary symbionts (S-endosymbionts). The major role of the former is to provide essential amino acids, vitamins and nutrients missing from the insect host’s diet. Buchnera aphidicola2,3 and Wigglesworthia glossinidia4, for example, complement the diet and metabolic capacity of aphids and tsetse flies respectively. In contrast, the roles of secondary endosymbionts are related to host adaptation, especially aspects of host survival, competitive capacity and providing resistance against natural enemies and pesticides5,6,7. Therefore, P-endosymbionts are generally always presented in a host species, while the prevalence of S-endosymbionts varies between populations within a species.
The whitefly, Bemisia tabaci (sensu Russell) (Hemiptera: Sternorrhyncha: Aleyrodidae) is a global agricultural pest and a species complex that comprises more than 37 cryptic species (as defined by species delimitation metrics)8,9. B. tabaci has both P- and seven genera of S-endosymbionts which have been recorded from the different member species in the previous 15 years: Hamiltonella10, Arsenophonus10, Cardinium11, Wolbachia12, Fritschea13, Rickettsia14 and Hemipteriphilus15. The diversity of S-endosymbionts, however, has not previously been mapped systematically across the entire B. tabaci group. Thus, the distributions of S-endosymbionts both at the cryptic species level and across evolutionary lineages within B. tabaci were unknown.
Many studies have surveyed the S-endosymbionts using 16S ribosomal RNA (rRNA) molecular markers by normal PCR in field-collected whiteflies16,17,18 and showed that the infection incidences of different S-endosymbionts differ. But most of these studies are mainly focused on the diversity of endosymbionts in B. tabaci with restricted region distributions and lack an angle of linking these diversities with its hosts. Also, a major limitation of these studies is the unreliability of the diagnostic PCR techniques used. Ji, et al.17, for example, found higher incidence of Wolbachia infections using nested-PCR than in previous studies using universal primers in end-point PCR16,17. Primers developed for Arsenophonus were also found to amplify Rickettsia18 thus making the currently available methods for S-endosymbionts diagnosis and diversity unreliable. We considered, therefore, that an accurate understanding of B. tabaci endosymbionts required both systematic sampling and robust evaluation with high-efficiency detection methods. Over the past 10 years, advances in sequencing technologies have enabled considerable progress in the field of microbial ecology and revolutionized the characterization of complex microbial communities19. In particular, one of the third generation sequencing technologies, the PacBio Single Molecule, Real-Time (SMRT) DNA sequencing platform has demonstrated that it can generate high quality, full-length reference sequences of 16SrRNA genes, provide a reliable adjunct, and enable more accurate phylogenetic resolution of microbial communities20. In this study, therefore, we chose the PacBio sequencing instrumentation for the first time to systematically study the endosymbionts diversity of B. tabaci cryptic species.
Aside from the complicated distribution patterns of the S-endosymbionts, all B. tabaci species also contain the P-endosymbiont (Candidatus Portiera aleyrodidarumin) bacteriocytes21, which is transmitted vertically from mother to offspring and have been shown to have evolved in parallel with their insect hosts for millions of years22. As a member of sap-feeding insects (suborder Sternorrhyncha), the characterization of this primary endosymbiont associated with its host always indicate coevolution between P-endosymbionts and their hosts, which have been observed in many cases, such as (i) between Portiera and whiteflies23,24, (ii) Buchnera aphidicola and aphids25,26, (iii) Carsonella ruddii and psyllids27,28, as well as (iv) Moranella endobia and Tremblaya princeps in mealybugs29. Co-evolution between B. tabaci complex species and Portiera, however, has not previously been studied systematically across the group.
The aims of this study, therefore, were to: (i) investigate whether PacBio SMRT technology can improve detection and accurate identification of the known and unknown symbionts in the B. tabaci species group; (ii) evaluate bacterial diversity in the B. tabaci species complex; (iii) determine co-phylogenetic patterns between P-endosymbionts and B. tabaci species on a global scale and better understand the evolutionary pattern(s) of P-endosymbionts and B. tabaci.
Results
PacBio DNA sequencing of bacterial community in the B. tabaci species
To accurately estimate the bacteria diversity across evolutionary lineages within B. tabaci, a reliable and promising PacBio DNA platform was applied to generate 16S rRNA sequences of 1,513-bp from 21 B. tabaci and two outgroup species of B. afer with collection information in Table 1. In summary, we obtained a total 70,938 high-quality reads, and the reads count per sample ranged from 2,123–4,184 (Table S1). After quality filtering and removal of chimeric sequences, 65,774 clean 16S rRNA reads were remained and clustered into 29 OTUs using ≥97% sequence identity as the cutoff (Fig. 1A), of which 28 were found in B. tabaci species. The alpha diversities for bacterial OTUs from different cryptic species ranged from 0.21–1.59 Shannon index and 0.27–0.97 Simpson index, which are low (Table 2) yet similar to the result obtained by Jing, et al.30. In addition, the most diverse bacterial OTUs sets were detected in Asia II 6, New World 1, Asia II 3, Asia II 5, SubSaharan Africa 1 and Asia I. Based on the rarefaction curve for every sample with a characterization of tending to saturation (Fig. S1A, Supporting information), we deduced that the OTUs detected were representative of the 16S amplicons in each sample. However, due to under-sampling of field population diversity, there are likely to be additional bacteria yet to be found in B. tabaci species (Fig. S1B, Supporting information).

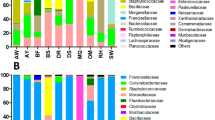
OTU profiles of the samples and phylogenetic analysis of all OTUs. (A) Heat map of OTU profiles (y-axis) inferred from PacBio. The x-axis shows 23 samples. The number of reads per OTU was normalized by total number of reads from the sample. For each sample, the number of reads per OTU represents total number of reads (the number of reads were transformed to log 2) from the sample. The asterisk denoted OTUs show the new OTUs found in this study. (B) The Bayesian unrooted phylogenetic tree of representative sequences OTUs. Red text labels represent the new OTUs. The scale bar at the base indicates number of substitutions per site.
The bacterial community of B. tabaci
The 29 bacterial OTUs represent a total of 21 genera, 18 families, 15 orders and nine classes (Fig. 1) and the distributions across the different cryptic species are shown in Fig. 2. As expected, and importantly, our findings included all previously reported endosymbionts of Bemisia (P-endosymbiont: Portiera; and seven S-symbionts: Arsenophonus, Cardinium, Fritschea, Hamiltonella, Hemipteriphilus, Rickettsia and Wolbachia) (Fig. 1A) which were representatives of the classes of Gamma-proteobacteria, Chlamydiae, Cytophagia and Alpha-proteobacteria (Fig. 1B).

Bacteria composition showed in tested samples. Different bubble colors represent different taxonomic levels, while the different bubble sizes represent the relative number of different bacteria. The species names of OTUs are in Fig. 1B.
Other than Porteria, which was present in every species, the bacteria diversity in various whitefly cryptic species differed substantially. At higher taxonomic levels, the community was dominated by Gamma-proteobacteria and Alpha-proteobacteria. At the ordinal level, Oceanospirillales was the most abundant, as this includes the P-endosymbiont, which was present in all B. tabaci species. PCoA based on weighted and unweighted UniFrac distances did not show any significant differences in proportions of bacteria among the samples (Fig. S2, Supporting information).
Several other bacteria not previously reported as endosymbionts of Bemisia were found (Fig. 1B, red colored species), such as Halomonas (also reported in the gut of many insects), Massilia, Pelagibacterium and Curtobacterium. The distribution of taxa among the B. tabaci examined was relatively non-specific (Fig. 2).
Validation of a new micro-organism in B. tabaci by next-generation sequencing and FISH
One of the novel bacteria Halomonas sp. has been reported previously from the gut of other insects, and so, to detect the reliability of the PacBio sequencing platform, through feasibility evaluation, this strain was chosen to determine whether it was also distributed in the midgut of B. tabaci. Two subsequent experiments of illumina midguts -target sequencing and FISH were conducted. MED and Asia II 1 species are picked as midgut resources, as it to be widely distributed in the MED B. tabaci species and extremely lower in Asia II 1 species. Illumina midguts-target sequencing results showed that Halomonas was clearly presented in the MED midguts (reads number: 1931, 521 and 424) in all the post-emergence time periods (Fig. 3A). Identically, the specific designed FISH probe showed it was abundant and distributed in the MED B. tabaci midgut (Fig. 3B). As a negative control, it was not apparent in the Asia II 1 midgut (Fig. 3C). At the last, to give a general idea of the relationships of this strain with the other finding species under the same genus, the positions of this Halomonas bacteria strain (OTU 25) amongst the published Halomonas strains was then deduced by including 1,529-bp 16S in a phylogenetic analysis (Fig. 3D).

Halomonas appeared in the midgut of MED B. tabaci verified by application of the v4-v5 region of using dissected midguts (A) and fluorescence in situ hybridization (B,C). (D) Phylogenetic tree revealed the relationship of this Halomonas strain with the other published ones. New bacteria-specific probes (green) conjugated to cy5 were used, and the nuclei were stained with DAPI (blue). Portiera is used as a control for Halomonas. Portiera-specific probe (red) is expected not to have occurred in the gut of B. tabaci. B, Halomonas was present in gut of MED; C, Halomonas was not detected in the gut of Asia II 1 due to its low content.
Distribution of the well-known endosymbionts in B. tabaci
To present the well-known endosymbionts profile in B. tabaci, the distribution of the eight previously reported endosymbionts amongst B. tabaci cryptic species is mapped against a phylogeny of host mitochondrial relationships (Fig. 4). Clearly, two different P-endosymbiont Portiera OTUs were found with one in all B. tabaci species (OTU9) and a second one in B. afer (OTU12). Of the seven S-endosymbionts, a total of five Arsenophonus OTUs were found in this study: OTU7 (A1), OTU11 (A2), OTU13 (A3), OTU16 (A4) and OTU28 (A5). Of these, OTU16 (A4) has more than 97% nucleotide identity to Arsenophonus_nasoniae (son-killer_infecting_Nasonia_vitripennis) and was detected in 11 different cryptic species of B. tabaci plus both B. afer samples. Only one Cardinium OTU (OTU26) was detected, occurring in seven B. tabaci cryptic species. One Fritschea OTU (OTU29) was found, restricted to New World 113. One Hamiltonella OTU (OTU24) was found, occurring in six B. tabaci cryptic species. Singh, et al.31 and Bing, et al.15 reported Hemipteriphilus/OLO(OTU27) from specimens of China 1 (collected from China) and Asia I and Asia II 1, both collected from north India. However, OTU27 was found in Asia II 3, Asia II 6, China 1, China 2, Indian Ocean and SubSaharan Africa 6 in our study. Two OTUs were detected from each of the two remaining S-endosymbionts: Rickettsia (OTU3 and OTU19) and Wolbachia (OTU10 and OTU21).

Relative abundance (the number of reads were transformed to log 2 and then multiplied by 10) of PacBio reads assigned to bacterial symbionts of B. tabaci cryptic species. R, A, W, H, C, OLO, F and P represent Rickettsia, Arsenophonus, Wolbachia, Hamiltonella, Cardinium, Hemipteriphilus, Fritschea and Portiera, respectively. The R1 have high sequence identity with Rickettsia described by Gottlieb et al., (2006). The W1 have a high sequence identity with Wolbachia described by Bing et al., (2014). Bubble sizes indicate read abundance of individual bacteria in each sample (see scale), with alternate colors to facilitate discrimination between rows; operational taxonomic units (OTUs) less than 10 reads were not included (the full list of OTUs is provided in Table S2).
Through comparing our data to that of previous studies (Table S3), although the strains from different studies are incomparable, a relatively higher strain diversity was reflected by PacBio and several newly discovered symbionts (OTU7, OTU11, OTU3, OTU28 and OTU19) were detected. To improve the systematic knowledge of symbionts in the B. tabaci group, we downloaded all 16S rRNA sequences from each symbiont genus and retained one unique sequence as a representative of each 97% identical cluster. Together with the 16S rRNA sequences detected from this study, they were all subjected to phylogenetic analyses, and the results clearly present all strains found for each genus so far in B. tabaci. Currently, one Porteria, two Cardinium, one Fritschea, one Hemipteriphilus, one Hamiltonella, 14 Arsenophonus, four Rickettsia and two Wolbachia species have been reported from B. tabaci (Fig. 5).

All known symbionts found in the B. tabaci cryptic species complex in this study combined with those available from Genbank (A–H). P represents the primary endosymbiont discovered from B. tabaci, while P1 represents the primary endosymbiont discovered from B. afer. The 16S rRNA lengths used to build the phylogenetic tree for A-H are 1,085 bp, 1,569 bp, 1,194 bp, 1,273 bp, 1,495 bp, 1,433 bp, 1,235 bp, 1,621 bp, respectively. The phylogenetic trees were conducted by Bayesian inference and the posterior probabilities were shown in the trees.
Co-evolution analysis of P-endosymbionts and B. tabaci hosts
According to previous studies, it has been hypothesized that P-endosymbionts co-evolve with their hosts, while S-endosymbionts do not23,32. For instance, there is absence of evidence to support this hypothesis in B. tabaci. To achieve this, the most popular and frequently used marker 16S rRNA for bacterial phylogeny and reconstruction of insect-symbiont coevolution23,29 were cloned from 23 cryptic species, and subsequently used for generating the phylogenetics of P-endosymbionts. In contrast, full mitogenomes markers were applied to construct phylogenetic topology of host B. tabaci.
Unfortunately, preliminary analysis of the 16S rRNA phylogeny lacked sufficient variation for OTU members from different cryptic species for resolution of evolutionary patterns (Fig. S3). To overcome this limitation and find an alternative marker, we looked for markers with sufficient sequence divergence through scanning the three available P-endosymbiont genomes from MEAM1, MED and Asia II 1 B. tabaci (“data under submission, DanTong Zhu” for the corresponding author of that study). Briefly, a sliding window approach was applied to determine the local identities of the three well-aligned Portiera genomes using a custom Perl script (available from the senior author on request). The well-aligned regions with relative high divergence among the three Portiera genomes were selected for further analyses. Totally, five regions with a relatively high evolutionary rate (RER), which were located between non-coding regions, were picked to generate PCR clones from each cryptic species. Eventually, one specific region (PCR primers: F (5′-CACTTGGCGGTGAGGT-3′) and R (5′-ACAATCTTCCATTCTTTCCA-3′)), defined as RER locus with 814 bp, was then chosen to address this question. Filtering process was showed in Fig. S4.
For the phylogenetic topologies generated from B. tabaci and P-endosymbiont, MrBayes and RAxML were applied to analyze the phylogenetic relationships of the B. tabaci species using the 14,599-bp mitochondrial sequences and of the P-endosymbiont from the 814 bp RER. Aside from a few branches with low support, the backbone topologies of the BI and ML trees were identical. Thus, only the BI tree is presented here and is used for further cophylogenetic analyses. High congruence between B. tabaci mitochondrial and Portiera RER phylogenies was presented (Fig. 6A). Similarly, both ParaFit and PACoanalyses provided evidence for significant co-divergence (ParaFitGlobal = 0.0021, P ≤ 0.001; a residual sum of squares (m2) = 0.1301, P = 0.000). Of the 23 host–endosymbiont associations, 21 were significant based on a ParaFit1 value of P ≤ 0.05, while 22 were significant based on a Parafit 2 value of P ≤ 0.05 (Fig. 6B). Overall, we here demonstrated that P-endosymbionts co-evolve with their B. tabaci hosts for the first time.

(A) Phylogenetic tree from MrBayes analyses of whitefly P-endosymbiont-combined specific DNA nucleotide sequences (left) and host mitochondrial genomes (right). Designations refer to B. tabaci species. (B) Jacknifed squared residuals (bars) and upper 95% confidence intervals (error bars) associated with each B. tabaci–Portiera link. PACo was applied to HKY85 genetic distances. The dashed line indicates the median squared residual value. Asterisks identify links significantly supported by ParaFitLink1&2 (a < 0.05).
Discussion
By using PacBio sequencing platform, 28 bacterial OTUs were discovered in the B. tabaci cryptic species complex for the first time, which enriches the bacterial reference database of whiteflies. In comparison, in whitefly research, only the 454 pyrosequencing platform had been used once to investigate the bacterial communities30. In that research, fewer than 10 bacterial OTUs were reported through screening representatives of seven cryptic species and three field populations. Apparently, this study adds more samples across the species complex of B. tabaci with a promising PacBio technology, which resulted in a reservoir of new information on the bacteria diversity of B. tabaci. This study significantly revolutionizes our knowledge on the bacteria diversity of an important economical pest. However, one of the constraints of the PacBio sequencing platform is that relatively high quantities of DNA are needed for library preparation33 and it is preferable to carry out PCR-free library preparation to avoid problems generated by amplification. The numbers of adult whitefly individuals we had for each species, however, made it difficult to get enough DNA for PCR-free library construction due to its tiny body size and so we utilized a non-PCR free library construction method to conduct the experiment which is a limitation for this study.
Novel OTUs found in this study
Fifteen new bacterial OTUs were identified in this study, which hugely increases our catalogue of bacterial diversity in B. tabaci species. Some of the new OTUs were close relatives of those from free-living bacteria, and soil or plant associated bacteria, such as Halomonas, Massilia34, Cupriavidus (OUT 8, OUT 18)35 and Sphingomonas36, as well as some related to occasional human and plant pathogens such as Acinetobacter (OUT 6, OUT 14, OUT 20)37 and Rhizobiaceae38.
Several OTUs, interestingly, could be attributed to known bacterial symbionts from other organisms. OUT 2, a Phyllobacterium species (Rhizobiales: Phyllobacteriaceae, Alpha-proteobacteria) has previously been recorded in leaf nodules of a tropical plant species39 and was found here in several cryptic species of B. tabaci, especially in New World1 (supported by the presence of 27 reads). Burkholderia sp. (OUT 17 and OUT 18) that have been reported previously as symbionts of the broad-headed bugs Riptortus clavatus and Leptocorisa chinensis (Heteroptera: Alydidae)40, were found in the New World 1 (OUT 17, 199 reads) and in Japan 2 (OTU 18, 170 reads) species. In addition, Paenibacillus (OUT 2, 27 reads) found in the New World is a genus of facultative anaerobic, endospore-forming bacteria that has been reported from a variety of different environments41,42.
Gut microbiota are of particular interest, because they are involved in many aspects of microbial pathogen action43, such as impacting the development, growth and survival of Hemiptera44,45,46. The gut microbiota of B. tabaci has not been investigated in detail—even in the present study, we were only able to show that Halomonas sp. was present in the midgut. Such limitations can be attributed to the fact that most of our whitefly samples had been stored in ethanol, from which makes it difficult to cleanly dissect out the midgut. As a first identified midgut associated bacteria in B. tabaci, the evidences presented here not only confirmed the reliability of the PacBio result, but also severed as the first steps required in understanding whether it is a gut resident species, as well as the origin and role of this bacterium and its relationship with B. tabaci species. Additionally, as Poddar et al.,47 suggested, Sphingomonas is a cultivable gut bacteria across multiple developmental stages of whitefly (Asia II 1 and Asia I). Similarly, Sphingomonas was also detected in the New World 1 and New World 2 cryptic species. Unfortunately, the lack of live lab colonies impacts on any further verification of its presence.
Known symbionts
As a primary endosymbiont, Portiera reads were assigned to two different OTUs, one from the 21 B. tabaci species and a second from B. afer—most probably due to the significant length of time since these clades diverged48. The nucleotide identity of the two Portiera OTUs representatives was 96%, just outside our range for OTU cutoff (3% divergence). OUT 12 was compared against all Portiera 16S rRNA sequences in GenBank, which were all less than or equal to 96% identical. These results suggest that the Portiera 16S rRNA might be useful as a distinguishing marker to delineate members of the B. tabaci complex from other Bemisia species. As such, it shall help draw a clear boundary line between other Bemisia clades within the genus.
As regard to the secondary endosymbionts, previously, a total of two Cardinium spp.11,30, three Rickettsia spp. (one unpublished was deposited in the GenBank_LN 8296931)31,49 and three Wolbachia spp.12,16,18 have been discovered in B. tabaci. In this study, more novel OTUs were assigned to Arsenophonus, Rickettsia and Wolbachia resulting in at least two Cardinium spp.11,30, four Rickettsia spp.31,49,50,51, three Wolbachia spp.12,16,18,52, six Arsenophonus spp.10, one Hamiltonella53, one Fritschea13 and one Hemipteriphilus15 present in the B. tabaci species group. This result significantly improves our understanding on the substantial diversity of these endosymbionts. The reasons forthe observed variety and variability of secondary endosymbionts might be as follow: (1) horizontal transmission occurred among phylogenic closely species54, (2) horizontal transmission happened through insect parasitoids55, (3) as observed for Rickettsia, plant-mediated horizontal transmission also could be responsible for other bacteria56. Further work is clearly required to examine the above hypothesis and elucidate the mechanisms of transmission and maintenance of this bacterium within and between insects. In addition, due to a range of field populations that have built various, strong associations with different host-plants, field colonies are likely to contain a more varied set of microorganisms than lab strains. In our study, for example, a new Rickettsia OTU was detected in the Asia II 7 B. tabaci, which was collected from the plant host, Hibiscus rosa-sinensis (L.). It is different from the Rickettsia OTUs from MEAM1, Asia II 3 and Asia II 6 which were collected from eggplant, cotton and sweet potato, respectively.
Implications from the overall microbial diversity with host speciation
Apart from the Portiera P-endosymbiont, which was presented in all species, there was no significant pattern of S-endosymbiont association with B. tabaci species across the group. Multiple comparisons including alpha bacterial diversities, Principal Coordinate Analysis, indistinct endosymbiotic communities harbored by different species and the co-divergence analyses demonstrate the lack of association between overall microbial/S-endosymbiont diversity and cryptic species. Additionally, the majority of newly discovered endosymbionts are mainly secondary endosymbionts, but not the primary endosymbionts. These results indicate that the hypothesis of secondary endosymbiont-mediated speciation is unlikely to have occurred in the B. tabaci species group. The primary endosymbiont seems to be more reliable source to study the co-evolutionary speciation. The previous studies of phylogenetic congruence had conducted between distantly related whiteflies (i.e. between genera) and their P-endosymbionts23, but no studies had ever performed regard to the co-evolution between a group of closely related cryptic species and its primary endosymbiont, such as B. tabaci. In the molecular markers choice, the classical Portiera 16S rRNA marker was not able to distinguish P-endosymbionts from different B. tabaci cryptic species, which prevented the use of this marker for co-speciation studies. By scanning the three P-endosymbiont genomes generated from three cryptic species, a novel locus was selected to enable the design primers to investigate possible co-divergence of the P-endosymbiont and the host. Finally, significant cospeciating congruence between B. tabaci and Portiera57,58 was confirmed. In all, Portiera might have been resident in its host, B. tabaci when they were still a single species and subsequently diverged subtly with the divergence of B. tabaci. With the developmet of next/third-generation high throughput sequencing to obtain the genomes of P-endosymbionts and B. tabaci is more feasible. Further comparison of phylogenomic trees of them with precise fossils and biogeographic time points will allow studying the evolutionary history of their association24. Deciphering the evolution of B. tabaci and its bacteria might provide far-reaching insights not only on B. tabaci-endosymbiont themselves but also on the evolutionary history of other host-parasite pairs. This piece of research, therefore, provides a model framework, tools and methods to further test co-evolution hypothesis for other insect groups and their bacterial associations.
Conclusion
There are five main conclusions we can draw from our research:
-
(1)
Additional bacterial diversity in B. tabaci, with 15 new bacteria were found, which increases our understanding of the diversity of symbionts present in B. tabaci species and could revolutionize bacteria-insect microbiology, and it is now apparent that the diversity of microorganisms is much greater than previously thought.
-
(2)
To detect whether the results reflected by the PacBio are reliable, one of the new micro-organisms (Halomonas) was further confirmed to be present in B. tabaci. This finding provides robust experimental evidence to credibly support the presence of novel bacteria found in this study.
-
(3)
One Portiera OTU was present in all B. tabaci species, with a second in B. afer, indicating Portiera 16S might be a highly-effective marker for delineating the B. tabaci group members from other whitefly species within the Genus Bemisia. Supposing this conclusion is true, it will provide a significant contribution for boosting the taxonomy of some cryptic species groups infected by primary endosymbionts.
-
(4)
The hypothesis that P-endosymbionts co-evolve with their cryptic species hosts was firstly confirmed through utilizing a novel developed marker with a more rapid evolutionary rate than the 16S.
-
(5)
The lack of association between overall microbial/S-endosymbiont diversity with cryptic species revealed by multiple comparisons (alpha bacterial diversities, Principal Coordinate Analysis, indistinct endosymbiotic communities harbored by different species) indicate that secondary endosymbiont-mediated speciation is unlikely to have occurred in the B. tabaci species group.
All in all, these results confirmed that PacBio SMRT technology can improve detection and accurate identification of the known and unknown bacteria in the B. tabaci species group. This implies it may be an effective tool for bacteria diversity investigations in other pests. What’s more, as many novel endosymbionts discovered here, the diversity of endosymbionts in the B. tabaci (Hemipterans) seems to be much larger than previously thought. This suggests that the endosymbionts diversity in other insects of Hemipterans might have been underestimated, and as a consequence, their roles in mediating the host adaptation are largely overlooked. Overall, this study provides important implications for taxonomy, diversity and applied identification of pests known under collective name.
Material and Methods
Collection and preservation of specimens
Table 1 lists the meta-data on the B. tabaci and B. afer specimens, together with available information on host plants, locations, and GenBank information. B. afer has a genetically close relationship with B. tabaci and provides a good reference to interpret the results. The whiteflies were stored in 100% ethanol at −80 °C until DNA extraction.
DNA extraction and PCR amplification
To test the purity of each sample, the 5′ partial mtCOI gene region for species identification was amplified from B. tabaci samples using the new design primer pairs FHL (5′-TGRTTYTTTGGTCATCCVGAAGT-3′) and RHL(5′-TTACTGCACTTTCTGCCACATTAG-3′). DNA was extracted from 10 adult females separately from each sample (whole insect extractions) using the DNeasy animal tissue kit (Qiagen, Germany) following the manufacturer’s protocol and pooled in equal proportions.
16S rRNA amplication
The almost full-length, bacterial 16S rRNA genes of 10 individual females mixture of each specimens were amplified by PCR (95 °C for 2 min, followed by 35 cycles at 95 °C for 30 s, 55 °C for 30 s, and 72 °C for 60 s and a final extension at 72 °C for 10 min) using the forward primer 16S-27F (5′-AGAGTTTGATCMTGGCTCAG-3′), and the reverse primer16S-1492R (5′-GGYTACCTTGTTACGACTT-3′) modified from general 16S rRNA gene primers59 containing a variable 8 bp barcode sequence. PCR reactions were performed in triplicate, with 20 μL mixture containing 4 μL of 5 × FastPfu Buffer, 2 μL of 2.5mM dNTPs, 0.8 μL of each primer (5 μM), 0.4 μL of FastPfu Polymerase and 10 ng of template DNA. PCR amplicons were extracted from 2% agarose gels and purified using the AxyPrep DNA Gel Extraction Kit (Axygen Biosciences, Union City, CA, USA) according to the manufacturer’s instructions. After that, purified products were quantified by QuantiFluor™-ST (Promega, Madison, WI, USA).
PacBio 16S library construction and sequencing
Purified PCR products were quantified by Qubit®3.0 (Life Invitrogen) and every 24 amplicons with different barcodes were mixed in equal quantities. Pooled DNA was sequenced using P6-C4 chemistry on a PacBio RS II instrument (Pacific Biosciences, USA) and the entire sequence preprocessing was performed as described by Mosher, et al.60.
Processing of sequencing data generated from PacBio platform
For PacBio 16S datasets, raw data processing was carried out using the protocol RS_ReadsOfInsert.1 available in SMRT Portal version 2.7 as described by Hou, et al.61. Operational Taxonomic Units (OTUs) were clustered with 97% similarity cutoff using UPARSE (version 7.1 http://drive5.com/uparse/), and chimeric sequences were identified and removed using UCHIME. The phylogenetic affiliation of each 16S rRNA gene sequence was analyzed by the tool of RDP Classifier (http://rdp.cme.msu.edu/) against the SILVA62 16S rRNA reference database using a confidence threshold of 70%63. To show the distribution of the OTUs along various cryptic species, network analyses was done by Cytoscape64.
Diversity analyses
Rarefaction, Chao, ACE and Shannon diversity indices were performed using a set of Mothur tool65. To examine dissimilarities in the composition of bacterial communities, a Principal Coordinate Analysis (PCoA) was performed with an operating principle of using a distance matrix to plot n samples in (n–1)-dimensional space66.
Sequence alignments and phylogenetic tree for each bacterium
Sequences for the toward full-length 16S rRNA, specific region of P-endosymbionts and their corresponding B. tabaci hosts were each aligned using default parameters in MUSCLE67 as implemented in MEGA 568 or ClustalW-MPI69. Alignments were inspected, and ends were trimmed, resulting in 14,599 base pairs (bp) for the B. tabaci mitogenome sequences and 795 bp for the Portiera genome region.
The 16S rRNA gene data sets of published bacterial communities were downloaded from GenBank for phylogenetic analyses. Multiple 16S rRNA nucleotide sequences were aligned and clustered with a common and an empirical similarity threshold (97%)70 by using CD-HIT tool71. The Bayesian inference (BI) method was used to build the 16S molecular phylogenetic tree for each bacterium.
Verification of the presence and distribution of one of the new micro-organisms in the MED B. tabaci species
Halomonas72 has been found in the gut of pine weevils and it was detected in our PacBio data in more than 13 species, including MED, with more than five reads per sample. To determine whether this bacterium was present in the gut of B. tabaci, we conducted two subsequent experiments, which were illumina midguts -target sequencing and Fluorescence in situ hybridization (FISH) analyses.
Twenty mid-guts of adults were dissected at different times post-emergence (0-day, 0–1 day, 4–5 days) from both lab colony of MED and Asia II 1 (where Halomonas reads were absent)73,74. DNA was extracted from the midgut and used for sequencing the universal V4-V5 hyper variable region (515F/907R primer set)of 16S rRNA genes75 using Illumina Hiseq2500 platform with PE250 mode at Biozeron (Shanghai, China). To aid distinguish samples after sequencing, sample-specific 12-bp barcodes were added to the reverse primers. Different from PacBio sequences processing pipeline, V4-V5 region sequences were analyzed with QIIME software package (Quantitative Insights Into Microbial Ecology) following UPARSE pipeline76. OTUs were identified with UCLUST at the 97% sequence similarity level77 and a representative sequence from each OTU was aligned using PyNAST76.
To visualize the presence or absence of the Halomonas in the gut, FISH analyses were performed on dissected midguts as described by Ghanim, et al.78 with a slight modification. To target the 16S rRNA of the new Halomonas micro-organism, the probe Ha-Cy5 (5′-Cy5- TCACCAACTAGCTAATCCGACAT-3′) for the Halomonas strain was designed using Primer3 software (http://fokker.wi.mit.edu/primer3/) based on the sequences of the new bacterium strain 16S rRNA. The specificity of the detection was confirmed using the following controls: a no-probe control, an unlabeled competitive suppression control and Halomonas-free whiteflies.
Co-divergence analysis
The global ParaFit79 statistic was used to conduct the co-divergence analysis. ParaFit measures two hypotheses: H0, evolution of the hosts and parasites occurred independently; and H1, the positions of the individual Host-Parasite (H-P) links are not random but associated with corresponding branches of the two evolutionary trees. A high fit to H1 is consistent with cophylogenetic history. In this case, B. tabaci represents the host while Portiera represents the parasite. They were used to quantify the degree of congruence between B. tabaci and P-endosymbiont topologies and identify the individual associations contributing to the co-phylogenetic structure80. To get reliable topologies for further analyses, two major phylogenetic methods, BI and Maximum Likelihood (ML) were used to compare phylogenetic structures of P-endosymbiont and B. tabaci. BI was conducted using MrBayes 3.281 and ML analyses with RAxMLv 7.0.4, implementing a fast bootstrapping algorithm82. Bayesian analysis was conducted in combination with an exact model (GTR + I + G) of molecular evolution, as well as a rapid approximation of a posterior probability tree using Markov Chain Monte Carlo83. MrBayes 3.2 was run using four incrementally heated chains and run for 10 million generations, sampling every 5000th generation with four heated chains and a burn-in length of 1 million. To corroborate the phylogenies, as determined through BI, the ML P-endosymbiont and B. tabaci phylogenies were generated and the substitution model GTR with CAT approximation was used to incorporate heterogeneity rate across sites. The B. tabaci Japan 2 species was chosen as an outgroup owing to this species having a more distant genetic relationship with the other members of the B. tabaci species.
Matrices of parasite and host distances were calculated from host and parasite phylogenies with an additional matrix of host–parasite links. Next, ParaFit analyses were performed in R using the package “ape”84 with 999 permutations to implement a global test as well as individual links. Each B. tabaci species and P-endosymbiont interaction was determined to be significant if either its ParaFitLink1 or ParaFitLink2 P-value ≤ 0.05. The recently developed, “Procrustean Approach to Co-phylogeny(PACo)” program85, implemented in R using packages ape and vegan86, was used to obtain and potentially corroborate, comparable global goodness-of-fit statistics with ParaFit global values. Although some specimens belonged to the same species, each terminal taxon was treated as an independent evolutionary unit as is common in co-speciation literature87.
Data Availability
The raw reads of PacBio sequencing results were deposited in the NCBI Sequence Read Archive (SRA) database (Accession Number: SRP117655).
Change history
18 June 2019
A correction to this article has been published and is linked from the HTML and PDF versions of this paper. The error has been fixed in the paper.
References
López-Sánchez, M. J. et al. Evolutionary convergence and nitrogen metabolism in Blattabacterium strain Bge, primary endosymbiont of the cockroach Blattella germanica. PLoS Genet. 5, e1000721, https://doi.org/10.1371/journal.pgen.1000721 (2009).
Shigenobu, S., Watanabe, H., Hattori, M., Sakaki, Y. & Ishikawa, H. Genome sequence of the endocellular bacterial symbiont of aphids Buchnera sp. APS. Nature 407, 81–86, https://doi.org/10.1038/35024074 (2000).
Douglas, A. E. Phloem-sap feeding by animals: problems and solutions. J. Exp. Bot. 57, 747–754, https://doi.org/10.1093/jxb/erj067 (2006).
Aksoy, S. Tsetse-a haven for microorganisms. Parasitol. Today 16, 114–118 (2000).
Oliver, K. M. et al. Parasitic wasp responses to symbiont-based defense in aphids. BMC Biol. 10, 11, https://doi.org/10.1186/1741-7007-10-11 (2012).
Scarborough, C. L., Ferrari, J. & Godfray, H. C. J. Aphid protected from pathogen by endosymbiont. Science 310, 1781–1781, https://doi.org/10.1126/science.1120180 (2005).
Hedges, L. M., Brownlie, J. C., O’Neill, S. L. & Johnson, K. N. Wolbachia and virus protection in insects. Science 322, 702–702, https://doi.org/10.1126/science.1162418 (2008).
Boykin, L. M. et al. Global relationships of Bemisia tabaci (Hemiptera: Aleyrodidae) revealed using Bayesian analysis of mitochondrial COI DNA sequences. Mol. Phylogenet. Evol. 44, 1306–1319, https://doi.org/10.1016/j.ympev.2007.04.020 (2007).
Dinsdale, A., Cook, L., Riginos, C., Buckley, Y. & Barro, P. D. Refined global analysis of Bemisia tabaci (Hemiptera: Sternorrhyncha: Aleyrodoidea: Aleyrodidae) mitochondrial cytochrome oxidase 1 to identify species level genetic boundaries. Ann. Entomol. Soc. Am. 103, 196–208, https://doi.org/10.1603/AN09061 (2010).
Zchori-Fein, E. & Brown, J. K. Diversity of prokaryotes associated with Bemisia tabaci (Gennadius) (Hemiptera: Aleyrodidae). Ann. Entomol. Soc. Am. 95, 711–718, https://doi.org/10.1603/AN09061 (2002).
Weeks, A. R., Velten, R. & Stouthamer, R. Incidence of a new sex-ratio-distorting endosymbiotic bacterium among arthropods. P. Roy. Soc. B-Biol. Sci. 270, 1857–1865, https://doi.org/10.1098/rspb.2003.2425 (2003).
Nirgianaki, A. et al. Wolbachia infections of the whitefly Bemisia tabaci. Curr. Microbiol. 47, 0093–0101,, https://doi.org/10.1007/s00284-002-3969-1 (2003).
Everett, K. D. E., Thao, M., Horn, M., Dyszynski, G. E. & Baumann, P. Novel chlamydiae in whiteflies and scale insects: endosymbionts ‘Candidatus Fritschea bemisiae’ strain Falk and ‘Candidatus Fritschea eriococci’ strain Elm. Int. J. Sys. Evol. Micr. 55, 1581–1587, https://doi.org/10.1099/ijs.0.63454-0 (2005).
Gottlieb, Y., Perlman, S. J., Chiel, E. & Zchori Fein, E. 10 Rickettsia get around. Manipulative Tenants: Bacteria Associated with Arthropods, 191 (2011).
Bing, X. L., Yang, J., Zchori-Fein, E., Wang, X. W. & Liu, S. S. Characterization of a newly discovered symbiont of the whitefly Bemisia tabaci (Hemiptera: Aleyrodidae). Appl. Environ. Microb. 79, 569–575, https://doi.org/10.1128/AEM.03030-12 (2013).
Bing, X. L., Ruan, Y. M., Rao, Q., Wang, X. W. & Liu, S. S. Diversity of secondary endosymbionts among different putative species of the whitefly Bemisia tabaci. Insect Sci. 20, 194–206, https://doi.org/10.1111/j.1744-7917.2012.01522.x (2013).
Ji, H. L., Qi, L. D., Hong, X. Y., Xie, H. F. & Li, Y. X. Effects of host sex, plant species, and putative host species on the prevalence of Wolbachia in natural populations of Bemisia tabaci (Hemiptera: Aleyrodidae): A modified nested PCR study. J. Econ. Entomol. 108, 210–218, https://doi.org/10.1093/jee/tou004 (2015).
Ghosh, S., Bouvaine, S. & Maruthi, M. N. Prevalence and genetic diversity of endosymbiotic bacteria infecting cassava whiteflies in Africa. BMC Microbiol. 15, 93, https://doi.org/10.1186/s12866-015-0425-5 (2015).
Degnan, P. H. & Ochman, H. Illumina-based analysis of microbial community diversity. ISME J. 6, 183–194, https://doi.org/10.1038/ismej.2011.74 (2012).
Singer, E. et al. High-resolution phylogenetic microbial community profiling. ISME J. 10, 2020–2032, https://doi.org/10.1038/ismej.2015.249 (2016).
Baumann, P., Moran, N. A. & Baumann, L. Bacteriocyte-associated endosymbionts of insects. The Prokaryotes: Volume 1: Symbiotic associations, Biotechnology, Applied Microbiology, 403–438 (2006).
Wilson, A. C. C. et al. Genomic insight into the amino acid relations of the pea aphid, Acyrthosiphon pisum, with its symbiotic bacterium Buchnera aphidicola. Insect Mol. Biol. 19, 249–258, https://doi.org/10.1111/j.1365-2583.2009.00942.x (2010).
Thao, M. L. & Baumann, P. Evolutionary relationships of primary prokaryotic endosymbionts of whiteflies and their hosts. Appl. Environ. Microb. 70, 3401–3406, https://doi.org/10.1128/AEM.66.7.2898-2905.2000 (2004).
Santos-Garcia, D., Vargas-Chavez, C., Moya, A., Latorre, A. & Silva, F. J. Genome evolution in the primary endosymbiont of whiteflies sheds light on their divergence. Genome Biol. Evol. 7, 873–888, https://doi.org/10.1093/gbe/evv038 (2015).
Clark, M. A., Moran, N. A., Baumann, P. & Wernegreen, J. J. Cospeciation between bacterial endosymbionts (Buchnera) and a recent radiation of aphids (Uroleucon) and pitfalls of testing for phylogenetic congruence. Evolution 54, 517–525, https://doi.org/10.1111/j.0014-3820.2000.tb00054.x (2000).
Funk, D. J., Helbling, L., Wernegreen, J. J. & Moran, N. A. Intraspecific phylogenetic congruence among multiple symbiont genomes. P. Roy. Soc. B: Biol. Sci. 267, 2517–2521, https://doi.org/10.1098/rspb.2000.1314 (2000).
Sloan, D. B. & Moran, N. A. Genome reduction and co-evolution between the primary and secondary bacterial symbionts of psyllids. Mol. Biol. Evol. 29, 3781–3792, https://doi.org/10.1093/molbev/mss180 (2012).
Spaulding, A. W. & Von Dohlen, C. D. Psyllid endosymbionts exhibit patterns of co‐speciation with hosts and destabilizing substitutions in ribosomal RNA. Insect Mol. Biol. 10, 57–67, https://doi.org/10.1046/j.1365-2583.2001.00231.x (2001).
Gruwell, M. E., Morse, G. E. & Normark, B. B. Phylogenetic congruence of armored scale insects (Hemiptera: Diaspididae) and their primary endosymbionts from the phylum Bacteroidetes. Mol. Phylogenet. Evol. 44, 267–280, https://doi.org/10.1016/j.ympev.2007.01.014 (2007).
Jing, X. et al. The bacterial communities in plant phloem-sap-feeding insects. Mol. Ecol. 23, 1433–1444, https://doi.org/10.1111/mec.12637 (2014).
Singh, S. T. et al. Diversity and phylogenetic analysis of endosymbiotic bacteria from field caught Bemisia tabaci from different locations of North India based on 16S rDNA library screening. Infect., Genet. Evol. 12, 411–419, https://doi.org/10.1016/j.meegid.2012.01.015 (2012).
Ahmed, M. Z., De Barro, P. J., Ren, S. X., Greeff, J. M. & Qiu, B. L. Evidence for horizontal transmission of secondary endosymbionts in the Bemisia tabaci cryptic species complex. PLoS ONE 8, e53084, https://doi.org/10.1371/journal.pone.0053084 (2013).
Quail, M. A. et al. A tale of three next generation sequencing platforms: comparison of Ion Torrent, Pacific Biosciences and Illumina MiSeq sequencers. BMC Genomics 13, 341, https://doi.org/10.1186/1471-2164-13-341 (2012).
Ofek, M., Hadar, Y. & Minz, D. Ecology of root colonizing Massilia (Oxalobacteraceae). PLoS ONE 7, e40117, https://doi.org/10.1371/journal.pone.0040117 (2012).
Lafi, F. F. et al. Draft genome sequence of the plant growth-promoting Cupriavidus gilardii strain JZ4 isolated from the desert plant Tribulus terrestris. Genome Announcements 4, e00678–00616, https://doi.org/10.1128/genomeA.00678-16 (2016).
White, D. C., Sutton, S. D. & Ringelberg, D. B. The genus Sphingomonas: physiology and ecology. Curr. Opin. Biotech. 7, 301–306 (1996).
Seifert, H. et al. Distribution of Acinetobacter species on human skin: comparison of phenotypic and genotypic identification methods. J. Clin. Microb. 35, 2819–2825 (1997).
Cooper, W. R., Sengoda, V. G. & Munyaneza, J. E. Localization of ‘Candidatus Liberibacter solanacearum’(Rhizobiales: Rhizobiaceae) in Bactericera cockerelli (Hemiptera: Triozidae). Ann. Entomol. Soc. Am. 107, 204–210, https://doi.org/10.1603/AN13087 (2014).
Mantelin, S. et al. Emended description of the genus Phyllobacterium and description of four novel species associated with plant roots: Phyllobacterium bourgognense sp. nov., Phyllobacterium ifriqiyense sp. nov., Phyllobacterium leguminum sp. nov. and Phyllobacterium brassicacearum sp. nov. Int. J. Syst. Evol. Micro. 56, 827–839, https://doi.org/10.1099/ijs.0.63911-0 (2006).
Kikuchi, Y., Meng, X. Y. & Fukatsu, T. Gut symbiotic bacteria of the genus Burkholderia in the broad-headed bugs Riptortus clavatus and Leptocorisa chinensis (Heteroptera: Alydidae). Appl. Environ. Microbiol. 71, 4035–4043, https://doi.org/10.1128/AEM.71.7.4035-4043.2005 (2005).
Ash, C., Priest, F. G. & Collins, M. D. Molecular identification of rRNA group 3 bacilli (Ash, Farrow, Wallbanks and Collins) using a PCR probe test. Antonie van Leeuwenhoek 64, 253–260 (1993).
da Mota, F. F., Gomes, E. A., Paiva, E. & Seldin, L. Assessment of the diversity of Paenibacillus species in environmental samples by a novel rpoB-based PCR-DGGE method. FEMS Microbiol. Ecol. 53, 317–328, https://doi.org/10.1016/j.femsec.2005.01.017 (2005).
Dillon, R. J. & Dillon, V. M. The gut bacteria of insects: nonpathogenic interactions. Ann. Rev. Entomol. 49, 71–92, https://doi.org/10.1146/annurev.ento.49.061802.123416 (2004).
Fukatsu, T. & Hosokawa, T. Capsule-transmitted gut symbiotic bacterium of the Japanese common plataspid stinkbug, Megacopta punctatissima. Appl. Environ. Microbiol. 68, 389–396, https://doi.org/10.1128/AEM.68.1.389-396.2002 (2002).
Douglas, A. E. The microbial dimension in insect nutritional ecology. Funct. Ecol. 23, 38–47, https://doi.org/10.1111/j.1365-2435.2008.01442.x (2009).
Prado, S. S. & Almeida, R. P. P. Role of symbiotic gut bacteria in the development of Acrosternum hilare and Murgantia histrionica. Entomol. Exp. Appl. 132, 21–29, https://doi.org/10.1111/j.1570-7458.2009.00863.x (2009).
Poddar, N., Ramakrishnan, B. & Subramanian, S. Culturable bacteria in Asia I and Asia II genetic groups of whitefly Bemisia tabaci (Gennadius) species complex. Indian J. Entomol. 78, 260–263, https://doi.org/10.5958/0974-8172.2016.00069.9 (2016).
Boykin, L. M., Bell, C. D., Evans, G., Small, I. & De Barro, P. J. Is agriculture driving the diversification of the Bemisia tabaci species complex (Hemiptera: Sternorrhyncha: Aleyrodidae)?: Dating, diversification and biogeographic evidence revealed. BMC Evol. Biol. 13, 228, https://doi.org/10.1186/1471-2148-13-228 (2013).
Gottlieb, Y. et al. Identification and localization of a Rickettsia sp. in Bemisia tabaci (Homoptera: Aleyrodidae). Appl. Environ. Microbiol. 72, 3646–3652, https://doi.org/10.1128/AEM.72.5.3646-3652.2006 (2006).
Rao, Q., Wang, S., Zhu, D. T., Wang, X. W. & Liu, S. S. Draft genome sequence of “Rickettsia sp. strain MEAM1”, isolated from the whitefly. Bemisia tabaci. J. Bacteriol. 194, 4741–4742, https://doi.org/10.1128/JB.00909-12 (2012).
Zhu, D. T. et al. Sequencing and comparison of the Rickettsia genomes from the whitefly Bemisia tabaci Middle East Asia Minor I. Insect Sci. 23, 531–542, https://doi.org/10.1111/1744-7917.12367 (2016).
Bing, X. L. et al. Diversity and evolution of the Wolbachia endosymbionts of Bemisia (Hemiptera: Aleyrodidae) whiteflies. Ecol. Evol. 4, 2714–2737, https://doi.org/10.1002/ece3.1126 (2014).
Rao, Q. et al. Draft genome sequence of “Candidatus Hamiltonella defensa”, an endosymbiont of the whitefly. Bemisia tabaci. J. Bacteriol. 194, 3558, https://doi.org/10.1128/JB.00069-12 (2012).
Russell, J., Latorre, A., Sabater-Muñoz, B., Moya, A. & Moran, N. Side-stepping secondary symbionts: widespread horizontal transfer across and beyond the Aphidoidea. Mol. Ecol. 12, 1061–1075, https://doi.org/10.1046/j.1365-294x.2003.01780.x (2003).
Benjamin, D. H., Robert, D. J., William, G. F. & Stephen, F. H. Horizontal transfer of Wolbachia between phylogenetically distant insect species by a naturally occurring mechanism. Curr. Biol. 9, 313–316, https://doi.org/10.1016/S0960-9822(99)80139-0 (1999).
Caspi-Fluger, A. et al. Horizontal transmission of the insect symbiont Rickettsia is plant-mediated. Proc. R Soc. B 279, 1791–1796, https://doi.org/10.1098/rspb.2011.2095 (2012).
Peek, A. S., Feldman, R. A., Lutz, R. A. & Vrijenhoek, R. C. Cospeciation of chemoautotrophic bacteria and deep sea clams. PNAS. 95, 9962–9966 (1998).
Thao, M. L. et al. Cospeciation of psyllids and their primary prokaryotic endosymbionts. Appl. Environ. Microbiol. 66, 2898–2905, https://doi.org/10.1128/AEM.70.6.3401-3406.2004 (2000).
Osborne, C. A., Galic, M., Sangwan, P. & Janssen, P. H. PCR-generated artefact from 16S rRNA gene-specific primers. FEMS Microbiol. Lett. 248, 183–187, https://doi.org/10.1016/j.femsle.2005.05.043 (2005).
Mosher, J. J., Bernberg, E. L., Shevchenko, O., Kan, J. & Kaplan, L. A. Efficacy of a 3rd generation high-throughput sequencing platform for analyses of 16S rRNA genes from environmental samples. J. Microbiol. Methods 95, 175–181, https://doi.org/10.1016/j.mimet.2013.08.009 (2013).
Hou, Q. et al. Evaluation of bacterial contamination in raw milk, ultra-high temperature milk and infant formula using single molecule, real-time sequencing technology. J. Dairy Sci. 98, 8464–8472, https://doi.org/10.3168/jds.2015-9886 (2015).
Pruesse, E. et al. SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res. 35, 7188–7196, https://doi.org/10.1093/nar/gkm864 (2007).
Amato, K. R. et al. Habitat degradation impacts black howler monkey (Alouatta pigra) gastrointestinal microbiomes. ISME J. 7, 1344–1353, https://doi.org/10.1038/ismej.2013.16 (2013).
Kohl, M., Wiese, S. & Warscheid, B. Cytoscape: software for visualization and analysis of biological networks. Data Mining in Proteomics: From Standards to Applications, 291–303, https://doi.org/10.1007/978-1-60761-987-1_18 (2011).
Schloss, P. D. et al. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 75, 7537–7541, https://doi.org/10.1128/AEM.01541-09 (2009).
Lozupone, C., Lladser, M. E., Knights, D., Stombaugh, J. & Knight, R. UniFrac: an effective distance metric for microbial community comparison. ISME J. 5, 169 (2011).
Edgar, R. C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797, https://doi.org/10.1093/nar/gkh340 (2004).
Tamura, K. et al. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 28, 2731–2739, https://doi.org/10.1093/molbev/msr121 (2011).
Li, K. B. ClustalW-MPI: ClustalW analysis using distributed and parallel computing. Bioinformatics 19, 1585–1586, https://doi.org/10.1093/bioinformatics/btg192 (2003).
Konstantinidis, K. T. & Tiedje, J. M. Genomic insights that advance the species definition for prokaryotes. PNAS. 102, 2567–2572 (2005).
Fu, L., Niu, B., Zhu, Z., Wu, S. & Li, W. CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics 28, 3150–3152, https://doi.org/10.1093/bioinformatics/bts565 (2012).
Ölander, T. Culture independent analysis of microbiota in the gut of pine weevils. PHD thesis. Royal Institute of Technology. Sweden (2013).
Wang, H. L. et al. Developing conversed microsatellite markers and their implications in evolutionary analysis of the Bemisia tabaci complex. Sci. Rep. 4, 6351, https://doi.org/10.1038/srep06351 (2014).
Zhu, D. T. et al. Methods for the extraction of endosymbionts from the whitefly Bemisia tabaci. J. Vis. Exp. 124, e55809, https://doi.org/10.3791/55809 (2017).
Biddle, J. F., Fitz-Gibbon, S., Schuster, S. C., Brenchley, J. E. & House, C. H. Metagenomic signatures of the Peru Margin subseafloor biosphere show a genetically distinct environment. PNAS. 105, 10583–10588, https://doi.org/10.1073/pnas.0709942105 (2008).
Caporaso, J. G. et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7, 335–336, https://doi.org/10.1038/nmeth.f.303 (2010).
Edgar, R. C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461, https://doi.org/10.1093/bioinformatics/btq461 (2010).
Ghanim, M., Brumin, M. & Popovski, S. A simple, rapid and inexpensive method for localization of Tomato yellow leaf curl virus and Potato leafroll virus in plant and insect vectors. J. Virological Methods 159, 311–314, https://doi.org/10.1016/j.jviromet.2009.04.017 (2009).
Legendre, P., Desdevises, Y. & Bazin, E. A statistical test for host–parasite coevolution. Syst. Biol. 51, 217–234, https://doi.org/10.1080/10635150252899734 (2002).
Desdevises, Y. Cophylogeny: Insights from fish-parasite systems. Parassitologia 49, 125–128 (2007).
Ronquist, F. et al. Mrbayes 3.2: Efficient bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 61, 539–542, https://doi.org/10.1093/sysbio/sys029 (2012).
Stamatakis, A., Hoover, P. & Rougemont, J. A rapid bootstrap algorithm for the RAxML web servers. Syst. Biol. 57, 758–771, https://doi.org/10.1080/10635150802429642 (2008).
Huelsenbeck, J. P., Larget, B., Miller, R. E. & Ronquist, F. Potential applications and pitfalls of Bayesian inference of phylogeny. Syst. Biol. 51, 673–688, https://doi.org/10.1080/10635150290102366 (2002).
Popescu, A. A., Huber, K. T. & Paradis, E. Ape 3.0: New tools for distance-based phylogenetics and evolutionary analysis in R. Bioinformatics 28, 1536–1537, https://doi.org/10.1093/bioinformatics/bts184 (2012).
Balbuena, J. A., Míguez-Lozano, R. & Blasco-Costa, I. PACo: a novel procrustes application to cophylogenetic analysis. PLoS ONE 8, e61048, https://doi.org/10.1371/journal.pone.0061048 (2013).
Philip, D. VEGAN, a package of R functions for community ecology. J. Veg. Sci. 14, 927–930, https://doi.org/10.1658/1100-9233(2003)014[0927:VAPORF]2.0.CO;2 (2003).
Page, R. D. et al. Phylogeny of “Philoceanus complex” seabird lice (Phthiraptera: Ischnocera) inferred from mitochondrial DNA sequences. Mol. Phylogenet. Evol. 30, 633–652, https://doi.org/10.1016/S1055-7903(03)00227-6 (2004).
Acknowledgements
This work was supported by the China Postdoctoral Science Foundation (Grant number 517000-X91502), the China Postdoc Science Special Foundation (Grant number 517000-X91609), the National Natural Science Foundation of China (Grant number 31501878) and by grants from the Bill & Melinda Gates Foundation (African cassava whitefly project, OPP1058938). Grants (AGL2013-48913-C2-1-R and AGL2016-75819-C2-2-R) from the Ministerio de Economía y Competitividad (MINECO, Spain), co-financed by ERDF. E.F.O. is a recipient of a ‘Juan de la Cierva-Incorporación’ postdoctoral contract from MINECO.
Author information
Authors and Affiliations
Contributions
Hua-Ling Wang contributed to the design, implementation and data collection. Hua-Ling Wang wrote the manuscript with contributions from all authors, Teng Lei, Wen-Qiang Xia, Stephen Cameron,Yin-Quan Liu, Zhen Zhang, Maruthi M N Gowda,Paul De Barro, Jesús Navas-Castillo, Christopher A Omongo, Hélène Delatte, Kyeong-Yeoll Lee, Mitulkumar V. Patel, Renate Krause-Sakate, James Ng, SanLing Wu, Elvira Fiallo-Olivé, Shu-Sheng Liu, John Colvin and Xiao-Wei Wang, in writing, producing the figures, analyzing the data and interpreting the results.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wang, HL., Lei, T., Xia, WQ. et al. Insight into the microbial world of Bemisia tabaci cryptic species complex and its relationships with its host. Sci Rep 9, 6568 (2019). https://doi.org/10.1038/s41598-019-42793-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-42793-8
This article is cited by
-
Composition of the whiteflies microbiome in populations with and without insecticide applications in Yucatan Mexico
Biologia (2024)
-
Development of a novel nuclear DNA marker to identify biotypes A and B of the stone leek leafminer, Liriomyza chinensis (Diptera: Agromyzidae)
Applied Entomology and Zoology (2022)
-
Bemisia tabaci in Java, Indonesia: genetic diversity and the relationship with secondary endosymbiotic bacteria
Symbiosis (2021)
-
The potential of nanobiopesticide based on zein nanoparticles and neem oil for enhanced control of agricultural pests
Journal of Pest Science (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
