
Abstract
Human milk represents a source of bacteria for the initial establishment of the oral (and gut) microbiomes in the breastfed infant, however, the origin of bacteria in human milk remains largely unknown. While some evidence points towards a possible endogenous enteromammary route, other authors have suggested that bacteria in human milk are contaminants from the skin or the breastfed infant mouth. In this work 16S rRNA sequencing and bacterial culturing and isolation was performed to analyze the microbiota on maternal precolostrum samples, collected from pregnant women before delivery, and on oral samples collected from the corresponding infants. The structure of both ecosystems demonstrated a high proportion of taxa consistently shared among ecosystems, Streptococcus spp. and Staphylococcus spp. being the most abundant. Whole genome sequencing on those isolates that, belonging to the same species, were isolated from both the maternal and infant samples in the same mother-infant pair, evidenced that in 8 out of 10 pairs both isolates were >99.9% identical at nucleotide level. The presence of typical oral bacteria in precolostrum before contact with the newborn indicates that they are not a contamination from the infant, and suggests that at least some oral bacteria reach the infant’s mouth through breastfeeding.
Similar content being viewed by others

Introduction
Maternal microbes exert important roles in determining the establishment and assembly of human microbiomes in early life and, therefore, have a notable impact on infant health in the short and long term1,2,3. Diverse factors including, among others, gestational age, delivery mode (vaginal delivery versus C-section), type of infant feeding (exclusive breastfeeding, infant formula, mixed feeding), time from birth to skin-to-skin contact, perinatal use of antibiotics or the health status of the mother (e.g., infections, diabetes, metabolic syndrome) significantly influence the process of acquisition and establishment of the infant microbiota1,4,5,6,7,8,9,10,11,12,13,14. Globally, such influences may have direct consequences on the infant metabolism, immunological and neuroendocrinological programming and ultimately on health outcomes later in life. In fact, those situations altering or disturbing the infant gut microbiota have been associated with an increased incidence of metabolic and inflammatory disorders later in adulthood15,16,17,18,19.
In this context, human milk seems to play a particularly relevant role in driving the establishment of the infant gut microbiota, attributed to its content in antimicrobial and immunological compounds, prebiotic substances (lactose, human milk oligosaccharides) and live microorganisms, among other biologically active components20,21,22,23. Indeed, breastfeeding has been demonstrated to represent a supply of bacteria for the infant gut and, the sharing of bacterial species and strains between maternal milk and the infant gut has been demonstrated in several occasions through both culture-independent and culture-dependent approaches13,14,24,25,26,27,28. While in the past, the presence of bacterial cells in human milk was considered the result of contamination originating from the infant’s oral cavity (streptococci) or the mother’s skin (staphylococci, propionibacteria)29, the detection of live bacterial cells and/or DNA from genera and species (including strict anaerobes) that are usually confined to the gut (Bifidobacterium, Bacteroides, Parabacteroides, Blautia, Clostridium, Collinsella, Coprococcus, Faecalibacterium, Roseburia, Subdoligranulum, …)24,30 has fueled a scientific debate on the origin of human milk bacteria31. In addition, human milk also contains many bacteria which are typical inhabitants of the oral cavity, ranging from Streptococcus to Veillonella, Leptotrichia, Prevotella or members of the TM7 phylum, whose presence in human milk has been generally attributed to contaminants originating in the infant mouth32. Therefore, although infant’s mouth and/or the maternal skin may provide some bacteria to the milk, this is not incompatible with the potential role of human milk as a source of bacteria to the infant33,34.
In this context, the aims of this work were first to analyze the microbiota of precolostrum, the biological fluid secreted by some women during late pregnancy, and thus before having had any contact with the infant’s mouth; and next, to determine the potential transfer of bacterial strains present in precolostrum at the end of pregnancy, to the infant mouth.
Material and Methods
Participating women and sample collection
A total of 17 mother-infant pairs were recruited for their participation in the study. To be eligible for participation, women had to meet the following criteria: (1) ≥18 year of age; (2) a healthy pregnancy ≥37 weeks; (3) secreting precolostrum; (4) intention to breastfeed their babies; and (5) normal body mass index (18.5 to 24.9 before pregnancy). Exclusion criteria included underlying conditions (including breast pain) and maternal use of antibiotics in the previous 30 days. The main characteristics of the recruited women are presented in Table 1.
Precolostrum samples were collected at week 38–40 of pregnancy by manual expression using gloved hands. For this purpose, the mammary areola and nipple were first cleaned with prepackaged castile soap towelettes (Professional Disposables International, Inc.; Orangeburg, NY) and, later, soaked in chlorhexidine (Cristalmina, Salvat, Spain). The first drops were discarded and, subsequently, a fraction of ~500 μL was absorbed into a sterile cotton swab. Infant oral samples were collected 5–7 days after delivery by rubbing the infant gums with a sterile cotton swab. Samples were immediately placed in cold box (4 °C) and, within 1 hour, they were frozen and kept at −20 °C until processed.
All mothers participating in the study gave written informed consent to the protocol (reference 10/017E), which had been previously approved by the Ethical Committee of Clinical Research of Hospital Clínico San Carlos, Madrid (Spain). The study was carried out in accordance with the Declaration of Helsinki.
Culture-dependent analysis
Both precolostrum and infant oral samples were retrieved from the cotton swabs through suspension into 1 ml of sterile peptone water and vigorous shaking. Then 50 μl of serial ten-fold dilutions of each sample were spread on Columbia Nadilixic Acid (BioMerieux) (CNA), MacConkey (BioMerieux) (MCK), and de Man, Rogosa and Sharpe (Oxoid) supplemented with 0.25% g L−1 L-cysteine hydrochloride (Sigma) (MRSc) agar plates. CNA and MCK plates were incubated aerobically at 37 °C for 2 days while MRSc plates were incubated anaerobically at 37 °C for 3 days, prior to colony picking. Representative colonies from each plate exhibiting different morphologies were stored in glycerol stocks and further identified by Matrix Assisted Laser Desorption Ionization-Time of Flight (MALDI-TOF) mass spectrometry using a Vitek-MS™ instrument (BioMérieux, Marcy l’Etoile, France). Briefly, a portion of a bacterial colony (~1 µL) was directly spotted onto a MALDI sample plate. Then, it was overlaid with 1 µL of a saturated solution of α-cyano-4-hydroxycinnamic acid in acetonitrile (28%) and allowed to dry at room temperature. For each isolate, a mean spectrum was constructed with at least 50 m/z spectra profiles and used for the identification by comparison with the spectra contained in the Myla database (Biomerieux). Identification was defined as a 99–100% match to the species-specific m/z values in the database.
In order to determine potential maternal-infant sharing of bacteria at the strain level, those isolates belonging to the same species and obtained from the precolostrum and infant oral samples of the same mother-infant pair were further genotyped by RAPD-PCR profiling following the procedure described by Martín and colleagues26 and whole genome sequencing as described below. Samples of mature milk and infant mouth from 3 additional mother-infant pairs collected 8 weeks after birth were also included in this part of the work. These were used as a kind of positive controls since potential sharing of strains between precolostrum and infant mouth was unknown at the beginning of this study while a high degree of strain sharing between mature milk and infant mouth was expected14,33,34.
DNA extraction, amplification and 16S rRNA sequencing
Both precolostrum and infant oral samples were preserved and processed identically as previously described35. Briefly, suspended cells were collected by centrifugation at 13,000 ×g for 10 min at 4 °C and suspended in 0.5 mL TE50 (10 mM Tris-HCl, 50 mM EDTA, pH 8). Then, they were subjected to enzymatic lysis by adding 100 µL of a mixture containing lysozyme (Sigma-Aldrich), mutanolysin (Sigma-Aldrich) and lysostaphin (Sigma-Aldrich) as previously described35 and incubating the samples for 1 h at 37 °C. Immediately afterwards, physical disruption was conducted by 3 bead-beating cycles of 1 min each, on a FastPrep instrument (QBioGene, Irvine, CA, USA) by using 0.1 mm zirconia/silica beads (Sigma). Following cell lysis, DNA was purified from the sample by using the QIAmp DNA Mini Kit (Qiagen) according to the manufacturer’s instructions.
Extracted DNA was eluted in 22 µL nuclease-free water and stored at −20 °C until further analysis. A dual-barcoded 2-step PCR was conducted to amplify a fragment of the V3–V4 hypervariable region of the bacterial 16S ribosomal RNA (rRNA) gene; by using equimolar concentrations of the universal primers S-D- Bact-0341-b-S-17 (ACACTGACGACATGGTTCTACACCTACGGGNGGCWGCAG) and S-D-Bact-0785-a-A-21 (TACGGTAGCAGAGACTTGGTCTGACTACHVGGGTATCTAATCC) as previously described36, generating ~464 bp amplicons from the V3 to V4 hypervariable regions. Barcodes used for Illumina sequencing were appended to 3′ and 5′ terminal ends of PCR amplicons to allow separation of forward and reverse sequences. A bioanalyzer (2100 Bionalyzer, Agilent) was used to determine the concentration of every sample in the region of interest.
Two negative control blanks, which included no sample, were subject to all steps of the DNA extraction and purification procedure described above. The concentration of DNA in the two blank preparations was approximately 0.01 ng µl−1 while that obtained from the biological samples that were included in the 16S rRNA sequencing analysis was, at least, 159.9 ng µl−1. In addition, no amplification was detected from the blank samples after the first PCR and, as a consequence, the two blank controls were not submitted to sequencing.
Barcoded PCR products from all samples were pooled at approximately equal molar DNA concentrations and run on a preparative agarose gel. The correct sized band was excised, the DNA was purified and one aliquot of pooled, purified, barcoded DNA amplicons was sequenced on an Illumina MiSeq machine using a pair-ends 2 × 250 bp protocol (Illumina Inc., San Diego, CA, USA) at the Unidad de Genómica of the Fundación Parque Científico de Madrid (Spain). All sequences generated in this work are deposited in the Short Reads Archive under bioproject number PRJNA489791.
Processing of 16S rDNA amplicon sequences
Raw sequencing reads of V3-V4 amplicons of the 16S rRNA gene amplicons were processed by using the version 1.9.1 of the Quantitative Insights Into Microbial Ecology (QIIME) software, using the open-reference OTU calling approach37,38. Briefly, reads were demultiplexed and primers and barcodes were trimmed using QIIME’s default settings. Following trimming, paired end sequence reads were merged with default QIIME settings and clustered into OTUs against the GreenGenes database (Version 13_8) by using the parallel uclust_ref method39. Reads with no hits in the reference sequence collection were clustered with de novo OTU method. Singletons and doubletons OTUs were filtered out, and only those OTUs representing more than 0.001% of the total filtered taxa were retained for diversity analyses40. For conducting statistical analyses at genus level, OTUs were collapsed into the corresponding taxonomic level by using the tax_glom functions in PhyloSeq41.
Whole genome sequencing of mother-child paired isolates and comparative genomics
DNA from 10 pairs of bacterial isolates (each pair represented by one isolate from precolostrum and one isolate from the corresponding infant’s oral cavity) that appeared to be identical by MALDI-TOF and RAPD-PCR profiles was extracted by a combination of physical and chemical lysis as described above. The 20 isolates were sequenced in the same run with a Miseq Illumina platform using standard protocols from the manufacturer, with a 2 × 300 bp paired-ends kit.
Illumina reads were trimmed using Trimmomatic 0.3642, removing adapters, short reads and leading bases with a quality below Q3. Trimmed reads were assembled using SPAdes 3.1043 and contigs shorter than 1 Kb were discarded. Due to the risk of contamination in Illumina assemblies44, we searched for and filtered out contigs with coverage values and GC content that significantly deviated from those expected for our genomes.
In order to contextualize each pair of isolates among the known species genomic diversity, we selected 3 to 6 assembled genomes of the same or close related species from the NCBI database, depending on availability of fully-sequenced isolates. Average Nucleotide Identity (ANI), a measure of nucleotide-level genomic similarity between the genomes of two bacterial isolates, was estimated between all the pairs of isolates of each species using the ANI method from pyani (https://github.com/widdowquinn/pyani). Next, for each of these groups, we identified core genes using Roary with default parameters45 and core genome alignments were constructed, which were then used to build ML trees with FastTree46. Finally, assembly comparisons between pairs of isolates and alignment graphs were constructed using circoletto47.
Statistical analysis
All the statistical analyses were performed in Rstudio (version 3.3.2.1). Microbiota profiles and study variable (Sample type) were included in the estimation of alpha diversity (Shannon, Chao1 and Simpson indexes) and beta diversity analysis with the Vegan R package48. Dissimilarities between pairs of samples were estimated with the Bray-Curtis dissimilarity index and Unifrac indexes49 and analyzed with Principal Coordinate Analysis (PCoA) in Vegan50. The Vegan envfit function was used to evaluate if the factor of study (Sample type) was associated to the PCoA ordinations by using permutational multivariate analysis of variance using distance matrices (adonis)51, which was employed to describe the strength and significance that a categorical factor has in determining variation of ecological distances. The significance of the fitted factor was estimated by using 999 permutations.
A minimum of 35,000 sequences per sample were used for standardizing the microbiota measures. Rarefaction curves indicated that the sequencing depth was enough since the samples reached the plateau phase (Supplementary Fig. 1). Thus for subsequent core-microbiota and statistical analysis of compositional data, relative abundance was estimated by total-sum scaling normalization. The extent of microbiota shared across all the analyzed mother-infant pairs was estimated as the overlap between the core microbiotas at each site (infant saliva and maternal precolostrum) at genus level. The core microbiota for each sample type was predicted as those genera present in ≥50% of the samples and presenting a mean relative abundance above 1% within the corresponding groups (infant saliva and maternal precolostrum), using the core function in the microbiome R package52. Sharing of bacterial taxa between maternal precolostrum and infant oral microbiota beyond the predicted core microbiotas was studied at OTU level within every mother-infant pair.
Differences in the relative abundances of specific taxa among the different sample types collected were analyzed statistically at phylum, family and genus levels, in those taxa exhibiting a mean relative abundance above 0.1%. Due to the lack of normality or homogenous variances in some of the variables analyzed the non-parametric U-Mann-Whitney test was used to asses taxa differentially represented in both sample types. All p values were corrected when appropriate by using the Benjamini-Hochberg false discovery rate (FDR) correction. Significance was established at a p < 0.05 threshold for all the statistical analysis conducted.
Results
Characteristics of the population
The study was conducted on samples collected from 17 healthy mothers (aged 29–45) and their corresponding infants, including a mother and a pair of twins. A sample of precolostrum was obtained from the pregnant mother at 38–40 weeks of pregnancy, before delivery, and a swab sample from the gums of the corresponding infant was obtained 5–7 days after birth. All infants were exclusively breastfed and received no specific medication. Only two mothers delivered by C-section and 12 of them were primiparous (Table 1).
Analysis of microbiome community structure in precolostrum and infant oral samples
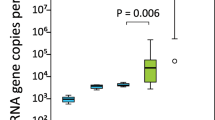
16S rRNA gene taxonomic profiling was conducted in samples collected from 15 of the 17 participant mother-infant pairs, as for the remaining two sample pairs, the quantity and quality of DNA yield was below the threshold required for the sequencing service (Unidad de Genómica of the Fundación Parque Científico de Madrid). Overall, this analysis resulted in the generation of a total of 3,683,325 reads, representing on average 81,060 (±33,469 SD) reads per sample in the precolostrum samples and 72,504 (±35,874 SD) reads per sample in the infant oral samples. After filtering out singletons and OTUs with low relative abundance, 1,877 OTUs were retained for diversity analyses. Sample richness Chao1 index, diversity Shannon index and evenness Simpson index were estimated across the two sample groups (precolostrum and infant mouth). Overall, precolostrum samples displayed higher alpha diversity values than the infant oral samples, as determined through Shannon and Simpson indexes (U Mann-Whitney p = 0.037 and p = 0.040, respectively). (Fig. 1A).

(A) Box plots of 3 different alpha diversity measures: the Chao I estimator, the Shannon index and Simpson index based on OTUs clustered at 97% similarity for precolostrum (red) and infant oral samples (blue). (B) Principal coordinates analysis of infant oral (blue) and maternal precolostrum (red) samples. Distances are based on the Bray-Curtis distances (upper right pannel), or weighted Unifrac distance metric (bottom right pannel), calculated using normalized data (PCoA based on unweighted Unifrac distance metrics provided very similar results to those calculated with weighted Unifrac).
In addition, a global principal coordinate analysis with all the available data evidenced that the type of sample (precolostrum vs infant mouth) significantly influenced samples ordination based both on Bray-Curtis dissimilarities and weighted and unweighted Unifrac distances (adonis: R2 = 0.27, p = 0.001; R2 = 0.18, p = 0.001; and R2 = 0.06, p = 0.013, respectively) (Fig. 1B). There was no separation between primiparous, delivery type or mother-infant pairs (data not shown). After filtering low abundance groups (mean relative abundance <0.001%), 21 phyla, 137 families and 244 genera were retrieved for further statistical analyses. From those groups, the dominant phylum across all the sample types analyzed was Firmicutes (mean relative abundance 77.22% ± 13.44), followed by Actinobacteria (mean relative abundance 10.51% ± 6.58) and Proteobacteria (mean relative abundance 6.15 ± 7.48). Of note, these phyla were detected in all the samples included in the study, accounting for more than 90% of the OTUs detected. At family and genus levels, precolostrum samples were dominated by Streptococcaceae [Streptococcus] and Staphylococcaceae [Staphylococcus], followed by groups typically associated with the oral cavity or skin including Micrococcaceae [Rothia and Kocuria], Corynebacteriaceae [Corynebacterium] or Veilonellaceae [Veillonella], in addition to groups typically associated with the intestinal ecosystem like Bifidobacteriaceae [Bifidobacterium]. Infant oral samples shared some of the dominant groups with precolostrum samples, as they were dominated by Streptococcus and Staphylococcus, followed by other groups typically associated with either the oral and nasopharingeal cavity (Rothia, Haemophilus, Gemellaceae, Veillonella or Fusobacterium) or even with the gut ecosystem (Bifidobacterium, Veillonella) (Fig. 2).

Relative abundances of most abundant taxa at phyla (left), family (middle) and genus (right) levels in both infant oral (upper pannels) and maternal precolostrum (bottom pannels) samples are represented.
Analysis of taxa shared or differentially represented in precolostrum and infant oral samples
Focusing on the differences and similarities of specific taxa between maternal precolostrum and infant oral samples at phylum level, Firmicutes were the dominant taxa in both sample types, followed by Proteobacteria and Actinobacteria, which were detected across all the samples analyzed. Bacteroidetes and Fusobacteria were only detected in a few of the infant oral samples (Fig. 2) although no statistically significant differences were detected for any of the phyla. When analyzed at the family and genus levels, the comparison between precolostrum and infant oral samples identified 11 families and 6 genera displaying statistically significant differences (FDR-value < 0.1, Table 2). Among them, significant differences were found in the top most abundant genera: Staphylococcus, which appeared overrepresented in precolostrum samples (FDR < 0.05); and Streptococcus, which appeared overrepresented in infant oral samples (FDR < 0.1). In addition, the most notable differences were a higher abundance of Kocuria, Acinetobacter, Delftia, and Corynebacterium in the precolostrum samples.
A major aim of this work was to identify the taxa shared between the maternal precolostrum and the infant oral ecosystems. For this purpose, the core microbiomes estimated for each sample type were compared to determine the extent of overlapping among them and to discern the composition of a putative shared core microbiota, consistently shared between maternal precolostrum and infant mouth in most mother-infant pairs. A core group of 18 genera was found to be shared between both sample types in more than 50% of the mother-infant pairs included in the study (Fig. 3), which include typical taxa of the nasopharyngeal cavity, like Haemophilus, Acinetobacter, members of the Gemellaceae family, Streptococcus, Granulicaella and Rothia, in addition to a number of genera frequently detected in maternal milk samples, such as Staphylococcus, Propionibacteria and Corynebacterium. Notably, other bacterial groups typically associated with the gut ecosystem, including Bifidobacterium, Veillonella, and Prevotella (the latter two also common oral inhabitants), were also detected as part of the common shared core between maternal precolostrum and infant mouth, despite of their low relative abundance in both ecosystems. In order to further characterize the taxa shared between maternal precolostrum and linked infant’s mouth, we also analyzed the OTUs within each mother-infant cluster (Supplementary Tables 1 and 2). The extent of OTU sharing within each pair ranged between 19.2 to 65.38% of the detected OTUs, and twelve of the pairs shared more than 45% of the OTUs. Remarkably, 26 taxonomic groups were detected to be shared between maternal precolostrum and infant oral ecosystem in at least 5 of the analyzed pairs. Among them, the most frequently shared OTUs included Streptococcus and Staphylococcus members, which were also the dominant groups in the corresponding samples types. In addition, other abundant OTUs not included within predicted abundant core groups, such as Bifidobacterium, Micrococcus, Blautia or Actinomyces, were also shared in a significant proportion of the analyzed pairs.

(A) Venn Diagram representing the overlap between the infant oral and the maternal precolostrum core microbiotas estimated at genus level. Core microbiotas were defined as taxa present at a relative abundance above 1% in at least 50% of the samples of the corresponding group. (B) Heatmap representing log2 relative abundances of the 43 genus comprising both precolostrum and infant oral core microbiotas. Samples and taxa clustering were performed on Euclidean distances using complete linkage.
Culture-dependent analysis
In combination to culture-independent metataxonomic analysis, an aliquot of each sample was used for culture-based analysis. The total bacterial load recovered from each sample ranged from 102 to 104 colony formation units per ml and a total number of 160 different colonies (58 from precolostrum samples and 79 from infant oral samples) were recovered, stocked and identified through MALDI-TOF (Fig. 4). The identification of bacteria recovered through culturing was consistent with the microbiota profiles previously described, with a higher number of Streptococcus and Staphylococcus isolates recovered from the oral and the precolostrum samples, respectively. In addition, it is also worth remarking that several isolates belonging to typical oral bacteria including Rothia, Gemella and Actinomyces species could be recovered from the infant mouth samples. Surprisingly, although most infants participating in the study were born vaginally, in this study a low proportion of lactobacilli species could be recovered through culturing from the infant oral samples, as opposed to previous reports based on culture-independent analysis53. Overall, in 11 of the analyzed mother-infant pairs, at least one isolate belonging to the same genus/species was recovered from both the precolostrum and the respective infant oral sample; it was also observed in 2 out of the 3 mature milk-infant mouth pairs (Fig. 4B). RAPD-PCR profiling revealed that in 9 cases, the isolates recovered from both sample types within the same pair could not be distinguished by RAPD-PCR genotype profiling. These isolates included four pairs classified as Streptococcus epidermidis, one pair as Staphylococcus auricularis, two pairs as Streptococcus mitis/oralis, one pair of Streptococcus salivarius and one pair as Streptococcus parasanguinis. Subsequently, such pairs of isolates were whole genome sequenced and genomic analyses were performed in order to confirm whether they were related strains. Only in one of the pairs (pair 8), Actinomyces odontolyticus were recovered from both the maternal and infant sample, and these strains were also subjected to whole genome sequencing despite they did not share RAPD-PCR profiles.

(A) Histogram representing the composition of the collection of isolates recovered from culturing infant oral and precolostrum samples. Identifications presented were performed through MALDI-TOF analysis. (B) Table summarising the mother-infant pairs in which bacteria sharing identical identification were recovered from both the precolostrum and the infant oral sample. (C) RAPD-PCR genotype profiling of those isolates that, belonging to the same genus/species were isolated from both samples from the same mother-infant pair and showed similar genotyping profile.
Comparative genomics of matched pairs of isolated strains
All isolates were sequenced at >50x coverage, facilitating the assembly of high-quality genomes. For each pair of isolates, a phylogenomic tree based on shared genes was constructed including 3 to 6 genomes from the same or closely related species, depending on availability in public repositories (Fig. 5, with precolostrum and infant oral isolates marked in blue and red, respectively). Next, we computed the Average Nucleotide Identity (ANI) values for each pair of isolates, a measure of DNA relatedness based on genomic similarity. Genomic synteny (i.e. conservation of gene order in the genome) is represented by lines joining homologous regions in the paired genomes. Notably, the pair 11 corresponds to a potential new streptococcal species, as evidenced by an ANI value to any described species well below the consensus of 95%, which is considered the species threshold54. In one of the 10 cases analyzed (pair 6), the two strains (belonging to Streptococcus mitis) appeared to be clearly different and in another case (the isolates pair 8, corresponding to Actinomyces odontolyticus) the two genomes showed an ANI of 99.76%. The other 8 pairs of analyzed genomes had a DNA identity over 99.9%, and in several instances, like pairs 9, 11 and 12, the genomes displayed a perfect synteny, strongly indicating that they correspond exactly to the same clone. It is also important to underline that the degree of phylogenomic similarity between the paired isolates is much higher than between any other available genome in public databases. Thus, these data indicate that the clones present in precolostrum appear a week later in the oral cavity of the corresponding infant, suggesting direct transmission from mothers to infants.

Whole genome sequencing comparison of mother-infant paired isolates. The genomes of each pair of isolates and 3–6 additional available genomes of the same species were compared by a phylogenomic tree constructed based on all shared genes. The blue and red dots indicate isolates from the precolostrum and oral samples of the baby, respectively. The average values of similarity (average nucleotide identity values) of the genomes for each mother-child pair are shown. Sinteny (conservation in gene order) is shown by lines that join the same fragments.
Discussion
In this study, the microbiota of precolostrum and infant oral swabs obtained from 15 mother-infant pairs were analyzed and compared using a combination of 16S rRNA sequencing, bacterial isolation, whole genome sequencing and comparative genomics (WGS) of those isolates that seemed to be shared by both types of samples within the same mother-infant pair. These analyses show that precolostrum, secreted by women at the end of pregnancy, contains a bacterial profile very similar to that encountered in mature milk, which is dominated by Staphylococcus species, but including also species of bacterial groups typically found in the oral cavity (such as Streptococcus, Fusobacterium, Veillonella or Porphyromonas), in accordance with previous studies on mature human milk30,32,55,56. Notably, precolostrum samples from our study had not been in contact with the infant, thus ruling out the possibility that the bacterial presence in human milk might exclusively originate as contamination from either the skin and/or infant oral cavity. In addition, in some of the analyzed mother-infant pairs, our study isolated identical strains from precolostrum and from the mouth of the respective infant, a finding that suggests that human milk may have a significant impact in the initial establishment of the infant oral microbiota. It must be highlighted that, for many of the recruited women, this was their first pregnancy and therefore presence of oral bacteria in precolostrum cannot be explained on the basis of oral contamination from a previous child through breast suckling.
The initial assembly of the infant microbiomes in early life has been recognized as a key factor impacting infant health in the short and long terms and thus, understanding the forces driving its establishment has received increasing scientific attention in recent years6,28. Among the factors influencing such process, maternal microbiomes and, particularly, feeding mode are recognized as some of the major forces shaping the infant gut microbiome. In fact, breastfeeding is known to provide the infant a vast array of bioactive compounds including, among others, human milk oligosaccharides, which can act as prebiotic substrates for specific commensal microorganisms, as well as a set of commensal microorganisms to initially colonize the infant gut21. However, until recently, intra-mammary human milk was assumed to be a sterile fluid under physiological conditions and thus, the presence of bacteria in human milk has remained controversial. On the one hand, the common presence of staphylococci, streptococci or corynebacteria in expressed human milk was explained as the result of contamination arising from either the skin (staphylococci, corynebacteria, propionibacteria) or the infant’s mouth (streptococci)57,58. In the last case, ultrasound imaging of milk ejection revealed a certain degree of milk flow back into the mammary ducts during suckling, suggesting that bacteria from the infant’s oral cavity may contaminate milk32,59. However, the detection of live bacterial cells and/or DNA from gut-related facultative and strict anaerobe species prompted to hypothesize the existence of an endogenous entero-mammary pathway during late pregnancy and lactation, likely mediated through complex interactions between bacteria, immune cells and epithelial cells60. Although the existence or not of such physiological and selective translocation has been controversial and remains as a subject of debate24,31,61,62, subsequent work provided evidence in support of such hypothesis63,64,65,66,67,68,69. More recently, two lactic acid bacteria strains (Lactococcus lactis MG1614 and Lactobacillus salivarius PS2) were transformed with a plasmid containing the lux genes and orally administered to pregnant mice. The murine model allowed the visualization, isolation, and PCR detection of the transformed bacteria in different body locations, including mammary tissue and milk, reinforcing the hypothesis that physiological translocation of maternal bacteria during pregnancy and lactation may contribute to the composition of the mammary and milk microbiota70.
Many transient physiological changes occur during pregnancy and lactation, which may favor an increased bacterial translocation during late pregnancy and early lactation. For instance, hormonal action induces relevant oral changes during pregnancy, affecting the pH and microbiota composition; the gums become hyperemic and edematous and tend to bleed and, under such circumstances, the transfer of some pathogenic bacterial species and inflammatory compounds to the bloodstream have been associated with the onset of premature birth71,72,73. In this context, although coagulase-negative staphylococci and streptococci (salivarius and mitis groups) have been traditionally associated to the skin and oral cavity, respectively, they are widespread in most, if not all, human mucosal surfaces, reaching high concentrations in the mucosal surfaces of the digestive and genitourinary tracts. Therefore, these maternal mucosal sites may, thus, represent possible origins for bacterial translocation to precolostrum and breastmilk during the end of pregnancy in healthy hosts33.
Our data show that most bacterial genera are shared between precolostrum and the oral cavity of the neonate, further supporting that breastmilk could be one of the first microbial sources inoculating the oral cavity during the first days of life. Similarly, initial oral colonization may arise from different sources, including amniotic fluid swallowing, the vaginal environment and precolostrum, colostrum and human milk intake. It has already been shown that buccal administration of human colostrum has an impact on the oral microbiome of very low birth weight premature infants74. In addition, breastfeeding has recently been found the most important factor influencing oral microbiome colonization, having a long-term impact on bacterial composition that extends to 7 years of age75. In the present work, the mother-infant sharing of microorganisms at strain level was unequivocally demonstrated by means of a culture-dependent and whole comparative genomics approach. Indeed, a recent study had already demonstrated the existence of a high degree of strains sharing between maternal microbiomes at different body sites, and infant oral and gut microbiomes, through a metagenome approach28. However, to our knowledge, our work is the first to study human precolostrum as a potential source of microbes for the infant mouth, and to demonstrate through a combination of culture-independent and culture-dependent approach, the sharing of strains between both ecosystems in healthy mother-infant pairs. While the colonization process of the infant gut microbiome has been studied extensively, the origin of the oral microbiota and the factors dictating initial oral microbiota development are far from elucidated and deserve research attention due to its relevant implications for human health. Although the human oral microbiome is still widely unknown, Streptococcus species (e.g., Streptococcus salivarius) are considered as pioneer microorganisms that predominate in the initial oral microbial load75,76,77,78,79,80, and stimulate changes in the oral cavity (such as production of extracellular polymers) that favor the attachment and growth of subsequent species81. Thus, the right, timely exposure to the prebiotic and probiotic components of breastmilk could be vital for oral microbiota development. In cohort studies where oral samples have been analyzed from birth through several years, other factors influencing microbiota development were delivery mode (with a short-term impact) and antibiotic treatment during the first 2 years of life, which appeared to have a long-term impact in bacterial composition75.
The study faced some limitations, such as the limited number of recruited women, a fact that may be explained by the nature of the type of sample studied (precolostrum), which is only available in a small subset of pregnant women and, usually, at small amounts. Besides, we acknowledge that sequencing low biomass samples, such as those analyzed in this study, is currently a challenging process that may result in skew results due to possible introduction of contaminants during the DNA manipulation and sequencing process. In this regards, no amplification was detected in the first PCR round from two blank controls submitted to the same extraction procedure that the biological samples analyzed in this work. Nevertheless, this study provides, to our knowledge, the first analysis on the microbiological composition of human precolostrum secreted by pregnant women during their last third of pregnancy. Besides, our results suggest that this fluid already harbors a microbiota quite similar to the mature milk microbiota, and has an important role in initiating the infant oral microbiota establishment. Further studies are required in order to discern the pathways by which typical oral bacteria reach the mammary gland before any contact with the infant mouth.
Data Availability
Sequences generated in this study have been deposited in the NCBI public repository under Accession Number #PRJNA489791.
References
Drell, T. et al. The influence of different maternal microbial communities on the development of infant gut and oral microbiota. Sci. Rep. 7, 9940, https://doi.org/10.1038/s41598-017-09278-y (2017).
Vuillermin, P. J. et al. The maternal microbiome during pregnancy and allergic disease in the offspring. Semin. Immunopathol. 1–7, https://doi.org/10.1007/s00281-017-0652-y (2017).
Rautava, S. Early microbial contact, the breast milk microbiome and child health. J. Dev. Orig. Health Dis. 7, 5–14, https://doi.org/10.1017/S2040174415001233 (2016).
Fan, W. et al. Diversity of the intestinal microbiota in different patterns of feeding infants by Illumina high-throughput sequencing. World J. Microbiol. Biotechnol. 29, 2365–2372, https://doi.org/10.1007/s11274-013-1404-3 (2013).
Timmerman, H. M. et al. Intestinal colonisation patterns in breastfed and formula-fed infants during the first 12 weeks of life reveal sequential microbiota signatures. Sci. Rep. 7, 8327, https://doi.org/10.1038/s41598-017-08268-4 (2017).
Milani, C. et al. The first microbial colonizers of the human gut: composition, activities, and health implications of the infant gut microbiota. Microbiol. Mol. Biol. Rev. 81, e00036–17, https://doi.org/10.1128/MMBR.00036-17 (2017).
Biesbroek, G. et al. The impact of breastfeeding on nasopharyngeal microbial communities in infants. Am. J. Respir. Crit. Care Med. 190, 298–308, https://doi.org/10.1164/rccm.201401-0073OC (2014).
Bäckhed, F. et al. Dynamics and stabilization of the human gut microbiome during the first year of life. Cell Host Microbe 17, 690–703, https://doi.org/10.1016/j.chom.2015.04.004 (2015).
Martin, R. et al. Early-life events, including mode of delivery and type of feeding, siblings and gender, shape the developing gut microbiota. PLoS One 11, e0158498, https://doi.org/10.1371/journal.pone.0158498 (2016).
Garcia-Mantrana, I. & Collado, M. C. Obesity and overweight: Impact on maternal and milk microbiome and their role for infant health and nutrition. Mol. Nutr. Food Res. 60, 1865–1875, https://doi.org/10.1002/mnfr.201501018 (2016).
Cerdó, T. et al. Maternal obesity is associated with gut microbial metabolic potential in offspring during infancy. J. Physiol. Biochem. 1–11, https://doi.org/10.1007/s13105-017-0577-x (2017).
Chu, D. M. et al. Maturation of the infant microbiome community structure and function across multiple body sites and in relation to mode of delivery. Nat. Med. 23, 314–326 (2017).
Biagi, E. et al. The Bacterial Ecosystem of Mother’s Milk and Infant’s Mouth and Gut. Front. Microbiol. 8, 1214, https://doi.org/10.1038/nm.4272 (2017).
Pannaraj, P. S. et al. Association between breast milk bacterial communities and establishment and development of the infant gut microbiome. JAMA Pediatr. 171, 647–654, https://doi.org/10.1001/jamapediatrics.2017.0378 (2017).
Walker, A. Initial intestinal colonization in the human infant and immune homeostasis. Ann Nutr Metab 63, 8–15, https://doi.org/10.1159/000354907 (2013).
Arrieta, M.-C., Stiemsma, L. T., Amenyogbe, N., Brown, E. M. & Finlay, B. The intestinal microbiome in early life: health and disease. Front. Immunol. 5, 427, https://doi.org/10.3389/fimmu.2014.00427 (2014).
Praveen, P., Jordan, F., Priami, C. & Morine, M. J. The role of breast-feeding in infant immune system: a systems perspective on the intestinal microbiome. Microbiome 3, 41, https://doi.org/10.1186/s40168-015-0104 (2015).
Stokholm, J. et al. Cesarean section changes neonatal gut colonization. J. Allergy Clin. Immunol. 138, 881–889.e2, https://doi.org/10.1016/j.jaci.2016.01.028 (2016).
Magne, F., Puchi Silva, A., Carvajal, B. & Gotteland, M. The elevated rate of cesarean section and its contribution to non-communicable chronic diseases in Latin America: the growing involvement of the microbiota. Front. Pediatr. 5, 192, https://doi.org/10.3389/fped.2017.00192 (2017).
Fernández, L. et al. The human milk microbiota: origin and potential roles in health and disease. Pharmacol. Res. 69, 1–10, https://doi.org/10.1016/j.phrs.2012.09.001 (2013).
Jeurink, P. V. et al. Human milk: a source of more life than we imagine. Benef. Microbes 4, 17–30, https://doi.org/10.3920/BM2012.0040 (2013).
Andreas, N. J., Kampmann, B. & Mehring Le-Doare, K. Human breast milk: A review on its composition and bioactivity. Early Hum. Dev. 91, 629–635, https://doi.org/10.1016/j.earlhumdev.2015.08.013 (2015).
Jost, T., Lacroix, C., Braegger, C. & Chassard, C. Impact of human milk bacteria and oligosaccharides on neonatal gut microbiota establishment and gut health. Nutr. Rev. 73, 426–437, https://doi.org/10.1093/nutrit/nuu016 (2015).
Jost, T., Lacroix, C., Braegger, C. P., Rochat, F. & Chassard, C. Vertical mother-neonate transfer of maternal gut bacteria via breastfeeding. Environ. Microbiol. 16, 2891–2904, https://doi.org/10.1111/1462-2920.12238 (2014).
Jost, T., Lacroix, C., Braegger, C. & Chassard, C. Assessment of bacterial diversity in breast milk using culture-dependent and culture-independent approaches, https://doi.org/10.1017/S0007114513000597 (2017).
Martín, V. et al. Sharing of bacterial strains between breast milk and infant feces. J. Hum. Lact. 28, 36–44, https://doi.org/10.1177/0890334411424729 (2012).
Milani, C. et al. Exploring vertical transmission of bifidobacteria from mother to child, https://doi.org/10.1128/AEM.02037-15.
Ferretti, P. et al. Mother-to-infant microbial transmission from different body sites shapes the developing infant gut microbiome. Cell Host Microbe 24, 133–145.e5, https://doi.org/10.1016/j.chom.2018.06.005 (2018).
West, P. A., Hewitt, J. H. & Murphy, O. M. Influence of methods of collection and storage on the bacteriology of human milk. J. Appl. Bacteriol. 46, 269–77 (1979).
Jiménez, E. et al. Metagenomic analysis of milk of healthy and mastitis-suffering women. J. Hum. Lact. 31, 406–415, https://doi.org/10.1177/0890334415585078 (2015).
Rodríguez, J. M. The origin of human milk bacteria: is there a bacterial entero-mammary pathway during late pregnancy and lactation? Adv. Nutr. 5, 779–84, https://doi.org/10.3945/an.114.007229 (2014).
Cabrera-Rubio, R. et al. The human milk microbiome changes over lactation and is shaped by maternal weight and mode of delivery. Am. J. Clin. Nutr. 96, 544–51, https://doi.org/10.3945/ajcn.112.037382 (2012).
Mira, A. & Rodriguez, J. In Prebiotics and Probiotics in Human Milk: Origins and Functions of Milk-Borne Oligosaccharides and Bacteria (eds McGuire, M., McGuire, M. & Bode, L.) 349–364 (Academic Press, 2017).
Mira, A., Simon-Soro, A. & Curtis, M. A. Role of microbial communities in the pathogenesis of periodontal diseases and caries. J. Clin. Periodontol. 44, S23–S38, https://doi.org/10.1111/jcpe.12671 (2017).
Williams, J. E. et al. Human milk microbial community structure is relatively stable and related to variations in macronutrient and micronutrient intakes in healthy lactating women. J. Nutr. 147, 1739–1748, https://doi.org/10.3945/jn.117.248864 (2017).
Klindworth, A. et al. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 41, e1, https://doi.org/10.1093/nar/gks808 (2013).
Caporaso, J. G. et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7, 335–6, https://doi.org/10.1038/nmeth.f.303 (2010).
Rideout, J. R. et al. Subsampled open-reference clustering creates consistent, comprehensive OTU definitions and scales to billions of sequences. PeerJ 2, e545, https://doi.org/10.7717/peerj.545 (2014).
McDonald, D. et al. An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J. 6, 610–8, https://doi.org/10.1038/ismej.2011.139 (2012).
Bokulich, N. A. et al. Quality-filtering vastly improves diversity estimates from Illumina amplicon sequencing. Nat. Methods 10, 57–9, https://doi.org/10.1038/nmeth.2276 (2013).
McMurdie, P. J. & Holmes, S. phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One 8, e61217, https://doi.org/10.1371/journal.pone.0061217 (2013).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–20, https://doi.org/10.1093/bioinformatics/btu170 (2014).
Bankevich, A. et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–77, https://doi.org/10.1089/cmb.2012.0021 (2012).
Jeong, H., Pan, J.-G. & Park, S.-H. Contamination as a major factor in poor Illumina assembly of microbial isolate genomes. bioRxiv 081885, https://doi.org/10.1101/081885 (2016).
Page, A. J. et al. Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics 31, 3691–3, https://doi.org/10.1093/bioinformatics/btv421 (2015).
Price, M. N., Dehal, P. S. & Arkin, A. P. FastTree 2–approximately maximum-likelihood trees for large alignments. PLoS One 5, e9490, https://doi.org/10.1371/journal.pone.0009490 (2010).
Darzentas, N. Circoletto: visualizing sequence similarity with Circos. Bioinformatics 26, 2620–2621, https://doi.org/10.1093/bioinformatics/btq484 (2010).
Oksanen, J. et al. Vegan: community ecology package. R package version 2.4-5, https://CRAN.R-project.org/package=vegan (2017).
Lozupone, C. A., Hamady, M., Kelley, S. T. & Knight, R. Quantitative and qualitative beta diversity measures lead to different insights into factors that structure microbial communities. Appl. Environ. Microbiol. 73, 1576–85, https://doi.org/10.1128/AEM.01996-06 (2007).
Bray, J. R. & Curtis, J. T. An ordination of the upland forest communities of Southern Wisconsin. Ecol. Monogr. 27, 325–349 (1957).
Anderson, M. J. A new method for non-parametric multivariate analysis of variance. Austral Ecol. 26, 32–46 (2001).
Lahti, L. & Shetty, S. Tools for microbiome analysis in R. Version 1.1.10013, http://microbiome.github.com/microbiome. (2017).
NELUN BARFOD, M. et al. Oral microflora in infants delivered vaginally and by caesarean section. Int. J. Paediatr. Dent. 21, 401–406, https://doi.org/10.1111/j.1365-263X.2011.01136.x (2011).
Richter, M. & Rosselló-Móra, R. Shifting the genomic gold standard for the prokaryotic species definition. Proc. Natl. Acad. Sci. USA 106, 19126–31, https://doi.org/10.1073/pnas.0906412106 (2009).
Kumar, H. et al. Distinct patterns in human milk microbiota and fatty acid profiles across specific geographic locations. Front. Microbiol. 7, 1619, https://doi.org/10.3389/fmicb.2016.01619 (2016).
Li, S.-W. et al. Bacterial composition and diversity in breast milk samples from mothers living in Taiwan and Mainland China. Front. Microbiol. 8, 965, https://doi.org/10.3389/fmicb.2017.00965 (2017).
Dewhirst, F. E. et al. The human oral microbiome. J. Bacteriol. 192, 5002–17, https://doi.org/10.1128/JB.00542-10 (2010).
Martín, V., Mediano, P., Del Campo, R., Rodríguez, J. M. & Marín, M. Streptococcal diversity of human milk and comparison of different methods for the taxonomic identification of Streptococci. J. Hum. Lact. 32, NP84–NP94, https://doi.org/10.1177/0890334415597901 (2016).
Ramsay, D. T., Kent, J. C., Owens, R. A. & Hartmann, P. E. Ultrasound imaging of milk ejection in the breast of lactating women. Pediatrics 113, 361–7 PMID: 14754950 (2004).
Martín, R. et al. The commensal microflora of human milk: new perspectives for food bacteriotherapy and probiotics. Trends Food Sci. Technol. 15, 121–127, https://doi.org/10.1016/j.tifs.2003.09.010 (2004).
Young, W., Hine, B. C., Wallace, O. A. M., Callaghan, M. & Bibiloni, R. Transfer of intestinal bacterial components to mammary secretions in the cow. PeerJ 3, e888, https://doi.org/10.7717/peerj.88 (2015).
McGuire, M. K. & McGuire, M. A. Got bacteria? The astounding, yet not-so-surprising, microbiome of human milk. Curr. Opin. Biotechnol. 44, 63–68, https://doi.org/10.1016/j.copbio.2016.11.013 (2017).
Schultz, M., Göttl, C., Young, R. J., Iwen, P. & Vanderhoof, J. A. Administration of oral probiotic bacteria to pregnant women causes temporary infantile colonization. J. Pediatr. Gastroenterol. Nutr. 38, 293–7 PMID: 15076629 (2004).
Jiménez, E. et al. Oral administration of Lactobacillus strains isolated from breast milk as an alternative for the treatment of infectious mastitis during lactation. Appl. Environ. Microbiol. 74, 4650–4655, https://doi.org/10.1128/AEM.02599-07 (2008).
Abrahamsson, T. R., Sinkiewicz, G., Jakobsson, T., Fredrikson, M. & Björkstén, B. Probiotic lactobacilli in breast milk and infant stool in relation to oral intake during the first year of life. J. Pediatr. Gastroenterol. Nutr. 49, 349–54, https://doi.org/10.1097/MPG.0b013e31818f091b (2009).
Arroyo, R. et al. Treatment of infectious mastitis during lactation: antibiotics versus oral administration of Lactobacilli isolated from breast milk. Clin. Infect. Dis. 50, 1551–8, https://doi.org/10.1086/652763 (2010).
Albesharat, R., Ehrmann, M. A., Korakli, M., Yazaji, S. & Vogel, R. F. Phenotypic and genotypic analyses of lactic acid bacteria in local fermented food, breast milk and faeces of mothers and their babies. Syst. Appl. Microbiol. 34, 148–55, https://doi.org/10.1016/j.syapm.2010.12.001 (2011).
Fernández, L. et al. Prevention of infectious mastitis by oral administration of Lactobacillus salivarius PS2 during late pregnancy. Clin. Infect. Dis. 62, 568–573, https://doi.org/10.1093/cid/civ974 (2016).
Perez, P. F. et al. Bacterial imprinting of the neonatal immune system: lessons from maternal cells? Pediatrics 119, e724–32, https://doi.org/10.1542/peds.2006-1649 (2007).
de Andrés, J. et al. Physiological translocation of lactic acid bacteria during pregnancy contributes to the composition of the milk microbiota in mice. Nutrients 10, https://doi.org/10.3390/nu10010014(2017).
Bearfield, C., Davenport, E. S., Sivapathasundaram, V. & Allaker, R. P. Possible association between amniotic fluid micro-organism infection and microflora in the mouth. BJOG 109, 527–33, PMID: 12066942 (2002).
Straka, M. Pregnancy and periodontal tissues. Neuro Endocrinol. Lett. 32, 34–8 (2011).
Cobb, C. et al. The oral microbiome and adverse pregnancy outcomes. Int. J. Womens. Health 9, 551–559, https://doi.org/10.2147/IJWH.S142730 (2017).
Sohn, K., Kalanetra, K. M., Mills, D. A. & Underwood, M. A. Buccal administration of human colostrum: impact on the oral microbiota of premature infants. J. Perinatol. 36, 106–111, https://doi.org/10.1038/jp.2015.157 (2016).
Dzidic, M. et al. Oral microbiome development during childhood: an ecological succession influenced by postnatal factors and associated with tooth decay. ISME J. 12, 2292–2306, https://doi.org/10.1038/s41396-018-0204-z (2018).
Smith, D. J., Anderson, J. M., King, W. F., van Houte, J. & Taubman, M. A. Oral streptococcal colonization of infants. Oral Microbiol. Immunol. 8, 1–4 (1993).
Pearce, C. et al. Identification of pioneer viridans streptococci in the oral cavity of human neonates. J. Med. Microbiol. 42, 67–72, https://doi.org/10.1099/00222615-42-1-67 (1995).
Caufield, P. W. et al. Natural history of Streptococcus sanguinis in the oral cavity of infants: evidence for a discrete window of infectivity. Infect. Immun. 68, 4018–23, https://doi.org/10.1128/IAI.68.7.4018-4023.2000 (2000).
Cephas, K. D. et al. Comparative analysis of salivary bacterial microbiome diversity in edentulous infants and their mothers or primary care givers using pyrosequencing. PLoS One 6, e23503, https://doi.org/10.1371/journal.pone.0023503 (2011).
Gomez-Arango, L. F. et al. Antibiotic treatment at delivery shapes the initial oral microbiome in neonates. Sci. Rep. 7, 43481, https://doi.org/10.1038/srep43481 (2017).
Sampaio-Maia, B. & Monteiro-Silva, F. Acquisition and maturation of oral microbiome throughout childhood: An update. Dent. Res. J. (Isfahan). 11, 291–301, PMID: 25097637 (2014).
Acknowledgements
This research was supported by grant AGL2016-75476-R. LR was funded by grant 624773 (FP-7-PEOPLE-2013-IEF, European Commission) and is currently supported by the Juan de la Cierva Postdoctoral Trainee Program of the Spanish Ministry of Economy and Competitiveness (MINECO; IJCI-2015-23196).
Author information
Authors and Affiliations
Contributions
J.M.R. and A.M. participated in the experimental design and study conception. M.A.C. coordinated recruitment of participants and samples collection. L.R., C.G.C., A.B.A. and C.B.S. conducted experimental work. L.R., R.B. and H.A. participated in data analysis and figures elaboration. L.R., J.M.R. and A.M. drafted the manuscript. All authors approved and reviewed the final version of the manuscript.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ruiz, L., Bacigalupe, R., García-Carral, C. et al. Microbiota of human precolostrum and its potential role as a source of bacteria to the infant mouth. Sci Rep 9, 8435 (2019). https://doi.org/10.1038/s41598-019-42514-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-42514-1
This article is cited by
-
From nitrate to NO: potential effects of nitrate-reducing bacteria on systemic health and disease
European Journal of Medical Research (2023)
-
Gut microbiome studies in CKD: opportunities, pitfalls and therapeutic potential
Nature Reviews Nephrology (2023)
-
Secretory carcinoma of the salivary gland is rich in lactoferrin: a possible lactational-like differentiation?
European Archives of Oto-Rhino-Laryngology (2023)
-
Centrifugation does not remove bacteria from the fat fraction of human milk
Scientific Reports (2021)
-
Differential analysis of the bacterial community in colostrum samples from women with gestational diabetes mellitus and obesity
Scientific Reports (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
