
Abstract
X chromosome inactivation (XCI) is a mammalian specific, developmentally regulated process relying on several mechanisms including antisense transcription, non-coding RNA-mediated silencing, and recruitment of chromatin remodeling complexes. In vitro modeling of XCI, through differentiation of embryonic stem cells (ESCs), provides a powerful tool to study the dynamics of XCI, overcoming the need for embryos, and facilitating genetic modification of key regulatory players. However, to date, robust initiation of XCI in vitro has been mostly limited to mouse pluripotent stem cells. Here, we adapted existing protocols to establish a novel monolayer differentiation protocol for rat ESCs to study XCI. We show that differentiating rat ESCs properly downregulate pluripotency factor genes, and present female specific Xist RNA accumulation and silencing of X-linked genes. We also demonstrate that RNF12 seems to be an important player in regulation of initiation of XCI in rat, acting as an Xist activator. Our work provides the basis to investigate the mechanisms directing the XCI process in a model organism different from the mouse.
Similar content being viewed by others

Introduction
In mammals, X chromosome inactivation (XCI) ensures dosage compensation of sex chromosomal genes between females (XX) and males (XY)1,2. The process of XCI occurs early during female embryonic development and is mediated by a multitude of epigenetic mechanisms that result in the complete transcriptional inactivation of one entire X chromosome within the nucleus of every female somatic cell. In eutherians, initiation of XCI is mediated by the long non-coding RNA Xist3,4,5,6. During XCI, Xist RNA spreads in cis along the entire length of the X chromosome and triggers chromosome-wide silencing of X-linked genes by recruitment of a plethora of chromatin remodelers7,8,9,10. The study of XCI relies both on in vivo and in vitro models that allow genetic manipulation of the factors involved, and the vast majority of our current knowledge has been achieved by using the mouse as a model organism. In vivo studies have shown that XCI starts around the 4–8 cell stage of female mouse embryonic development and is initially imprinted (iXCI), resulting in exclusive inactivation of the paternal X chromosome (Xp)11,12,13,14. Later in development, at the blastocyst stage (~E4.5), the Xp becomes reactivated in the inner cell mass (ICM) of the embryo, whereas iXCI persists in the extra-embryonic lineages12,13. Reactivation of Xp in the ICM is then followed by random inactivation (rXCI) of either the paternal or maternal X chromosome in cells of the developing epiblast. In vitro, mouse embryonic stem cells (mESCs) have been extensively used to model rXCI. In fact, undifferentiated mESCs carry two active X chromosomes and faithfully mimic the pluripotent environment of the ICM, whereas their differentiation results in random inactivation of one of the two X chromosomes. Mouse ESC-based in vitro studies have led to the discovery of the long non-coding gene Tsix, which is transcribed antisense to Xist and represents the major repressor of Xist up-regulation at the onset of XCI15,16,17,18. XCI is tightly linked to loss of the pluripotent state19,20 and several pluripotency factors including NANOG, SOX2, OCT4, REX1 and PRDM14 have been described to function as XCI-inhibitors either by directly inhibiting Xist expression or by enhancing Tsix21,22,23,24. Activation of XCI is mediated by the X-linked E3 ubiquitin ligase RNF12 involved in dose-dependent degradation of REX125,26. Since Rnf12−/− mouse ESCs fail to upregulate Xist upon differentiation, Rnf12 has been suggested to be essential for the initiation of XCI27. In vivo, female embryos that maternally inherited an Rnf12 null allele fail to initiate iXCI on the paternal X chromosome and die in utero28, whereas RNF12 has been shown to be dispensable for rXCI to occur29. Thus, further research is needed to better understand the observed discrepancies between in vivo and in vitro studies. Interestingly, the study of XCI in female pre-implantation embryos from different species suggested that the epigenetic processes that mediate XCI might be more heterogeneous than expected. Indeed, iXCI occurs in the extra-embryonic lineages of rat and cow30,31,32 whereas in other species such as human, monkey, horse, pig and rabbit, rXCI has been exclusively observed in both embryonic and extra-embryonic tissues33,34. Comparative analysis of Xist RNA expression dynamics and X-linked gene silencing between rabbit and human pre-implantation embryos confirmed substantial diversity in the timing and regulation of XCI initiation among mammals, with cells of the human ICM showing two active X chromosomes regardless of Xist RNA coating30. In addition, the overall Xist gene structure appears to be conserved in all placental mammals32. For instance, comparative analysis of mouse and rat Xist sequence have shown that most exons are conserved between the two species, including repeat A, the 5′ element which is required to achieve proper gene silencing during XCI35,36. On the other hand, Tsix’s sequence evolved rapidly and differs between species37,38,39,40. Finally, Tsix antisense transcription through the Xist promoter is absent in human41,42, but appears to be conserved in rodents43. Interestingly, differentiation of mouse-rat allodiploid ES cells leads to specific primary inactivation of the mouse X chromosome44. This mouse allele-biased expression of Xist has been proposed to result from the higher expression of Tsix from the rat allele, interfering with expression of Xist in cis44.
Clearly, the development of novel in vitro systems derived from different species is necessary to reach a comprehensive understanding of the XCI process. However, although the induced pluripotent stem cell (iPSC) technology has allowed the generation of several ES cell-like lines from different species45,46,47, except for mouse iPSCs48, the characterization of the X chromosome status and the generation of in vitro differentiation protocols that recapitulate XCI have proven to be challenging49,50,51. Specific culture conditions that allow studying dosage compensation in human ES cells (hESCs) have also been established52,53. These studies provided in vitro systems that resemble the pre-XCI state of human pre-implantation embryos, but also showed that hESCs do not undergo random XCI upon differentiation. Thus, these observations suggest the presence of an epigenetic memory that may affect the faithful recapitulation of XCI as it occurs upon early human development, and highlight the need to further optimize cell culture conditions53. In this context, rat ES cells (rESCs)54,55,56,57,58,59,60,61,62, represent a powerful alternative to mESC to study the dynamics of XCI. Although closely related, mouse and rat diverged ~12–24 million years ago63, and there are significant differences between the two species. The large-scale structure of the rat genome is closer to the human genome than the mouse genome64, and the X chromosome has undergone a higher number of rearrangements in the rodent lineages compared to other mammalian orders65,66. Furthermore, compared to the mouse, the rat offers several advantages as a model system for modelling human diseases67,68. In functional genetic studies, the use of rat as a model system has been limited by the lack of genome engineering tools that allow efficient genetic manipulation of rESC. In this context, the development of novel genomic resources that followed the sequencing of the rat genome69 and the establishment of the CRISPR/Cas9 technology for genome editing rapidly enhanced the generation of transgenic rat models70,71 providing the basis to perform genetic studies.
Here, we set out to generate a robust in vitro system that could faithfully mimic the dynamics of XCI in rat. By developing a neuronal monolayer differentiation protocol for rESCs adapted from Vaskova and colleagues72, we were able to follow several aspects of XCI regulation in rat. Similar to mouse, we were able to observe (I) Xist up-regulation at an early stage of rESC differentiation followed by (II) transcriptional inactivation of X-linked genes and (III) H3K27me3 accumulation on the inactive X chromosome (Xi). In addition, (IV) overexpression experiments in rESCs confirmed that the REX1-RNF12 axis of Xist regulation is most likely conserved between rat and mouse. Thus, our data has established the technical basis to study the dynamics of XCI in a different system from the mouse and suggests that specific aspects of XCI may be conserved in mouse and rat.
Results
In vitro neuronal differentiation of rESCs
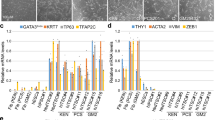
In vitro differentiation of mESCs towards different functional cell types including neurons, cardiomyocytes, hepatocytes and pancreatic cells can be efficiently achieved by several established protocols73. Usually, differentiation strategies are based on the formation of embryoid bodies (EB) followed by growth-factor-mediated induction of early progenitor cells to differentiate into their respective lineages. Despite of the growing list of differentiation protocols for mESCs, differentiation of rESCs is extremely difficult to achieve in vitro. To date, only two strategies have been described in which rESCs were triggered to differentiate into either cardiomyocytes or neuronal precursors and in these differentiation protocols MEK and GSK3β inhibitors, that are commonly used for ESC culture, are always present in low concentrations in the differentiation media74,75. XCI is closely linked to loss of pluripotency, and the presence of an inactive X chromosome provides a powerful readout for cell differentiation. Several rESCs derived from different rat inbred strains were differentiated, including three pure Lewis lines (LEW) (A4p20, A9p20, A10p20), and two lines of a mixed background of dark agouti (DA) and Sprague-Dawley (SD) (135-7, 141-6). All ESC lines displayed dome shaped morphology and expressed the pluripotency factors Rex1, Prdm14, and Esrrb (Fig. 1A,B). We initially set out to assess rat XCI after inducing rESCs differentiation according to protocols, which included MEK and GSK3β inhibitors. Although female cells appeared to be morphologically differentiated into neuronal precursors, we observed enrichment of the H3K27me3 histone modification (hallmark of gene silencing upon XCI) in only 20% of the cell population, (Supplementary Fig. 1A). Xist RNA FISH analysis, detecting Xist and Tsix, revealed absence of Xist accumulation, and confirmed the presence of small pinpoint transcription signals on both X chromosomes in most cells, likely representing Tsix transcription signals, as observed in mouse (Supplementary Fig. 1B). These results indicated our rESCs to be compromised to initiate XCI. We therefore set out to optimize the protocol in such a way that differentiation would include proper initiation of XCI. The absence of Xist accumulation and XCI might be related to the presence of MEK and GSK3β inhibitors, stabilizing the pluripotent state, and potentially resulting in expression of factors that repress Xist20,22,23,24,57. We therefore adapted the neuronal differentiation protocol initially described by Peng et al. and Vaskova et al.72,75 as follows: (I) both 2i inhibitors were completely eliminated starting from day 1 of neuronal differentiation, (II) the concentration of ROCK (rho-associated protein kinase) inhibitor, shown to prevent dissociation-induced apoptosis in cultured human ES cells46,76, was increased, (III) cells were seeded on laminin, (IV), a greater number of rESCs was used for differentiation and finally (V) differentiation cultures were serum and FGF2 free. Using these modified conditions, we were able to maintain viable differentiating male and female rESCs in the absence of 2i inhibitors (Fig. 1A). Importantly, qPCR analysis of both pluripotency and differentiation marker expression levels at different time points upon differentiation confirmed efficient downregulation of the pluripotency factors Esrrb, Prdm14 and Rex1, and parallel up-regulation of the neuronal precursor marker Nestin (Fig. 1B).

Neuronal differentiation of rESCs. (a) Schematic representation of our neuronal differentiation strategy. Brightfield images of female (A10p20, upper panel) and male (A8p20, lower panel) rESCs at day 0 and day 2, 3, 4 and 6 of differentiation are shown. Scale bars represent 80 μm. (b) qPCR analysis of Esrrb, Prdm14, Rex1 (pluripotency factors) and Nestin (neuronal differentiation marker) expression levels normalised to Actinb in female (A10p20, A4p20) and male (A8p20) differentiating rESCs. Error bars represent standard deviations of two technical replicates from one experiment.
Female rESCs undergo XCI upon in vitro neuronal differentiation
We then addressed the question of whether differentiating rESCs without the supplement of 2i inhibitors would facilitate XCI. To this end, four independent female rESC lines were differentiated and the Xist RNA expression level was assessed by qPCR analysis at different time points upon neuronal differentiation. Importantly, in order to assess the sex-specific regulation of Xist RNA, one male rESC line was also included in our analysis. As in mouse, we found that Xist upregulation occurs exclusively in female rat cells between day 2 and day 4 of differentiation (Fig. 2A). In contrast, Tsix pinpoint signals decrease upon differentiation in all differentiating rESC lines that we tested (Fig. 2A, and Supplementary Fig. 2). Next, we addressed the dynamics of Xist expression by performing Xist RNA FISH analysis at different time points upon neuronal differentiation. In undifferentiated rESCs, Xist RNA pinpoint signals were observed within the nuclei of both female and male cells (Fig. 2B). However, since the Xist RNA FISH probe can hybridize to both Xist and Tsix RNA, the pinpoint signal might represent Tsix expression instead of Xist. Around day 2 of neuronal differentiation, Xist RNA starts to accumulate exclusively on a single X chromosome within female nuclei, whereas Xist RNA accumulation was never observed in differentiating male rESCs (Fig. 2B, lower panel). Importantly, upon differentiation of A10p20 and A4p20 rESC female lines, more than 60% of the nuclei showed an Xist RNA-coated X chromosome at day 6 of differentiation (Fig. 2C). Taken together, these observations show that neuronal differentiation of rESCs in absence of 2i inhibitors allows Xist RNA to be upregulated and spread in cis from a single X chromosome in female cells.

Monoallelic upregulation of Xist RNA upon female rESCs differentiation. (a) Xist and Tsix qPCR expression analysis in female (A10p20, A4p20, 135-7, 141-6) and male (A8p20) differentiating rESCs. Expression levels of Xist and Tsix at different time points upon neuronal differentiation are shown. In the Xist panel error bars represent standard deviation of technical replicates from one experiment. In the Tsix panel error bars in A10P20, A4P20 and A8P20 represent standard deviations of technical replicates from one experiment and in 135-7 and 141-6 standard deviations of technical replicates from two experiments. (b) Representative images of Xist RNA FISH (green) analysis upon differentiation of female (A10p20-upper panel) and male (A8p20-lower panel) rESCs. DNA is stained with DAPI (blue). Scale bars represent 10 μm. (c) Quantification of relative number of Xist RNA signals (pinpoints or clouds) in A10p20 (left graph) and A4p20 (right graph) female rESCs at day 0, 2, 3, 4 and 6 upon neuronal differentiation. Xist clouds were never observed in male nuclei. More than 100 nuclei were counted for each time point and for each experiment. Error bars represent standard deviation of two experimental replicates. Asterisks indicate significant difference between the two groups (2 pinpoints versus cloud with or without pinpoint) within each differentiation time point. P-values are provided: p-valueA10-d0 < 0.0001, p-valueA10-d2 = 0.02, p-valueA10-d3 = 0.03, p-valueA10-d4 = 0.03 and p-valueA4-d0 < 0.0001, p-valueA4-d2 = 0.03, p-valueA4-d4 = 0.05, p-valueA4-d6 = 0.03.
In mouse, the H3K27me3 histone modification associated with gene silencing represents one of the earliest histone modifications that accumulates on the Xi during XCI77,78,79. Therefore, we monitored enrichment of H3K27me3 by immunofluorescence analysis upon differentiation of both male and female rESCs. In undifferentiated rESCs, no H3K27me3 domains were observed in neither male nor female cells (Fig. 3A). However, starting from day 2 of differentiation and in line with female-specific upregulation of Xist RNA, H3K27me3 started to accumulate into specific nuclear domains within female cells. By day 6, more than 60% of the female nuclei showed one H3K27me3 domain, thus confirming that XCI is efficiently initiated upon female rESCs differentiation (Fig. 3B). Finally, to precisely assess the dynamics of X-linked gene silencing, we followed the Xist-mediated inactivation of the X-linked genes Pgk1 and Rnf12 by two-colour RNA-FISH analysis at different time points upon rESCs differentiation. While the single copy of Pgk1 and Rnf12 in male cells remains actively transcribed throughout differentiation, the transcriptional inactivation of one copy of both X-linked genes in female cells starts around day 2 of differentiation (Fig. 3C and Supplementary Fig. 3). At day 6 of differentiation inactivation of X-linked genes is reached in up to 70% of the female nuclei (Fig. 3C and Supplementary Fig. 3).

Xist-mediated silencing of X-linked genes. (a) Representative images of H3K27me3 (green) immunofluorescence analysis in female (A10p20–upper panel) and male (A8p20-lower panel) rESC at different time points upon neuronal differentiation. DNA is stained with DAPI (blue). Scale bars represent 10 μm. (b) Quantification of relative number of cells carrying a H3K27me3 domain at day 0, 2, 3, 4 and 6 of neuronal differentiation. Data of A10p20 (left graph) and A4p20 (right graph) female rESC lines are shown. H3K27me3 domains were never noticed in male nuclei. More than 100 nuclei were counted for each time point and in each experimental replicate. Error bars represent standard deviations between two experiments. Asterisks indicate significant differences between the two groups (absence versus presence of domain) within each differentiation time point. The p-values are the given: p-valueA10-d0 < 0.0001, p-valueA10-d2 = 0.003 and p-valueA4-d0 < 0.0001, p-valueA4-d2 = 0.03, p-valueA4-d4 = 0.04. (c) Representative images of Xist (green)/Pgk1 (red) two-colour RNA-FISH in both female (A10p20-upper panel) and male (A8p20-lower panel) D0 and D6 differentiating rat cells are shown. DNA is stained with DAPI (blue). Scale bars represent 10 μm. (d) Xist (green)/Pgk1 (red) two-colour RNA-FISH quantitative analysis at different time points during neuronal differentiation of female (A10p20-upper panel) and male (A8p20-lower panel) rESCs. The relative number of cells showing either biallelic or monoallelic Pgk1 expression is quantified, together with the relative number of cells carrying Xist pinpoints or cloud signals. More than 100 nuclei were counted for each time point and in each experiment, with exception of A10p20 experimental replicate 2, where at day 2, 50 nuclei were counted. Error bars represent standard deviation of two experimental replicates. Asterisks indicate significant differences between groups (biallelic expression versus biallelic expression and Xist cloud versus monoallelic expression) within each differentiation time point. P-values are provided: p-valueA10-d0bial. VS A10-d0bial.&cloud < 0.0001, p-valueA10-d0bial. VS A10-d0monal. < 0.0001, p-valueA10-d2bial. VS A10-d2monal. = 0.049, p-valueA10-d6bial. VS A10-d6monal. < 0.0001, p-valueA10-d6bial.&cloud VS A10-d6monal. < 0.0001, p-valueA10-d6bial. VS A10-d6bial&cloud. = 0.00076 and p-valueA4-d0bial. VS A4-d0bial.&cloud < 0.0001, p-valueA4-d0bial. VS A4-d0monal. < 0.0001, p-valueA4-d6bial. VS A4-d6monal. = 0.0047, p-valueA4-d6bial.& VS A4-d6bial.&cloud = 0.003 and p-valueA8-d0monal. VS A8-d0monal.&cloud < 0.0001 p-valueA8-d2monal. VS A8-d2monal.&cloud < 0.0001, p-valueA8-d6monal. VS A8-d6monal.&cloud < 0.0001.
Overexpression of RNF12 leads to Xist activation
The X-linked E3 ubiquitin ligase RNF12 has been previously shown to activate Xist transcription at the onset of XCI25,27. Importantly, the pluripotency factor REX1 has been identified as a key target of RFN12, and dose-dependent degradation of REX1 by RNF12 has been proposed to act as a crucial mechanism directing initiation of XCI upon differentiation of female mESCs26. Since the RNF12-REX1 axis represents an important pathway for XCI initiation in mouse, we asked whether these factors play similar roles in rat XCI. To this end, we transiently overexpressed Rnf12 and Rex1 in rESCs, and determined the impact of overexpression on Xist RNA regulation. Based on findings in mouse, we expected REX1 overexpression to result in the inhibition of Xist transcription whereas overexpressing RNF12 would lead to Xist up-regulation26,27,80. Since the catalytic ring finger domain of RNF12 shows 100% of amino acid sequence identity between mouse and rat, and human RNF12 transgenes induce ectopic XCI in mouse ESCs25, we overexpressed the mouse RNF12 protein (mRNF12) in rESCs. Contrarily, as the zinc finger domain of REX1 is less conserved between the two species, overexpression of REX1 was achieved by transfecting rESCs with rat REX1 cDNA (rREX1). Xist RNA expression levels were determined by qPCR analysis, and the experiment was performed in three independent undifferentiated rESC lines as well as upon neuronal differentiation (Fig. 4A,B). Overexpression of mRNF12 consistently resulted in upregulation of Xist RNA in both male and female rESCs prior to and during rESCs differentiation, thus indicating RNF12 to act as important trans-acting activator of Xist in rat (Fig. 4A,B). Xist RNA FISH analysis performed at day 2 of neuronal differentiation upon mRNF12 overexpression further confirmed the impact of RNF12 on XCI initiation (Fig. 4C). However, the impact of rREX1 overexpression on Xist regulation, prior to and upon differentiation of rESCs, appeared less consistent (Fig. 4A–C). Although RNA-FISH and qPCR analysis indicated a decrease in Xist expression when rREX1 is overexpressed for one rESC line at day 2 of neuronal differentiation (line 135-7), Xist down-regulation was not significant for most comparisons. These results may suggest that Rex1 plays a less prominent role in XCI in rat, but could also be explained by our experimental setup, as previously observed in mouse ESCs26, where upregulation of Xist mediated by Rnf12 over-expression was also easier to detect than Rex1 over-expression mediated down-regulation of Xist. Although the role of Rex1 in XCI in rat needs further investigation, our findings indicate a role for Rnf12 in XCI in rat, providing a powerful new model system to elucidate the complex mechanisms directing initiation of XCI.

RNF12 and REX1 overexpression in rESCs and differentiating rESCs. (a) qPCR analysis of Xist, Rex1 and Rnf12 expression levels after overexpression of rat REX1 and mouse RNF12 proteins in undifferentiated rat ESCs. The controls (overexpression of empty vectors) were set to 1 for every gene analysed in each cell line used. Error bars represent standard deviation between technical replicates of three experiments. Asterisks and hashtags indicate significant differences, with the following p-values: p-value_A10Xist-Control VS Xist-OE_Rnf12 = 0.02, p-value_A10Xist-OE_Rex1 VS Xist-OE_Rnf12 = 0.03, p-value_A10Rnf12-Control VS Rnf12-OE_Rnf12 < 0.001, p-value_A10Rnf12-OE_Rex1 VS Rnf12-OE_Rnf12 < 0.001, p-value_A10Rnf12-OE_Rnf12 VS Rnf12-OE_Rex1 < 0.001, p-value_A10Rex1-Control VS Rex1-OE_Rex1 < 0.001, p-value_A10Rex1-OE_Rex1 VS Rex1-OE_Rnf12 < 0.001, (#)p-value_A10Rnf12-Control VS Rnf12-OE_Rex1 = 0.01, p-value_A4Xist-Control VS Xist-OE_Rnf12 < 0.001, p-value_A4Xist-OE_Rex1 VS Xist-OE_Rnf12 = 0.001, (#)p-value_A4Xist-Control VS Xist-OE_Rex1 = 0,002, p-value_A4Rnf12-Control VS Rnf12-OE_Rnf12 < 0.001, p-value_A4Rnf12-OE_Rex1 VS Rnf12-OE_Rnf12 < 0.001, (#)p-value_A4Rnf12-Control VS Rnf12-OE_Rex1 = 0.06, p-value_A4Rex1-Control VS Rex1-OE_Rex1 < 0.001, p-value_A4Rex1-OE_Rex1 VS Rex1-OE_Rnf12 < 0.001, p-value_A8Xist-Control VS Xist-OE_Rnf12 = 0.02, p-value_A8Xist-OE_Rex1 VS Xist-OE_Rnf12 = 0.03, p-value_A8Rnf12-Control VS Rnf12-OE_Rnf12 = 0.02, p-value_A8Rnf12-OE_Rex1 VS Rnf12-OE_Rnf12 = 0.02, p-value_A8Rex1-Control VS Rex1-OE_Rex1 = 0.02, p-value_A8Rex1-OE_Rex1 VS Rex1-OE_Rnf12 = 0.02. (b) qPCR analysis of Xist, Rex1 and Rnf12 expression levels after overexpression of rat REX1 and mouse RNF12 proteins in day 2 differentiating rat ESCs to neurons. The controls (overexpression of empty vectors) were set to 1 for every gene analysed in each cell line used. Error bars represent standard deviation between technical replicates from two experiments. Asterisks indicate significant differences with the following p-values: p-value_A10Rnf12-Control VS Rnf12-OE_Rnf12 = 0.007, p-value_A10Rnf12-OE_Rex1 VS Rnf12-OE_Rnf12 = 0.02, p-value_A10Rex1-Control VS Rex1-OE_Rex1 < 0.001, p-value_A10Rex1-OE_Rex1 VS Rex1-OE_Rnf12 < 0.001, p-value_A4Xist-Control VS Xist-OE_Rnf12 < 0.001, p-value_A4Xist-OE_Rex1 VS Xist-OE_Rnf12 < 0.001, p-value_A4Rnf12-Control VS Rnf12-OE_Rnf12 = 0.02, p-value_A4Rnf12-OE_Rex1 VS Rnf12-OE_Rnf12 < 0.001, p-value_A4Rex1-Control VS Rex1-OE_Rex1 < 0.001, p-value_A4Rex1-OE_Rex1 VS Rex1-OE_Rnf12 < 0.001, p-value_135-7Xist-Control VS Xist-OE_Rnf12 < 0.01, p-value_135-7Xist-OE_Rex1 VS Xist-OE_Rnf12 < 0.001, (#)p-value_135-7Xist-Control VS Xist-OE_Rex1 = 0.007, p-value_135-7Rnf12-Control VS Rnf12-OE_Rnf12 < 0.001, p-value_135-7Rnf12-OE_Rex1 VS Rnf12-OE_Rnf12 < 0.001, p-value_135-7Rex1-Control VS Rex1-OE_Rex1 < 0.001, p-value_135-7Rex1-OE_Rex1 VS Rex1-OE_Rnf12 < 0.001, p-value_A8Rnf12-Control VS Rnf12-OE_Rnf12 = 0.03. (c) Fold quantification of number of cells with Xist RNA clouds in 135-7 female line after overexpression of empty vector (control, set to 1), rRex1 and mRnf12 in rESCs at day 2 after initiation of differentiation towards neurons. Error bars represent standard variation between technical replicates from two experiments. The asterisks indicates significant difference with the following p-values: p-valueControl VS mRnf12 = 0,002 and p-valuerRex1 VS mRnf12 = 0,01.
Discussion
Our knowledge concerning the regulation of XCI in developing rat embryos is limited and relies on conservation of the key regulators Xist and Tsix between mouse and rat and a few studies in which, similar to mouse, iXCI has been proposed to occur in early rat embryonic development30,37,38,39,40. Studying the XCI process in rESCs offers the opportunity to explore species-specific epigenetic features and will help to reach a more comprehensive understanding of the XCI process in mammals. Although rESCs in vitro differentiation protocols have been previously established72,74,75, XCI studies were limited, only examining Xist expression and accumulation of chromatin modifications. Here, we confirm the accumulation of XCI-related epigenetic features in rESCs during differentiation according to the previously established protocols, and further show that this is accompanied by proper down-regulation of pluripotency markers81. We also showed that transcriptional inactivation of X-linked genes directly follows Xist RNA accumulation on one of the two X chromosomes. In fact, the exclusive enrichment of H3K27me3 loci in female nuclei starts around day 3 of neuronal differentiation, and Xist-mediated silencing of Pgk1 and Rnf12 occurs with similar dynamics.
Overexpression of mRNF12 protein in rESCs efficiently recapitulates RNF12 function in mouse as an activator of Xist expression. RNF12 is highly conserved among mammals82. The observed up-regulation of rat Xist upon mRNF12 overexpression is in line with our previous findings that overexpression of human RNF12 leads to ectopic Xist expression and XCI in mESCs25, indicating that the role of RNF12 in XCI is highly conserved in mammals. In contrast, rREX1 over-expression does not lead to consistent down-regulation of Xist levels as was expected based on our mouse studies. This result may indicate the presence of an alternative pathway by which RNF12 activates XCI in rat, a theory that is supported by the lower level of conservation in the REX1 zinc finger domain. On the contrary, REX1 represents the only target of RNF12 in mouse ESCs identified so far. In addition, measuring down-regulation of Xist expression in the present setting after transient transfection is technically more challenging than detecting up-regulation of Xist, and may require more sensitive approaches to experimentally define the exact role of Rex1 in XCI in rat. Additional alternative approaches, such as inducible over-expression of Rex1, and ChIP sequencing studies should shed more light on the exact role of Rex1 in XCI regulation.
In conclusion, we were able to set up a robust in vitro system to study the regulation of XCI in differentiating rESCs and our results suggest that the main steps of XCI in our rat in vitro system are highly similar to those of mouse XCI. The generation of hybrid F1 polymorphic rESCs together with the application of the CRISPR/Cas9 technology for genomic editing to the rat system will increase the use of rat as a model organism in basic epigenetic and biomedical research.
Methods
Cell culture and DNA transfection
rESCs were derived as previously described56 and subsequently maintained in N2B27 medium supplemented with 3 μM CHIR99021 (Stemgent), 1 μM PD0325901 and 1000 U/ml mouse LIF on mouse feeders.
For monolayer differentiation culture plates were coated with 100 μg/ml laminin (Sigma-Aldrich) for at least 4 hours at 37 °C, followed by three PBS washes. Single rESCs were plated at a density of 105/cm2 for the female cell lines and 2 × 104/cm2 for the male cell lines in N2B27 supplemented with 10 μM of ROCK inhibitor (Sigma-Aldrich) for the first three days. Thereafter, the ROCK inhibitor was eliminated. Medium was refreshed daily.
For overexpression experiments, the mRex1, rRex1 and mRnf12 coding sequences were subcloned into pCAG-Flag, a CAG-driven expression vector containing a Flag-tag. RESCs were transfected using lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions, followed by 48 hours of puromycin selection (1,5 μg/ml). Overexpression in differentiating cells was performed as follows: rESCs were trypsinised and plated at a density of 1,3*105/cm2 in gelatinized 6-well plates in 2i media supplemented with 10 μM of ROCK inhibitor (Sigma-Aldrich) without feeders. The next day cells were transfected using lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions. Cells were left to recover in 2i media overnight and then a 48 hour-puromycin selection (0,25 μg/ml for the A10p20, A4p20 and A8p20 cell lines and 1 μg/ml for the 135-7 cell line) was initiated in N2B27 differentiation media.
Probe preparation and Fluorescent in Situ Hybridization (FISH)
For preparing probes detecting Xist, Pgk1 and Rnf12 mRNAs, BACs harboring these genes were labelled as a whole, with digoxigenin and biotin (Roche) respectively, by nick translation following the manufacturer’s instructions.
For RNA-FISH at different time points of neuronal differentiation and after overexpression experiments during differentiation, cells were grown on glass coverslips and then fixed with 3% PFA for 10 minutes on ice, followed by three washes in PBS. Next, cells were permeabilised with 0.5% Triton and washed again three times in PBS. Cytoplasm was removed by treating the cells with 0.025% pepsin in 0.01 N HCL for 3 minutes at 37 °C. Subsequently, cells were dehydrated with sequential ethanol washes (70%, 85% and 100% 2 minutes each) and air-dried. Finally, probes were applied on the samples overnight at 37 °C in a 50% Formamide/2XSSC humid chamber. The next day, slides were washed two times, 5 minutes each, in 50% Formamide/2xSSC pH = 7.4 at 37 °C, followed by two washes, 5 minutes each in 2xSSC at 37 °C and cells were blocked for 30 minutes at room temperature with TSBSA (2 mg/ml bovine serum albumin in 0.1 M Tris and 0.15 M NaCl) in a humid chamber at room temperature. Detection was performed by incubation with anti-digoxigenin FITC (Boehringer, 1:250) and streptavidin alexa fluor 555 (Thermofisher Scientific, 1:400) in TSBSA for 30 minutes at room temperature. Slides were then washed two times, 5 minutes each with TS (0.1 M Tris, 0.15 M NaCl) and mounted with ProLong® Gold Antifade Mountant with Dapi (ThermoFisher Scientific). Imaging was performed on a Zeiss LSM700 microscope (Carl Zeiss, Jena).
Expression analysis
Cells were lysed by direct addition of 500 μl of TRIZOL and total RNA was extracted according to the manufacturer’s instructions (Invitrogen). To remove genomic DNA contamination, samples were treated 15 minutes at 37 °C with DNaseI (Invitrogen). Next, 1 μg of RNA was reverse transcribed by Superscript II reverse transcriptase with random hexamers (Invitrogen). For quantitative PCR (qPCR) gene expression levels were quantified using 2x SYBR Green PCR Master Mix (Applied Biosystems) in a CFX384 Real-Time machine (Bio-Rad). Expression levels were normalized to Actinb using the Δ-CT method. The primers used were as follows: Xist forward TGCCTGGATTTAGAGGAG and reverse CTCCACCTAGGGATCGTCAA, Tsix forward CGGACTATCTCCGCTTCTTG and reverse CACCTCTCTGGCTTCTTCCA, Nanog forward TAGCCCTGATTCTTCTAGCA and reverse TTTGCTGCAACGGCACATAA, Essrb forward GGCGTTCTTCAAGAGAACCA and reverse CCCACTTTGAGGCATTTCAT, Prdm14 forward AGGAACTGCGCTTCGTTCT and reverse GGCATCACCAAAAGCTGTCT, Rex1 forward AAATCATGACGAGGCAAGGC and reverse TGAGTTCGCTCCAACAGTCT, Nestin forward CTCTGCTGGAGGCTGAGAAC and reverse TGGTATCCCAAGGAAATTCG, Actinb forward GCTGGCCTTAGAGACCACAG and reverse AAGCAATTCAGCAACACCAA.
Immunocytochemistry
For immunofluorescence analysis of different time points of neuronal differentiation and of overexpression experiments, cells were grown on glass coverslips and then fixed with 3% PFA for 10 minutes at room temperature followed by three washes in PBS (3 × 5′). Thereafter, cells were permeabilised with 0.5% Triton, washed with PBS (3 × 5′) and blocked with 2% BSA, 5% donkey serum in PBS (blocking solution) for 30 minutes at room temperature. This was followed by anti-H3K27me3 rabbit (Diagenode, 1:500) incubation, diluted in blocking solution, at 4 °C overnight in a humid chamber. The next day, slides were washed in PBS (3 × 5′) and blocked with donkey anti-rabbit alexa fluor 488 (ThermoFischer Scientific, 1:250) secondary antibody, diluted in blocking solution for 1 hour at room temperature in a humid chamber. Slides were then washed in PBS (3 × 5′) and mounted with ProLong® Gold Antifade Mountant with Dapi (ThermoFisher Scientific). Confocal imaging was performed on a Zeiss LSM700 microscope (Carl Zeiss, Jena).
Statistical analysis
Statistical significance between the different groups was assessed by student t-test. Statistical significance was set at p < 0.05.
References
Gendrel, A.-V. & Heard, E. Noncoding RNAs and Epigenetic Mechanisms During X-Chromosome Inactivation. Annu. Rev. Cell Dev. Biol. 30, 561–580 (2014).
van Bemmel, J. G., Mira-Bontenbal, H. & Gribnau, J. Cis- and trans-regulation in X inactivation. Chromosoma 125, 41–50 (2016).
Marahrens, Y., Panning, B., Dausman, J., Strauss, W. & Jaenisch, R. Xist-deficient mice are defective in dosage compensation but not spermatogenesis. Genes Dev. 11, 156–66 (1997).
Penny, G. D., Kay, G. F., Sheardown, S. A., Rastan, S. & Brockdorff, N. Requirement for Xist in X chromosome inactivation. Nature 379, 131–137 (1996).
Borsani, G. et al. Characterization of a murine gene expressed from the inactive X chromosome. Nature 351, 325–329 (1991).
Brockdorff, N. et al. Conservation of position and exclusive expression of mouse Xist from the inactive X chromosome. Nature 351, 329–331 (1991).
Plath, K. et al. Developmentally regulated alterations in Polycomb repressive complex 1 proteins on the inactive X chromosome. J. Cell Biol. 167, 1025–35 (2004).
Schoeftner, S. et al. Recruitment of PRC1 function at the initiation of X inactivation independent of PRC2 and silencing. EMBO J. 25, 3110–3122 (2006).
Zhao, J., Sun, B. K., Erwin, J. A., Song, J.-J. & Lee, J. T. Polycomb Proteins Targeted by a Short Repeat RNA to the Mouse X Chromosome. Science (80-.). 322, 750–756 (2008).
da Rocha, S. T. et al. Jarid2 Is Implicated in the Initial Xist-Induced Targeting of PRC2 to the Inactive X Chromosome. Mol. Cell 53, 301–316 (2014).
Huynh, K. D. & Lee, J. T. Inheritance of a pre-inactivated paternal X chromosome in early mouse embryos. Nature 426, 857–862 (2003).
Mak, W. Reactivation of the Paternal X Chromosome in Early Mouse Embryos. Science (80-.). 303, 666–669 (2004).
Heard, E., Chaumeil, J., Masui, O. & Okamoto, I. Mammalian X-Chromosome Inactivation: An Epigenetics Paradigm. Cold Spring Harb. Symp. Quant. Biol. 69, 89–102 (2004).
Patrat, C. et al. Dynamic changes in paternal X-chromosome activity during imprinted X-chromosome inactivation in mice. Proc. Natl. Acad. Sci. 106, 5198–5203 (2009).
Lee, J. T. & Lu, N. Targeted mutagenesis of Tsix leads to nonrandom X inactivation. Cell 99, 47–57 (1999).
Navarro, P., Page, D. R., Avner, P. & Rougeulle, C. Tsix-mediated epigenetic switch of a CTCF-flanked region of the Xist promoter determines the Xist transcription program. Genes Dev. 20, 2787–2792 (2006).
Sado, T., Hoki, Y. & Sasaki, H. Tsix Silences Xist through Modification of Chromatin Structure. Dev. Cell 9, 159–165 (2005).
Ohhata, T., Hoki, Y., Sasaki, H. & Sado, T. Crucial role of antisense transcription across the Xist promoter in Tsix-mediated Xist chromatin modification. Development 135, 227–35 (2008).
Wutz, A. & Jaenisch, R. A shift from reversible to irreversible X inactivation is triggered during ES cell differentiation. Mol. Cell 5, 695–705 (2000).
Schulz, E. G. et al. The Two Active X Chromosomes in Female ESCs Block Exit from the Pluripotent State by Modulating the ESC Signaling Network. Cell Stem Cell 14, 203–216 (2014).
Ma, Z., Swigut, T., Valouev, A., Rada-Iglesias, A. & Wysocka, J. Sequence-specific regulator Prdm14 safeguards mouse ESCs from entering extraembryonic endoderm fates. Nat. Struct. Mol. Biol. 18, 120–127 (2011).
Navarro, P. et al. Molecular Coupling of Xist Regulation and Pluripotency. Science (80-.). 321, 1693–1695 (2008).
Navarro, P. et al. Molecular coupling of Tsix regulation and pluripotency. Nature 468, 457–460 (2010).
Payer, B. et al. Tsix RNA and the Germline Factor, PRDM14, Link X Reactivation and Stem Cell Reprogramming. Mol. Cell 52, 805–818 (2013).
Jonkers, I. et al. RNF12 is an X-Encoded dose-dependent activator of X chromosome inactivation. Cell 139, 999–1011 (2009).
Gontan, C. et al. RNF12 initiates X-chromosome inactivation by targeting REX1 for degradation. Nature 485, 386–390 (2012).
Barakat, T. S. et al. RNF12 Activates Xist and Is Essential for X Chromosome Inactivation. PLoS Genet. 7, e1002001 (2011).
Shin, J. et al. Maternal Rnf12/RLIM is required for imprinted X-chromosome inactivation in mice. Nature 467, 977–81 (2010).
Shin, J. et al. RLIM is dispensable for X-chromosome inactivation in the mouse embryonic epiblast. Nature 511, 86–89 (2014).
Wake, N., Takagi, N. & Sasaki, M. Non-random inactivation of X chromosome in the rat yolk sac. Nature 262, 580–1 (1976).
Dindot, S. V. Epigenetic and Genomic Imprinting Analysis in Nuclear Transfer Derived Bos gaurus/Bos taurus Hybrid Fetuses. Biol. Reprod. 71, 470–478 (2004).
Xue, F. et al. Aberrant patterns of X chromosome inactivation in bovine clones. Nat. Genet. 31, 216–220 (2002).
Okamoto, I. et al. Eutherian mammals use diverse strategies to initiate X-chromosome inactivation during development. Nature 472, 370–374 (2011).
Moreira de Mello, J. C. et al. Random X Inactivation and Extensive Mosaicism in Human Placenta Revealed by Analysis of Allele-Specific Gene Expression along the X Chromosome. PLoS One 5, e10947 (2010).
Yen, Z. C., Meyer, I. M., Karalic, S. & Brown, C. J. A cross-species comparison of X-chromosome inactivation in Eutheria. Genomics 90, 453–463 (2007).
Wutz, A., Rasmussen, T. P. & Jaenisch, R. Chromosomal silencing and localization are mediated by different domains of Xist RNA. Nat. Genet. 30, 167–174 (2002).
Chureau, C. et al. Comparative sequence analysis of the X-inactivation center region in mouse, human, and bovine. Genome Res. 12, 894–908 (2002).
Duret, L. The Xist RNA Gene Evolved in Eutherians by Pseudogenization of a Protein-Coding Gene. Science (80-.). 312, 1653–1655 (2006).
Nesterova, T. B. et al. Characterization of the Genomic Xist Locus in Rodents Reveals Conservation of Overall Gene Structure and Tandem Repeats but Rapid Evolution of Unique Sequence. Genome Res. 11, 833–849 (2001).
Elisaphenko, E. A. et al. A Dual Origin of the Xist Gene from a Protein-Coding Gene and a Set of Transposable Elements. PLoS One 3, e2521 (2008).
Migeon, B. R., Chowdhury, A. K., Dunston, J. A. & McIntosh, I. Identification of TSIX, Encoding an RNA Antisense to Human XIST, Reveals Differences from its Murine Counterpart: Implications for X Inactivation. Am. J. Hum. Genet. 69, 951–960 (2001).
Migeon, B. R., Lee, C. H., Chowdhury, A. K. & Carpenter, H. Species Differences in TSIX/Tsix Reveal the Roles of These Genes in X-Chromosome Inactivation. Am. J. Hum. Genet. 71, 286–293 (2002).
Shevchenko, A. I. et al. Variability of Sequence Surrounding the Xist Gene in Rodents Suggests Taxon-Specific Regulation of X Chromosome Inactivation. PLoS One 6, e22771 (2011).
Li, X. et al. Generation and Application of Mouse-Rat Allodiploid Embryonic Stem Cells. Cell 164, 279–292 (2016).
Takahashi, K. & Yamanaka, S. Induction of Pluripotent Stem Cells from Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors. Cell 126, 663–676 (2006).
Watanabe, K. et al. A ROCK inhibitor permits survival of dissociated human embryonic stem cells. Nat. Biotechnol. 25, 681–686 (2007).
Friedrich Ben-Nun, I. et al. Induced pluripotent stem cells from highly endangered species. Nat. Methods 8, 829–831 (2011).
Maherali, N. et al. Directly Reprogrammed Fibroblasts Show Global Epigenetic Remodeling and Widespread Tissue Contribution. Cell Stem Cell 1, 55–70 (2007).
Tchieu, J. et al. Female Human iPSCs Retain an Inactive X Chromosome. Cell Stem Cell 7, 329–342 (2010).
Mekhoubad, S. et al. Erosion of Dosage Compensation Impacts Human iPSC Disease Modeling. Cell Stem Cell 10, 595–609 (2012).
Pasque, V. & Plath, K. X chromosome reactivation in reprogramming and in development. Curr. Opin. Cell Biol. 37, 75–83 (2015).
Theunissen, T. W. et al. Molecular Criteria for Defining the Naive Human Pluripotent State. Cell Stem Cell 0, 150–162 (2016).
Sahakyan, A. et al. Human Naive Pluripotent Stem Cells Model X Chromosome Dampening and X Inactivation. Cell Stem Cell 20, 87–101 (2017).
Meek, S., Sutherland, L. & Burdon, T. Tuning Differentiation Signals for Efficient Propagation and In Vitro Validation of Rat Embryonic Stem Cell Cultures. in 73–85, https://doi.org/10.1007/7651_2014_121 (2014).
Meek, S. et al. Tuning of β-catenin activity is required to stabilize self-renewal of rat embryonic stem cells. Stem Cells 31, 2104–2115 (2013).
Meek, S. et al. Efficient Gene Targeting by Homologous Recombination in Rat Embryonic Stem Cells. PLoS One 5, e14225 (2010).
Buehr, M. et al. Capture of Authentic Embryonic Stem Cells from Rat Blastocysts. Cell 135, 1287–1298 (2008).
Li, P. et al. Germline Competent Embryonic Stem Cells Derived from Rat Blastocysts. Cell 135, 1299–1310 (2008).
Hirabayashi, M. et al. Establishment of rat embryonic stem cell lines that can participate in germline chimerae at high efficiency. Mol. Reprod. Dev. n/a–n/a, https://doi.org/10.1002/mrd.21123 (2009).
Kawamata, M. & Ochiya, T. Generation of genetically modified rats from embryonic stem cells. Proc. Natl. Acad. Sci. 107, 14223–14228 (2010).
Kawamata, M. & Ochiya, T. Establishment of Embryonic Stem Cells from Rat Blastocysts. in 169–177, https://doi.org/10.1007/978-1-60327-389-3_12 (2010).
Men, H., Bauer, B. A. & Bryda, E. C. Germline Transmission of a Novel Rat Embryonic Stem Cell Line Derived from Transgenic Rats. Stem Cells Dev. 21, 2606–2612 (2012).
Gibbs, R. A. et al. Genome sequence of the Brown Norway rat yields insights into mammalian evolution. Nature 428, 493–521 (2004).
Zhao, S. et al. Human, Mouse, and Rat Genome Large-Scale Rearrangements: Stability Versus Speciation. Genome Res. 14, 1851–1860 (2004).
Bourque, G., Pevzner, P. A. & Tesler, G. Reconstructing the genomic architecture of ancestral mammals: lessons from human, mouse, and rat genomes. Genome Res. 14, 507–16 (2004).
Jensen-Seaman, M. I. et al. Comparative Recombination Rates in the Rat, Mouse, and Human Genomes. Genome Res. 14, 528–538 (2004).
Iannaccone, P. M. & Jacob, H. J. Rats! Dis. Model. Mech. 2, 206–10 (2009).
Bryda, E. C. The Mighty Mouse: the impact of rodents on advances in biomedical research. Mo. Med. 110, 207–11.
Aitman, T. J. et al. Progress and prospects in rat genetics: a community view. Nat. Genet. 40, 516–522 (2008).
Shao, Y. et al. CRISPR/Cas-mediated genome editing in the rat via direct injection of one-cell embryos. Nat. Protoc. 9, 2493–2512 (2014).
Guan, Y., Shao, Y., Li, D. & Liu, M. Generation of Site-Specific Mutations in the Rat Genome Via CRISPR/Cas9. in 297–317, https://doi.org/10.1016/B978-0-12-801185-0.00014-3 (2014).
Vaskova, E. A. et al. Transcriptome Characteristics and X-Chromosome Inactivation Status in Cultured Rat Pluripotent Stem Cells. Stem Cells Dev. 24, 2912–2924 (2015).
Schroeder, I. S., Wiese, C., Truong, T. T., Rolletschek, A. & Wobus, A. M. Differentiation Analysis of Pluripotent Mouse Embryonic Stem (ES) Cells In Vitro. in 219–250, https://doi.org/10.1007/978-1-59745-471-1_12 (2009).
Cao, N. et al. In vitro differentiation of rat embryonic stem cells into functional cardiomyocytes. Cell Res. 21, 1316–1331 (2011).
Peng, X. et al. Conversion of rat embryonic stem cells into neural precursors in chemical-defined medium. Biochem. Biophys. Res. Commun. 431, 783–787 (2013).
Ishizaki, T. et al. Pharmacological properties of Y-27632, a specific inhibitor of rho-associated kinases. Mol. Pharmacol. 57, 976–83 (2000).
Chaumeil, J. A novel role for Xist RNA in the formation of a repressive nuclear compartment into which genes are recruited when silenced. Genes Dev. 20, 2223–2237 (2006).
Silva, J. et al. Establishment of histone h3 methylation on the inactive X chromosome requires transient recruitment of Eed-Enx1 polycomb group complexes. Dev. Cell 4, 481–95 (2003).
Plath, K. Role of Histone H3 Lysine 27 Methylation in X Inactivation. Science (80-.). 300, 131–135 (2003).
Loos, F. et al. Xist and Tsix Transcription Dynamics Is Regulated by the X-to-Autosome Ratio and Semistable Transcriptional States. Mol. Cell. Biol. 36, 2656–2667 (2016).
Magaraki, A. M. (Aristea M., Gribnau, J. H. & Ridderprint). Epigenetic regulation in rodent primordial germ cells and pre-implantation embryos = Epigenetische regulatie in primordiale kiemcellen en pre-implantatie embryo’s van knaagdieren. (Erasmus University Rotterdam, 2017).
Bach, I. et al. RLIM inhibits functional activity of LIM homeodomain transcription factors via recruitment of the histone deacetylase complex. Nat. Genet. 22, 394–9 (1999).
Acknowledgements
We would like to thank all lab members for helpful discussions. Part of this manuscript was published as a chapter in the thesis of AM.
Author information
Authors and Affiliations
Contributions
A.L., A.M. and J.G. conceived the work and designed the experiments. C.G.P. contributed to the design of the experiments. A.M., A.L., C.G.P., S.M., E.S.L. performed experiments, acquired and analysed the data. A.L., A.M. and J.G. wrote the draft of the manuscript. S.M., T.B., W.M.B. critically revised the manuscript. All authors are accountable for all aspects of the work and have approved the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Magaraki, A., Loda, A., Gontan, C. et al. A novel approach to differentiate rat embryonic stem cells in vitro reveals a role for RNF12 in activation of X chromosome inactivation. Sci Rep 9, 6068 (2019). https://doi.org/10.1038/s41598-019-42246-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-42246-2
This article is cited by
-
Dihydrotestosterone induces arterial stiffening in female mice
Biology of Sex Differences (2024)
-
CTCF chromatin residence time controls three-dimensional genome organization, gene expression and DNA methylation in pluripotent cells
Nature Cell Biology (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
