
Abstract
Chlorine, an extremely hydrophilic volatile element, provides important information regarding the origin of intrinsic volatiles in the Moon. Lunar apatite was found to have a wider spread of δ37Cl (from −1 to +40‰ versus standard mean ocean chloride) than most terrestrial and chondritic ones (0 ± 0.5‰). However, the provenance of the elevated lunar δ37Cl is still enigmatic. Here we report new isotopic data for H and Cl in apatite from three lunar meteorites and discuss possible mechanisms for Cl isotopic fractionation of the Moon. The apatite grain in Dhofar 458 has an average δ37Cl value of +76‰, indicative of an extremely heavy Cl reservoir in the Moon. Volatile loss associated with the Moon-forming Giant Impact and the formation of lunar magma ocean could account for the large Cl isotopic fractionation of the Moon. The observed H2O contents (220–5200 ppm), δD (−100 to +550‰) and δ37Cl values (+3.8 − +81.1‰) in lunar apatite could be understood if late accretion of hydrous components were added to the Moon after the fractionation of Cl isotopes. The heterogeneous distribution of lunar Cl isotopes is probably resulted from complex lunar formation and differentiation processes.
Similar content being viewed by others


Introduction
By virtue of recent analytical advances, considerable amounts of volatiles (H, C, F, S, and Cl) have been detected in lunar samples1,2,3,4,5,6, posing challenges to the favored Moon formation model7 in which most volatiles were thought to have been lost due to the Giant Impact between a Mars-sized body and the proto-Earth. On the other hand, a large variation of δ37Cl (from −1‰ to +40‰)8,9,10,11,12,13,14 was observed in the lunar samples. This appears to contradict the elevated volatile contents present in the lunar samples as the large Cl isotopic fractionation was ascribed to metal chlorides degassing from anhydrous magmas8. An alternative model argued that the residual melt of the lunar magma ocean (LMO), known as urKREEP (K, potassium; REE, rare earth elements; P, phosphorus), was the 37Cl-rich reservoir, with δ37Cl of ~+30‰9,10. Nevertheless, the correlation between high δ37Cl values and a KREEP component has been questioned14,15,16. A recent model14 proposed that the elevated δ37Cl values of apatite were partially inherited from vapor-phase metasomatism. However, the provenance of the 37Cl-rich gas is still enigmatic12,14. To better understand the origin and fractionation mechanism of lunar Cl, we conducted SIMS (secondary ion mass spectrometry) analyses on apatite [Ca5(PO4)3(F,Cl,OH)] from three lunar meteorites on the basis of detailed mineralogical and petrographic studies, and assess possible fractionation mechanisms and potential reservoirs of lunar Cl isotopes.
Results
In the mare basalt Miller Range (MIL) 05035 and the KREEP-assimilated gabbro Northwest Africa (NWA) 2977, subhedral to anhedral apatite (20–150 µm) commonly occurs in mesostasis along with other late-stage minerals (Fig. S1). Apatite contains REEs (0.09–1.98 wt%) that are positively correlated with its molar Fe/(Mg + Fe) and Cl/(Cl + F) ratios (Fig. S2), which is in agreement with the incompatibility of REEs and Cl over F during magmatic evolution. In the feldspathic breccia Dhofar 458, one subhedral apatite grain (60 × 100 µm, Fig. 1a) partially corroded by anorthite-augite intergrowths was found in an anorthositic troctolite clast (Fig. S1c). The apatite grain contains only trace amounts of REEs (Ce2O3, 0.08 wt%; Nd2O3, 0.05; Y2O3, 0.04; Table S1). All apatite grains studied here are F-rich (1.9–3.4 wt%; Table 1), indicative of a magmatic origin12,17.

Petrographic and mineralogic characterization of the apatite grain in the feldspathic breccia Dhofar 458. (a) Back-scattered electron (BSE) image of the apatite grain, showing that it was subhedral and partially corroded by recrystallized anorthite-augite intergrowths. The rectangular pits are from NanoSIMS analyses. The spot numbers are labelled beside the pits. (b) Raman spectra of the apatite grain, exhibiting characteristic bands of F,Cl-apatite at 963, 429, 580, 591, and 1050 cm−1. Abbreviations: Ap, apatite; An, anorthite; Aug, augite; Ol, olivine.
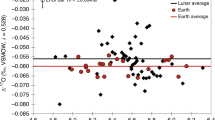
Eight apatite grains from MIL 05035 and NWA 2977 yielded δ37Cl values from +3.8 ± 3.1 (2σ) to +26.2 ± 1.8‰ and H2O contents from 1100 ± 120 to 5200 ± 280 ppm (Table 1, Fig. 2), which are comparable with previous reports2,3,4,8,9,10,11,12,13,14. Cl isotope compositions are homogeneous within individual grains. In Dhofar 458, the only apatite grain found has extremely high δ37Cl values of +67.2 ± 2.0 to +81.1 ± 2.0‰ and a relatively low H2O content averaging 240 ± 40 ppm (Table 1). The mean δD values of the apatite vary from −110 ± 30‰ in NWA 2977 to +400 ± 80‰ in MIL 05035, and to +510 ± 130‰ in Dhofar 458 (Table 1). No apparent correlation is observed among δ37Cl, Cl content, and δD in the apatite (Fig. 2).

Plots of (a) H2O content, (b) Cl content, and (c) δD versus δ37Cl for lunar apatite from MIL 05035, NWA 2977, and Dhofar 458. Data from the literature (hollow symbols)8,9,10,11,12 are also plotted for comparison. Red and magenta symbols represent mare basalts; blue and cyan symbols are KREEP-bearing basalts; black and grey symbols are highland rocks; and 14304 is a very high-K basalt. No apparent correlation can be observed among δ37Cl, δD, and Cl content.
Discussion
Most terrestrial rocks have a very restricted δ37Cl variation (0 ± 0.5‰)18. The uniformity of terrestrial δ37Cl has been attributed to HCl degassing from hydrous magmas, in which only negligible Cl fractionation occurred8. The elevated lunar δ37Cl and its wide dispersion therefore have been ascribed to metal chlorides degassing from anhydrous magmas, and the lunar interior was suggested to be extremely depleted in hydrogen (lower than Earth by a factor of ~104 to 105)8. However, studies of volatiles in lunar volcanic glasses, melt inclusions, and anorthosites have found robust evidence indicating that the lunar interior contains, at least partially, considerable amounts of water1,5,6. Our analyses of lunar apatite yield high H2O contents (220–5200 ppm) consistent with previous data2,3,4,9,10,11,12,14, but it is not trivial to determine the H2O content of the parental magma because the simple Nernst partition is not applicable to F, Cl, and OH in apatite19.
Because in some mare and highland samples, the δ37Cl values of apatite are positively correlated with Cl contents and bulk compositions of trace incompatible elements, it was inferred that the Cl isotopes were mixtures from two distinct reservoirs: one is the lunar mantle with δ37Cl of ~0‰ and the other is urKREEP with δ37Cl of ~+30‰9,10. The 37Cl enrichment of lunar samples was ascribed to the contamination of KREEP components which is enriched in heavy Cl and incompatible elements9,10. However, this relationship seems to be only valid within some low-Ti and KREEP-rich basalts14,15,16. The δ37Cl values of apatite from Apollo 14 high-Al samples do not show any correlation with bulk La/Lu ratios14, and the results of high-Ti basalts are not apparently correlated9,10,16. This might be understandable because the high-Al mare basalts could have assimilated a mixture of KREEP and granite20, and the high-Ti basalts might have sampled their trace elements from a reservoir distinct from urKREEP21. Our results show that although the apatite from NWA 2977 and Dhofar 458 has a rough positive correlation between the Cl content and δ37Cl value, no correlation has been observed in the apatite of MIL 05035 (Fig. 2b). More importantly, in spite of the extremely high δ37Cl values (+67.2 to +81.1‰), the apatite in Dhofar 458 has only trace amounts of REEs (Table S1), and the whole-rock incompatible element abundances are relatively low (e.g., 0.37 µg/g of Th, 3.06 µg/g of La, and 1.22 µg/g of Sm)22. It is apparent that the high δ37Cl of Dhofar 458 is decoupled from contamination of KREEP components. The highly fractionated Cl isotopes in Dhofar 458 might have originated from a 37Cl-enriched reservoir distinct from the previously suggested urKREEP9,10.
In order to understand the fractionation mechanism of Cl isotopes in the Moon, especially the extremely high δ37Cl of the apatite in Dhofar 458, all relevant physiochemical processes are considered: (i) solar nebula condensation; (ii) vaporization, re-condensation, and accretion during the hypothesized giant impact; (iii) massive degassing of the LMO; (iv) partial melting of mantle sources and crystallization of minerals; (v) evaporation at fumaroles; (vi) magma degassing; (vii) shock-induced melting, evaporation, and condensation; and (viii) surface alterations.
Initially, the pristine solar nebula had a δ37Cl of ~−5 to −3‰23. The HCl hydration that followed caused a small degree of Cl isotope fractionation (3–6‰)23. Thus, most chondritic materials would eventually have a δ37Cl around 0‰18. Processes (ii) and (iii) would greatly enhance the fractionation of Cl isotopes and will be discussed later. Partial melting and crystallization at igneous temperatures would hardly fractionate Cl isotope composition24.
Fumarole processes in terrestrial system could produce Cl isotopic fractionation up to 16‰ via a distillation-like mechanism, which requires >99% evaporated HCl (g) to recondense to Cl−aq in the liquid water along the fumarole conduit25. However, for lunar fumarole processes, the distillation-like mechanism could hardly proceed due to the extremely low pressure on the near-surface of the Moon26,27.
Anhydrous magmatic degassing of Cl-bearing species would lead to kinetic fractionation of Cl isotopes, i.e. 35Cl escapes more readily than 37Cl8. During this process, Cl concentration of the residual melt decreases gradually and δ37Cl value increases. Therefore, apatite grains crystallized from the magma are expected to show a negative correlation between δ37Cl and the Cl content. To the contrary, we found that the apatite with lower Cl content has lighter Cl isotopes in NWA 2977 (Table 1). Apatite grains in MIL 05035 have a large variation in Cl contents (4100–14300 ppm) but display almost identical δ37Cl (from +3.8 ± 3.1‰ to +7.8 ± 3.3‰). It might be possible that magmatic degassing had caused the Cl fractionation before apatite saturation. In this case, the Cl isotopes incorporated into apatite would not have to be correlated with the Cl content. However, one must be aware that an elevated δ37Cl could only be produced through extreme Cl loss8. Under the condition of Rayleigh distillation, +80‰ fractionation of Cl isotopes would require >99% volatilization of Cl in the magma (Fig. S3). However, most apatite grains analyzed here and reported in literatures8,9,10,11,12,14 contain considerable amounts of Cl. If apatite had crystallized from a magma with a high δ37Cl, a mechanism must be invoked to concentrate the highly fractionated Cl isotopes, e.g., the reservoir of urKREEP9,10. Therefore, we argue that magmatic degassing alone could not account for the large Cl isotope fractionation in the lunar samples.
The δ37Cl values are not correlated with the extent of shock metamorphism among lunar samples. The mare basalts MIL 05035 and NWA 2977 experienced a similar degree of shock metamorphism (refer to SI), but apatite in MIL 05035 has much lower δ37Cl than that of NWA 2977 (Table 1). For the Mg-suite highland rocks Apollo 76535 and 78235, the former is unshocked28 and the latter had been heavily shocked to ~50 GPa29, but their apatites have nearly identical δ37Cl values (~+30‰)10. Therefore, shock metamorphism would not be responsible for the lunar Cl fractionation as illustrated further in the later section.
Surface alteration, including micrometeorite bombardment and solar wind sputtering, could cause evaporation and isotopic fractionation of some volatile elements30,31 (e.g., S, C, and K). Studies of Cl isotopes of lunar regolith have been limited so far. Available data suggests no correlation between δ37Cl and the maturity of the soil8. Proton bombardment experiment simulating solar wind implantation yields no Cl isotopic fractionation8. Therefore, surface alteration might have played an insignificant role, but would definitely not be a major process in the Cl isotopic fractionation for most lunar samples.
Considering all of the scenarios discussed above, it is still not apparent what process is responsible for the fractionation of lunar Cl isotope, but there is a hint that the elevated lunar Cl isotope could be endogenous, developed during the Moon-forming processes. We are left with a scenario of the Giant Impact event in which the large Cl isotope fractionation could be induced during the energetic impact and massive evaporation of the LMO9. As previously suggested32, the formation of Moon involves the presence of a LMO which accreted from the vapor phases generated by the Giant Impact on the proto-Earth. Large fractions of volatile elements (e.g., K, Zn, and Cl) were lost at extremely high temperatures (up to ~1800 °C)32, and the vapor would enrich 37Cl through preferentially losing 35Cl to space (Rayleigh distillation). A similar scenario was suggested to account for the K and Zn isotope fractionations in the Moon31,33,34. The LMO remained in a liquidus state for about 10 to 200 Myr after the Giant Impact event, followed by the formation of mantle cumulates and an early anorthosite crust a few kilometers thick. These early formed lunar rocks are expected to carry a high 37Cl signature (37Cl/35Cl: ~0.344) as observed in Dhofar 458. It is possible that urKREEP, the last dreg of LMO crystallization, had concentrated volatiles and elevated δ37Cl values due to metal chloride evaporation, but our data is consistent with more than one highly fractionated Cl reservoir in the Moon. For the future work, it is highly desired to analyze Cl isotope compositions from primitive lunar anorthosites.
The reported ages for Dhofar 458 and its paired stone Dhofar 026 range from 0.57 ± 0.01 Ga to 3.43 ± 0.01 Ga35,36,37. Taken at face value, this stone is not a primitive lunar crust rock. However, these ages do not represent the formation time of Dhofar 458 but record a strong shock impact event at ~3.4 Ga35.
Because Dhofar 458 had been severely shocked and experienced extensive partial melting38, it is vital to ascertain the shock influence on the Cl isotopes of apatite. Lines of petrographic evidence support that the apatite was not crystallized from a shock-induced melt, but rather a relict grain. It has a large grain size (60 × 100 µm) and subhedral morphology (Fig. 1a) distinct from the skeletal crystals and fine-grained intergrowths crystallized from the shock-induced melt38. The edge of the apatite grain had been partially corroded by the recrystallized plagioclase-augite intergrowth (Fig. 1a), indicating that the formation of apatite preceded the formation of the intergrowth. If apatite was crystallized from a shock-induced melt, it should crystallize after the augite-plagioclase intergrowth. Therefore, the apatite grain is a relict grain partially corroded by shock-induced melt. It has been noted that under elevated temperatures (1100–2300 °C) and pressures (10–15 GPa), apatite could lose its volatile components and transform into a high-pressure polymorph tuite [γ-Ca3(PO4)2] through solid-state phase transition, decomposition, or crystallization from a shock-induced melt39. However, mineralogical evidence demonstrates that the shock-induced devolatilization and thus Cl isotope fractionation is minimal for the apatite in Dhofar 458. First, Raman spectra of the apatite exhibit characteristic bands of F,Cl-apatite at 963, 429, 580, 591, and 1050 cm−1 (Fig. 1c); the sharp narrow peaks indicate almost intact crystallinity. Second, this apatite grain has abundant F and Cl with a virtually ideal structural formula (Table S1). If the apatite experienced considerable solid-state devolatilization, lattice vacancy and decrease of F and Cl contents should be observed. Actually, the shock pressure and post-shock temperature were unevenly distributed within Dhofar 458, and the shock-induced melt had cooled and crystallized rapidly40. Therefore, we conclude that the shock process had only led to negligible halogen loss for the apatite grain in Dhofar 458. The high δ37Cl of the relict apatite was most likely inherited from its parental melt. There might be a reservoir in the Moon with δ37Cl value above +80‰, which was induced from the Moon-forming processes.
Experimental results have shown that under low pressure (<1 bar) and low oxygen fugacity, H-rich vapors evaporate rapidly and efficiently from the magma26. We speculate that immediately after the birth of the LMO, H-rich vapors, such as H2, HCl, HF, and H2S, might have lost from the surface or sub-surface of the LMO. After the fractionation of the Cl isotopes, hydrous components would be imported, which is compatible with the model that volatiles were accreted to the Moon as a “late veneer” during the crystallization period (~10–200 Myr) of the LMO4,41,42,43.
Our results show that the H2O and Cl contents and δ37Cl and δD values of lunar apatite exhibit distinct inter- and/or intra-sample variations (Table 1), which are basically compatible with previous reports1,2,3,4,5,6,7,8,9,10,11,12,13,14 that water and Cl are heterogeneously distributed in the Moon. The mechanism for the heterogeneity of lunar volatiles is not fully understood44,45. The variations of water content and D/H ratio of lunar samples have been ascribed to, at least, magmatic degassing, regolith assimilation, and delivery of impactors4,44,45,46. Nevertheless, our knowledge on the Cl signature of asteroid and comet impactors is limited. Available data from several carbonaceous chondrites exhibit a narrow range of δ37Cl (from −2.1 to +1.2‰)18. Surface alterations are believed to have negligible effect on the lunar Cl isotope ratio8. Therefore, although contributions from impactors cannot be ruled out completely, it is conceivable that the heterogeneity of lunar Cl is mainly due to endogenous mechanism, i.e. the complex lunar formation and differentiation processes47 including the secondary metasomatism suggested by Potts et al.14
Methods
Three polished thin sections of MIL 05035, 31 (loaned from MWG), Dhofar 458 (PMO-1026), and NWA 2977 (PMO-0095), and one polished specimen of NWA 2977 (PMO-0244) mounted in indium were studied. Apatite grains were identified with a Hitachi S-3400N scanning electron microscope (SEM) equipped with an Oxford X-Max 20 energy dispersive spectroscope, housed at Purple Mountain Observatory (PMO).
The chemical compositions of the minerals were measured with an electron microprobe (JEOL JXA-8230) at PMO. The accelerating voltage was 15 kV. The beam current was 20 nA. To minimize the halogen migration, F and Cl were measured first in each channel, and the integral times were halved to 10 s/5 s at the peak and background positions. Both synthetic and natural mineral standards were used, and ZAF and XPP (for apatite only) corrections were applied. The Durango apatite was analyzed as a secondary standard during analytical sessions. After measuring the major and minor elements, the REEs (Ce, Pr, and Nd detected by qualitative analysis) and Y in apatite were analyzed separately, using a beam current of 40 nA, beam diameters of 3–8 μm, and a collecting time of 30 s/15 s. The concentrations of the major and minor elements obtained were input as set compositions for accurate data correction. The detection limit of the REEs and Y was 80–160 ppm.
The Raman spectra of the apatite were obtained at PMO with a Thermo Fisher DXR micro-Raman spectrometer, using a 532 nm laser, 2 mW laser power, and 25 μm pinhole aperture. The estimated spot size was 0.7 μm and the spectral resolution was 2 cm−1.
Volatile contents and Cl isotope compositions of the apatite were measured with the Cameca NanoSIMS 50 L ion microprobe at Caltech. A primary Cs+ beam of 8 keV and ~12 pA was used, with a normal-incidence electron gun for sample charging compensation. Ion images of 12C−, 16O1H−, and 35Cl− were used to locate grain boundaries and to monitor any cracks or hot spots in grains before and after each analysis. Samples were pre-sputtered for ~5 min. Secondary ions of 12C−, 16O1H−, 18O−, 19F−, 32S−, 35Cl−, and 37Cl− were simultaneously detected with electron multipliers (EMs). The raster size of the primary beam was 3 × 3 µm2, but only secondary ions from the center area (1.5 × 1.5 µm2) were collected in order to avoid edge contamination. The mass resolving powers (MRPs) were ~10,000, sufficient to separate all significant interferences from the masses of interest, such as 17O− from 16O1H−, 31P1H− from 32S−, and 19F16O− from 35Cl−. Secondary ions were integrated for 200–2000 cycles (1 sec/cycle) based on volatile abundances. The mass peaks were auto-centered after every 100–200 cycles.
Calibrations for OH (reported as H2O equivalent), F, Cl, and S contents were based on natural apatites from Durango (Ap-3), Colorado (Ap-5), and Russia (Ap-18). Apatite standards and a San Carlos olivine grain were polished and embedded in an indium disk. The chemical compositions of the apatite standards were analyzed by Francis M. McCubbin at the University of New Mexico, Albuquerque, using an electron microprobe and H manometry3 (Table S3). At the beginning of an analytical session, apatite standards were analyzed and calibration curves were obtained (Fig. S4). To reduce instrumental background, all samples were kept in the sample chamber overnight before analysis. The chamber vacuum during analysis was (1–8) × 10–10 torr. Instrumental background was removed by measuring San Carlos olivine for apatite standards and adjacent plagioclase for lunar apatite. The reported uncertainties include both counting statistics and errors for the calibration curves. The δ37Cl values were corrected to the Durango apatite (+0.4‰).
After NanoSIMS analysis, all sections were re-polished and re-carbon-coated for measurement of H isotopes using the Cameca 7f-Geo ion microprobe at Caltech. The primary 16O− beam of 3–7 nA and −13 keV was focused to a diameter of 10–15 µm. The sample was pre-sputtered over 10 × 10 µm areas for ~5 min, and then the beam raster size was reduced to 1 × 1 µm area for data collection. Secondary ions, 1H+ and 2H+, were collected in peak-jumping mode with an EM. A 90% e-gate was applied to avoid edge contamination. The MRP was 1500. All samples were kept in the sample chamber overnight before analysis. Liquid nitrogen and a Ti-sublimation pump were used to maintain the chamber vacuum below 8 × 10−10 torr. The instrumental background was estimated from the San Carlos olivine and lunar plagioclase and was determined to contribute to the 1H+ count rate by <1% if the apatite had ~5000 ppm water. For the Durango apatite (~300 ppm water), the background contribution was ~10%. The δD values of apatite were standardized using the Durango apatite (δD = −120 ± 5‰). Due to the short cosmic ray exposure ages of our samples (<12 Ma, Supplementary Information) and the relatively high H2O content of apatite, spallogenic D only accounts for negligible proportions of the deuterium (<1%). Therefore, no corrections for spallogenic D were applied here.
In an independent session, we measured the water contents of the apatite in a different thin section of NWA 2977 (PMO-0095) using the Cameca 7f-Geo ion microprobe. The results were similar to those obtained from the section (PMO-0244) embedded in indium. Working conditions were as follows: primary Cs+ beam of 4 nA and 10 keV, impact energy of 19 keV, and an electron-gun was applied. Samples were pre-sputtered over 25 × 25 µm areas for 120 s. The primary beam, with a diameter of ~20 µm, was rastered over 10 × 10 µm areas for data collection. 16O1H− and 31P− ions were detected with an EM, using 31P− as a reference mass. The MRP was 6000, which is high enough to separate 17O− from 16O1H−. An 80% e-gate was used. The calibration curve and background corrections were made using the same procedure as those from the NanoSIMS analysis.
After each working session, all craters on the sample surface were examined carefully with an SEM. Only data from clean apatite regions were adopted.
Data Availability
All data analyzed during this study are included in this published article (and its Supplementary Information files).
References
Saal, A. E. et al. Volatile content of lunar volcanic glasses and the presence of water in the Moon’s interior. Nature 454, 192–195 (2008).
Boyce, J. W. et al. Lunar apatite with terrestrial volatile abundances. Nature 466, 466–469 (2010).
McCubbin, F. M. et al. Nominally hydrous magmatism on the Moon. Proc. Natl. Acad. Sci. USA 107, 11223–11228 (2010).
Greenwood, J. P. et al. Hydrogen isotope ratios in lunar rocks indicate delivery of cometary water to the Moon. Nat. Geosci. 4, 79–82 (2011).
Hauri, E. H., Weinreich, T., Saal, A. E., Rutherford, M. C. & Van Orman, J. A. High pre-eruptive water contents preserved in lunar melt inclusions. Science 333, 213–215 (2011).
Hui, H., Peslier, A. H., Zhang, Y. & Neal, C. R. Water in lunar anorthosites and evidence for a wet early moon. Nat. Geosci. 6, 177–180 (2013).
Canup, R. M. & Asphaug, E. Origin of the Moon in a giant impact near the end of the Earth’s formation. Nature 412, 708–712 (2001).
Sharp, Z. D., Shearer, C. K., McKeegan, K. D., Barnes, J. D. & Wang, Y. Q. The chlorine isotope composition of the Moon and implications for an anhydrous mantle. Science 329, 1050–1053 (2010).
Boyce, J. W. et al. The chlorine isotope fingerprint of the lunar magma ocean. Sci. Adv. 1, e1500380 (2015).
Barnes, J. J. et al. Early degassing of lunar urKREEP by crust-breaching impact(s). Earth Planet. Sci. Lett. 447, 84–94 (2016).
Tartèse, R., Anand, M., Joy, K. H. & Franchi, I. A. H and Cl isotope systematics of apatite in brecciated lunar meteorites Northwest Africa 4472, Northwest Africa 773, Sayh al Uhaymir 169, and Kalahari 009. Meteorit. Planet. Sci. 49, 2266–2289 (2014).
Treiman, A. H. et al. Phosphate-halogen metasomatism of lunar granulite 79215: Impact-induced fractionation of volatiles and incompatible elements. Am. Mineral. 99, 1860–1870 (2014).
Shearer, C. K. et al. Chlorine distribution and its isotopic composition in “rusty rock” 66095. Implications for volatile element enrichments of “rusty rock” and lunar soils, origin of “rusty” alteration and volatile element behavior on the Moon. Geochim. Cosmochim. Acta 139, 411–433 (2014).
Potts, N. J., Barnes, J. J., Tartèse, R., Franchi, I. A. & Anand, M. Chlorine isotopic compositions of apatite in Apollo 14 rocks: Evidence for widespread vapor-phase metasomatism on the lunar nearside ~4 billion years ago. Geochim. Cosmochim. Acta 230, 46–59 (2018).
Barnes, J. J., Anand, M. & Franchi, I. A. Chlorine in lunar basalts. 48th Lunar Planet. Sci. Conf. abstr, 1724 (2017).
Barnes, J. J., McCubbin, F. M., Boyce, J. W., Nguyen, A. N. & Messenger, S. Volatiles in high titanium basalts from the Moon. 48th Lunar Planet. Sci. Conf. abstr, 1727 (2017).
PatiñoDouce, A. E., Roden, M. F., Chaumba, J., Fleisher, C. & Yogodzinski, G. Compositional variability of terrestrial mantle apatites, thermodynamic modeling of apatite volatile contents, and the halogen and water budgets of planetary mantles. Chem. Geol. 288, 14–31 (2011).
Sharp, Z. D. et al. Chlorine isotope homogeneity of the mantle, crust and carbonaceous chondrites. Nature 446, 1062–1065 (2007).
Boyce, J. W., Tomlinson, S. M., McCubbin, F. M., Greenwood, J. P. & Treiman, A. H. The lunar apatite paradox. Science 344, 400–402 (2014).
Neal, C. R. & Kramer, G. Y. The petrogenesis of the Apollo 14 high-Al mare basalts. Am. Mineral. 91, 1521–1535 (2006).
Jerde, E. A. & Taylor, L. A. Searching for neuKREEP: An EMP study of Apollo 11 group A basalts. 24th Lunar Planet. Sci. Conf. abstr. 717–718 (1993).
Warren, P. H., Ulff-Møller, F. & Kallemeyn, G. W. “New” lunar meteorites: Impact melt and regolith breccias and large-scale heterogeneities of the upper lunar crust. Meteorit. Planet. Sci. 40, 989–1014 (2005).
Sharp, Z., Williams, J., Shearer, C., Agee, C. & McKeegan, K. The chlorine isotope composition of Martian meteorites 2. Implications for the early solar system and the formation of Mars. Meteorit. Planet. Sci. 51, 2111–2126 (2016).
Bonifacie, M. et al. The chlorine isotope composition of Earth’s mantle. Science 319, 1518–1520 (2008).
Sharp, Z. D., Barnes, J. D., Fischer, T. P. & Halick, M. An experimental determination of chlorine isotope fractionation in acid systems and applications to volcanic fumaroles. Geochim. Cosmochim. Acta 74, 264–273 (2010).
Ustunisik, G., Nekvasil, H., Lindsley, D. H. & McCubbin, F. M. Degassing pathways of Cl-, F-, H-, and S-bearing magmas near the lunar surface: Implications for the composition and Cl isotopic values of lunar apatite. Am. Mineral. 100, 1717–1727 (2015).
Vaniman, D., Reedy, R., Heiken, G., Olhoeft, G. & Mendell, W. The lunar environment. In Lunar Sourcebook: A user’s guide to the Moon. (eds Heiken, G. H., Vaniman, D. T. & French, B. M.) 27–60 (Lunar and Planetary Institute and Cambridge University Press, 1991).
Warren, P. H. A concise compilation of petrologic information on possibly pristine nonmare Moon rocks. Am. Mineral. 78, 360–376 (1993).
Sclar, C. B. & Bauer, J. F. Shock-induced subsolidus reduction-decomposition of orthopyroxene and shock-induced melting in norite 78235. 6th Lunar Planet. Sci. Conf. abstr. 730–732 (1975).
McCubbin, F. M. et al. Magmatic volatiles (H, C, N, S, Cl) in the lunar mantle, crust, and regolith: Abundances, distributions, processes, and reservoirs. Am. Mineral. 100, 1668–1707 (2015).
Wang, K. & Jacobsen, S. B. Potassium isotopic evidence for a high-energy giant impact origin of the Moon. Nature 538, 487–490 (2016).
Warren, P. H. The magma ocean concept and lunar evolution. An. Rev. Earth Planet. Sci. 13, 201–240 (1985).
Paniello, R. C., Day, J. M. D. & Moynier, F. Zinc isotopic evidence for the origin of the Moon. Nature 490, 376–379 (2012).
Kato, C., Moynier, F., Valdes, M. C., Dhaliwal, J. K. & Day, J. M. D. Extensive volatile loss during formation and differentiation of the Moon. Nat. Commun. 6, 7617 (2015).
Zhang, A. et al. Impact melting of lunar meteorite Dhofar 458: Evidence from polycrystalline texture and decomposition of zircon. Meteorit. Planet. Sci. 46, 103–115 (2011).
Cohen, B. A., Swindle, T. D., Taylor, L. A. & Nazarov, M. A. 40Ar-39Ar ages from impact melt clasts in lunar meteorites Dhofar 025 and Dhofar 026. 33rd Lunar Planet. Sci. Conf. abstr. 1252 (2002).
Fernandes, V. A., Anand, M., Burgess, R. & Taylor, L. A. Ar-Ar studies of Dhofar clast-rich feldspathic highland meteorites: 025, 026, 280, 303. 35th Lunar Planet. Sci. Conf. abstr. 1514 (2004).
Cohen, B. A., James, O. B., Taylor, L. A., Nazarov, M. A. & Barsukova, L. D. Lunar highland meteorite Dhofar 026 and Apollo sample 15418: Two strongly shocked, partially melted, granulitic breccias. Meteorit. Planet. Sci. 39, 1419–1447 (2004).
Xie, X., Zhai, S., Chen, M. & Yang, H. Tuite, γ-Ca3(PO4)2, formed by chlorapatite decomposition in a shock vein of the Suizhou L6 chondrite. Meteorit. Planet. Sci. 48, 1515–1523 (2013).
James, O. B., Cohen, B. A., Taylor, L. A. & Nazarov, M. A. Comment on: “New” lunar meteorites: Impact melt and regolith breccias and large-scale heterogeneities of the upper lunar crust, by P. H. Warren, F. Ulff-Møller, and G. W. Kallemeyn. Meteorit. Planet. Sci. 42, 1029–1032 (2007).
Barnes, J. J. et al. An asteroidal origin for water in the Moon. Nat. Commun. 7, 11684, https://doi.org/10.1038/ncomms11684 (2016).
Albarède, F. Volatile accretion history of the terrestrial planets and dynamic implications. Nature 461, 1227–1233 (2009).
Saal, A. E., Hauri, E. H., Van Orman, J. A. & Rutherford, M. J. Hydrogen isotopes in lunar volcanic glasses and melt inclusions reveal a carbonaceous chondrite heritage. Science 340, 1317–1320 (2013).
Robinson, K. L. & Taylor, G. J. Heterogeneous distribution of water in the Moon. Nat. Geosci. 7, 401–408 (2014).
Treiman, A. H. et al. D-poor hydrogen in lunar mare basalts assimilated from lunar regolith. Am. Mineral. 101, 1596–1603 (2016).
Gibson, E. K. Jr. & Moore, G. W. Volatile-rich lunar soil: Evidence of possible cometary impact. Science 179, 69–71 (1973).
Warren, P. H. The Moon. In Meteorites, comets, and planets. Treatise on Geochemistry, Vol. 1. (eds Davis, A. & Holland H. D.) 559–600 (Elsevier, 2014).
Acknowledgements
We acknowledge Francis M. McCubbin for conducting analysis of apatite standards. We are grateful to Profs. Timothy Swindle and Francis Albarède for their editorial handling of this manuscript. We also want to thank three anonymous reviewers for their constructive comments that helped improve the quality of this paper. This work was supported by the National Natural Science Foundation of China (Grant No. 41573060, 41773059 and 41573059), the Open Foundation of the State Key Laboratory of Lunar and Planetary Science, MUST (FDCT No. 039/2013/A2), the Macau FDCT (005/2017/A1, 119/2017/A3, 0079/2018/A2), and the Minor Planet Foundation of Purple Mountain Observatory.
Author information
Authors and Affiliations
Contributions
Y.W. and W.H. designed the research and wrote the manuscript; Y.W. and Y.G. performed the research; all authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wang, Y., Hsu, W. & Guan, Y. An extremely heavy chlorine reservoir in the Moon: Insights from the apatite in lunar meteorites. Sci Rep 9, 5727 (2019). https://doi.org/10.1038/s41598-019-42224-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-42224-8
This article is cited by
-
Detection of apatite in ferroan anorthosite indicative of a volatile-rich early lunar crust
Nature Astronomy (2024)
-
Evidence of a hydrated mineral enriched in water and ammonium molecules in the Chang’e-5 lunar sample
Nature Astronomy (2024)
-
Geochemical Constraints on the Origin of the Moon and Preservation of Ancient Terrestrial Heterogeneities
Space Science Reviews (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
