
Abstract
Because of its ability to expedite specimen identification and species delineation, the barcode index number (BIN) system presents a powerful tool to characterize hyperdiverse invertebrate groups such as the Acari (mites). However, the congruence between BINs and morphologically recognized species has seen limited testing in this taxon. We therefore apply this method towards the development of a barcode reference library for soil, poultry litter, and nest dwelling mites in the Western Palearctic. Through analysis of over 600 specimens, we provide DNA barcode coverage for 35 described species and 70 molecular taxonomic units (BINs). Nearly 80% of the species were accurately identified through this method, but just 60% perfectly matched (1:1) with BINs. High intraspecific divergences were found in 34% of the species examined and likely reflect cryptic diversity, highlighting the need for revision in these taxa. These findings provide a valuable resource for integrative pest management, but also highlight the importance of integrating morphological and molecular methods for fine-scale taxonomic resolution in poorly-known invertebrate lineages.
Similar content being viewed by others

Introduction
DNA barcoding1 alleviates many of the challenges associated with morphological specimen identification by comparing short, standardized fragments of DNA – typically 648 bp of the cytochrome c oxidase I (COI) gene for animals – to a well-curated reference library. The success of this method relies on the presence of a clearly defined ‘barcode gap’, where intraspecific divergences are much more constrained than interspecific divergences. Its presence not only enables rapid specimen identification, but also facilitates species delineation through molecularly defined taxonomic units, a process automated through the barcode index number (BIN) system2. BINs correspond well with morphologically recognized species in lineages with well-curated taxonomy2,3,4 and can improve taxonomic resolution by elucidating hidden diversity5,6. Consequently, BINs are a powerful tool for characterizing diversity in poorly-known, hyperdiverse, invertebrates7,8,9, but have seen limited validation in these taxa.
The mites (Acari) may exceed one million species, but remain poorly known because of their small size and cryptic morphology10. While BIN–based surveys have expedited surveys of this hyperdiverse group7,11,12, the rapidly growing collection of mite barcodes generally lack lower-level taxonomy. For example, just 18% of the >12,400 mite BINs (from nearly 120,000 DNA barcode sequences) on the Barcode of Life Data System (BOLD, v4.boldsystems.org) are linked with a species name (accessed August 2018). Nonetheless, successful species delineation through DNA barcodes has been documented in several mite lineages, including the Ixodida13, Mesostigmata14, Sarcoptiformes15, and Trombidiformes16. DNA barcodes have also helped resolve issues like lumping due to cryptic morphology17, and splitting due to heteromorphy18. However, concordance between species and BINs has only been tested in a single mite lineage: medically important ticks from Canada19.
While many species of mites have detrimentally impacted human health and agriculture20,21, others are recognized for their benefits as biological control agents22. The poultry red mite (PRM; Dermanyssus gallinae (De Geer, 1778), for example, is a widespread pest with significant economic costs23. Since the PRM is now resistant to most acaricides, the need for novel biocontrol methods is greater than ever24,25. From this perspective, natural mite communities in soil and bird nests may provide novel predators for conservation biological control of the PRM, but have seen limited investigation26,27,28. In the present study we begin the development of a DNA barcode reference library for the identification of poultry litter, soil, and nest dwelling mites in the Western Palearctic. Specifically, we test the correspondence between BINs and traditionally recognized species, and analyze intraspecific divergences at COI to identify potentially cryptic taxa.
Methods
Specimen Collection and Preparation
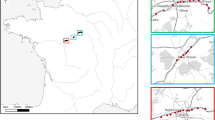
Samples of poultry litter and soil from the vicinity of poultry houses, as well as wild bird nests, were collected between 2015 and 2016 from 53 locations in Croatia, Belgium, France, Israel, Poland, and Spain (Fig. 1, Table 1). Mites were extracted from approximately 0.5 kg of substrate into 99% ethanol (EtOH) using modified Berlese-Tullgren funnels for five days. From each unique collection event (denoted by exact site and collection date), all mites, regardless of life stage or sex, were sorted to morphotype and identified to order using a standard stereomicroscope setup and keys in Krantz and Walter29. Up to five specimens per morphotype were selected for molecular analysis. Each specimen was imaged using a Leica DVM6 microscope and arrayed into a 96-well microplate (Eppendorf) containing 30 µL of 99% EtOH, with one blank well serving as a negative control. The museum identification code (Sample ID), collection details, order level taxonomy, and specimen images were uploaded to BOLD, available in the dataset DS-SMRPM through at https://doi.org/10.5883/DS-SMRPM.

Map of the 53 sampling sites in seven countries across the Western Palearctic. The location markers correspond with site numbers specified in Table 1; sample type (bird nest, poultry house, soil) is indicated by the colour of the marker.
Molecular Analysis
The specimens were sequenced for the barcode region of COI using standard invertebrate DNA extraction30,31, amplification32 and sequencing protocols33 at the Canadian Centre for DNA barcoding (CCDB; http://ccdb.ca/). However, DNA extraction was modified following Porco et al.34 to facilitate the recovery of voucher specimens. A cocktail (1:1 ratio) of LepF1/LepRI1 and LCO1490/HCO219835 primers were chosen to amplify and sequence a 652 bp fragment of DNA from the barcode region of COI because of their prior success in a broad array of mite taxa11. The DNA extracts were archived in −80 °C freezers at the Centre for Biodiversity Genomics (CBG; biodiversitygenomics.net), and the specimen vouchers were stored in 95% EtOH and returned to the Newe-Ya’ar Research Center and the Centre d’Ecologic Functionnelle & Evolutine for morphological preparations.
The forward and reverse chromatograms were assembled into consensus sequences for each specimen and edited using CodonCode Aligner v. 4.2.7 and uploaded to BOLD. Each sequence meeting minimum quality criteria (≥500 base pairs, <1% ambiguous nucelotides, free of contamination and stop codons) was assigned a BIN by BOLD. The sequences were further validated by inspecting their placement in a Neighbor-Joining tree (K2P distance model, BOLD alignment) and corresponding specimen images using the ‘Taxon ID Tree’ function in BOLD (Supplementary Figs 1 and 2). Taxa with unexpected placement in the tree (i.e. conflicting identifications within a cluster, conspecifics forming outgroups, etc.) were blasted against all barcode records on BOLD using the ‘Identification Engine’ tool whereupon instances of contamination (i.e. bacteria, Insecta, etc.) were flagged and filtered from the reference library.
Specimen Identification
Following BIN assignment, up to five vouchers per BIN were prepared for light microscopy by either mounting the specimens directly into Hoyer’s medium, or in the case of Oribatida, placing the specimen in lactic acid on a cavity slide. Since the specimens were sufficiently cleared during the tissue lysis stage of DNA extraction, the typical clearing procedures were not necessary. All remaining vouchers were prepared for SEM imaging on a Hitachi TM3000 TableTop Scanning Electron Microscope, with standard drying and coating procedures.
Each specimen was identified to the lowest possible level of taxonomy, and compared to identifications of other members of the same BIN. Some specimens were not slide mounted because of redundancy, or morphologically identified when precluded by their life stage, sex or voucher quality, and were thus assigned the lowest level of taxonomy in agreement with other members in the BIN. Specimens identified in this way were denoted by ‘BIN Taxonomy Match’ in the Identification Method field.
Data Analysis
Sampling completeness was assessed by constructing a BIN accumulation curve and by estimating total BIN richness using the incidence coverage estimator (ICE) in EstimateS36. Maximum intraspecific and minimum interspecific p-distances were calculated for all morphologically identified specimens using the ‘Barcode Gap Analysis’ tool on BOLD. Species correspondence with BINs were characterized by one of four categories: matches (perfect correspondence between one species and one BIN), splits (one species is represented by more than one BIN), merges (two or more species are assigned to a single BIN), and mixtures (a combination of splits and merges) as described in Ratnasingham and Hebert2.
Results
Sequence Recovery
Barcode compliant sequences were recovered from 298 of the 652 specimens analysed, with an overall PCR success rate of 76.5% and sequencing success rate of 45.7%. Success varied greatly among the major lineages. PCR success, for example, ranged from a high of 85% in the Trombidiformes, to a low of 45% in the Astigmatina (Sarcoptiformes). Sequencing success, on the other hand, ranged from a high of 56% in the Mesostigmata to a low of 0% in the Astigmatina (Sarcoptiformes) and Opilioacarida (Table 2). Non-target amplification was detected in 28 sequences, including cross-mite contamination, insects, and occasionally bacteria. These sequences were flagged on BOLD, removed from the BOLD identification engine, and excluded from subsequent analyses.
DNA Barcode Reference Library and Sample Completeness
Minimum quality requirements for BIN assignment were met by 298 sequences representing 70 BINs in total (\(\bar{x}\,=\,\)4.2 specimens/BIN). Of these 70 BINs, 48 (68.6%) were morphologically identified to the species level, while genus was the lowest identification for six BINs (8.6%), family for 15 BINs (21.4%), and one BIN was identified only to the order level (1.4%). In total, 35 species, 27 genera, 24 families, and three orders were identified in our barcode reference library (Table 3). The slope of the BIN accumulation curve remains steep, indicating incomplete sampling of the fauna (Fig. 2), and the estimate of total BIN richness was more than double the current observations (ICE = 172 BINs).

The observed (solid line) and estimated (dashed line) accumulation of BINs with increasing sample size for the 298 specimens with BIN assignments.
Barcode Gap and BIN Analysis
Of the 35 morphologically identified species with BINs, 19 (61%) perfectly corresponded with BIN assignments, while eight (26%) resulted in BIN splits, and two cases of BIN mixtures affecting four species (13%) were detected (Fig. 3, Table 3). The barcode gap analysis revealed nine species in which maximum interspecific p-distance exceeded minimum intraspecific p-distance (Fig. 3), all of which were involved in BIN splits or mixtures. Maximum intraspecific p-distances averaged 7.7%, and dropped to 0.9% when BIN splits and mixtures were excluded from analyses.

Comparison of maximum intraspecific and minim interspecific divergences (p-distances) of the 35 morphologically identified species. Data points are colourized based on species correspondence with BINs, and the diagonal red line indicates the 1:1 ratio of divergences. The barcode gap is present in species that fall above the line, and absent in those below.
Discussion
Through the integration of morphological and molecular taxonomic methods, we provide DNA barcode coverage for 35 described species and 70 mite BINs from soil, bird nest, and poultry house-associated assemblages in the Western Palearctic. The integrity of most vouchers was sufficiently maintained for morphological identification, and SEM imaging of diagnostic characters (see the following BIN page for example: BOLD:ADA3054). While only 13 of these species have been previously associated with the poultry red mite27,37, additional species are undoubtedly present in our dataset but remain undetected because of low sequencing success combined with several BINs lacking identifications. Our failure to generate any sequences for Astigmatina (Sarcoptiformes) may be explained by low primer affinity, considering amplification rates were also lowest in this group. Primer affinity, however, does not justify the low successes in other lineages with higher amplification rates. Comparable methods, for example, have yielded much higher successes (77%) among soil and leaf litter mites (including Astigmatina) from subarctic Canada11, demonstrating the broad applicability of these primers among a diverse array of taxa. Since 40% of the amplification products generated uninterpretable chromatograms, poor quality DNA template may be responsible for low sequencing successes among taxa.
The concordance between BINs and mite species was much lower than in some well-studied invertebrates (e.g. perfect concordance in 92% of beetles4 and ticks19). However, similar concordance levels have been reported for many taxa including geometrid moths38 (67%), true bugs39 (70%), and spiders5 (54%). Low concordance is mainly driven by species with large intraspecific divergences (>3% p-distance) resulting in the assignment of two or more BINs. While this does not preclude accurate barcode-based identification, it highlights potentially cryptic species because most BIN splits formed widely separated clades (e.g. >15% p-distance) lacking intermediate haplotypes. In fact, 16S and 18S rRNA gene topologies for Androlaelaps casalis (Berlese, 1887) and Proctolaelaps scolyti Evans, 1958 were congruent with BIN splits, further supporting our cryptic species hypothesis in these taxa27. Rhodacarellus silesiacus Willmann, 1936, on the other hand, also formed two distinct but narrowly separated clades (<3% divergence), with divergences similar to those in species with concordant BINs (e.g. Dermanyssus gallinae and Gamasodes spiniger (Oudemans, 1936)), such that additional sampling may reveal intermediate haplotypes causing the BINs to collapse into one2.
More problematic for the barcode based identification of mites are the two cases of shared barcodes confounded by BIN splits (BIN mixtures) affecting four species: Dendrolaelaps longiusculus (Leitner, 1949)/D. presepum (Berlese, 1918), and Proctolaelaps parascolyti Costa, 1963/P. pygmaeus (Müller, 1859). Since multiple species are assigned to the same BIN, mixtures impede accurate identifications, but may also represent taxonomic errors2. Misidentification is unlikely, since procedures were in place to evaluate and correct such errors. However, both cases of BIN mixtures involve closely allied congenerics which may be subjected to hybridization or incomplete lineage sorting40. Given the large intraspecific divergences observed, though, a more probable explanation is the presence of cryptic diversity compounded by inadequate species descriptions. Future work should scrutinize the morphology of genetic clusters from both mixtures and splits for more effective characters to discriminate these potentially cryptic species.
This study represents the first step towards development of a DNA barcode reference library for the identification of poultry litter, soil, and nest dwelling mites from the Western Palearctic, which may in turn reveal natural enemies key to the control of PRM. Although sequencing success rates should be improved, we demonstrate that nearly 80% of the species analysed can be accurately identified through DNA barcodes. Our BIN analysis, however, indicates a high proportion of cryptic diversity and some potential taxonomic confusion. This method consequently presents a powerful tool not only for the identification of unknown specimens, but as the foundation for integrative taxonomy and diversity estimation in hyperdiverse invertebrates such as mites.
Data Availability
All specimen and sequence data is available in the BOLD dataset DS-SMRPM through the following, https://doi.org/10.5883/DS-SMRPM. Valid sequences were also deposited in GenBank under the following accessions: MH983560-MH983861.
References
Hebert, P. D. N., Cywinska, A., Ball, S. L. & deWaard, J. R. Biological identifications through DNA barcodes. Proc. R. Soc. Lond. B. 270, 313–321 (2003).
Ratnasingham, S. & Hebert, P. D. N. A DNA-based registry for all animal species: The barcode index number (BIN) system. PLoS ONE 8, e66213 (2013).
Huemer, P., Mutanen, M., Sefc, K. M. & Hebert, P. D. N. Testing DNA barcode performance in 1000 species of European Lepidoptera: Large geographic distances have small genetic impacts. PLoS ONE 9, e115774 (2014).
Hendrich, L. et al. A comprehensive DNA barcode database for Central European beetles with a focus on Germany: adding more than 3500 identified species to BOLD. Mol. Ecol. Resour. 15, 795–818 (2015).
Blagoev, G. A. et al. Untangling taxonomy: a DNA barcode reference library for Canadian spiders. Mol. Ecol. Resour. 16, 325–341 (2016).
Ren, J.-M. et al. Barcode index numbers expedite quarantine inspections and aid the interception of nonindigenous mealybugs (Pseudococcidae). Biol. Invasions 20(2), 449–460 (2017).
Telfer, A. et al. Biodiversity inventories in high gear: DNA barcoding facilitates a rapid biotic survey of a temperate nature reserve. Biodivers, Data J. 3, e6313 (2015).
Hebert, P. D. N., Hollingsworth, P. M. & Hajibabaei, M. From writing to reading the encyclopedia of life. Philos. Trans. Royal Soc. B. 371, 20150321 (2016).
D’Souza, M. L. & Hebert, P. D. N. Stable baselines of temporal turnover underlie high beta diversity in tropical arthropod communities. Mol. Ecol. 27, 2447–2460 (2018).
Walter, D. E. & Proctor, H. C. Mites and biological diversity. in Mites: Ecology, evolution & behaviour 447–459 (Springer, 2013).
Young, M. R., Behan-Pelletier, V. M. & Hebert, P. D. N. Revealing the hyperdiverse mite fauna of subarctic Canada through DNA barcoding. PLoS ONE 7, e48755 (2012).
Beaulieu, F. et al. Acari of Canada. ZooKeys 819, 77–168 (2019).
Zhang, R. L. & Zhang, B. Prospects of using DNA barcoding for species identification and evaluation of the accuracy of sequence databases for ticks (Acari: Ixodida). Ticks Tick Borne Dis. 5, 352–358 (2014).
Li, J.-B. et al. COI barcoding as a molecular assay for the identification of phytoseiid mites. Sys. Appl. Acarol. 17 (2012).
Dabert, J., Ehrnsberger, R. & Dabert, M. Glaucalges tytonis sp. n. (Analgoidea, Xolalgidae) from the barn owl Tyto alba (Strigiformes, Tytonidae): compiling morphology with DNA barcode data for taxon descriptions in mites (Acari). Zootaxa 1719(11) (2008).
Pesic, V. & Smit, H. Neumania kyrgyzica sp. nov. a new water mite from Kyrgyzstan based on morphological and molecular data (Acari, Hydrachnidia: Unionicolidae). Sys. Appl. Acarol. 22, 885 (2017).
Stalstedt, J., Bergsten, J. & Ronquist, F. “Forms” of water mites (Acari: Hydrachnidia): intraspecific variation or valid species? Ecol. Evol. 3, 3415–3435 (2013).
Glowska, E., Dragun-Damian, A., Broda, L., Dabert, J. & Dabert, M. DNA barcodes reveal female dimorphism in syringophilid mites (Actinotrichida: Prostigmata: Cheyletoidea): Stibarokris phoeniconaias and Ciconichenophilus phoeniconaias are conspecific. Folia Parasit. 61, 272–276 (2014).
Ondrejicka, D. A., Morey, K. C. & Hanner, R. H. DNA barcodes identify medically important tick species in Canada. Genome 60, 74–84 (2017).
Lipsker, D., Jaulhac, B. (eds.) Lyme borreliosis: biological and clinical aspects. Vol. 37. (Karger, 2009).
Dhooria, M. S. Fundamentals of applied acarology. (Springer, 2016).
Mcmurtry, J. A., Moraes, G. J. D. & Sourassou, N. F. Revision of the lifestyles of phytoseiid mites (Acari: Phytoseiidae) and implications for biological control strategies. Sys. Appl. Acarol. 18, 297 (2013).
Sparagano, O. et al. Prevalence and key figures for the poultry red mite Dermanyssus gallinae infections in poultry farm systems. Exp. Appl. Acarol. 48, 3–10 (2009).
Marangi, M. et al. Evaluation of the poultry red mite, Dermanyssus gallinae (Acari: Dermanyssidae) susceptibility to some acaricides in field populations from Italy. Exp. Appl. Acarol. 48, 11–18 (2009).
Sparagano, O. A. E., George, D. R., Harrington, D. W. J. & Giangaspero, A. Significance and control of the poultry red mite, Dermanyssus gallinae. Annu. Rev. Entomol. 59, 447–466 (2014).
Lesna, I., Sabelis, M. W., van Niekerk, T. G. C. M. & Komdeur, J. Laboratory tests for controlling poultry red mites (Dermanyssus gallinae) with predatory mites in small ‘laying hen’ cages. Exp. Appl. Acarol. 58, 371–383 (2012).
Roy, L. et al. Arthropod communities of laying hen houses: an integrative pilot study toward conservation biocontrol of the Poultry Red Mite Dermanyssus gallinae. Biol. Control 114, 176–194 (2017).
Tamara, B. H., Granich, J., Körbes, J. H., da Silva, G. L. & Ferla, N. J. Mite fauna (Acari) associated with the poultry industry in different laying hen management systems in Southern Brazil: a species key. Acarologia 58, 140–158 (2018).
Krantz, G. W. & Walter, D. E. (eds) A Manual of Acarology. 3rd Edition. (Texas Tech University Press, 2009).
Ivanova, N. V., Dewaard, J. R. & Hebert, P. D. N. An inexpensive, automation-friendly protocol for recovering high-quality DNA: Technical Note. Mol. Ecol. Notes 6, 998–1002 (2006).
Ivanova, N. V., deWaard, J. R. & Hebert, P. D. N. CCDB protocols, glass fiber plate DNA extraction. ccdb.ca/site/wp-content/uploads/2016/09/CCDB_DNA_Extraction.pdf (2007).
Ivanova, N. V. & Grainger, C. M. CCDB protocols, COI amplification. ccdb.ca/site/wp-content/uploads/2016/09/CCDB_Amplification.pdf (2007).
Ivanova, N. V. & Grainger, C. M. CCDB protocols, sequencing. ccdb.ca/site/wp-content/uploads/2016/09/CCDB_Sequencing.pdf (2007).
Porco, D., Rougerie, R., Deharveng, L. & Hebert, P. Coupling non-destructive DNA extraction and voucher retrieval for small soft-bodied Arthropods in a high-throughput context: the example of Collembola. Mol. Ecol. Resour. 10, 942–945 (2010).
Folmer, O., Hoesh, W. R., Black, M. B. & Vrijenhoek, R. C. Conserved primers for PCR amplification of mitochondrial DNA from different invertebrate phyla. Mol. Mar. Biol. Biotechnol. 3, 294–299 (1994).
Colwell, R. K. EstimateS: Statistical estimation of species richness and shared species from samples. viceroy.eeb.uconn.edu/estimates/ (2013).
Brady, J. The Mites of Poultry Litter: Observations on the Bionomics of Common Species, with a Species List for England and Wales. J. Appl. Ecol. 7, 331 (1970).
Zahiri, R. et al. Probing planetary biodiversity with DNA barcodes: The Noctuoidea of North America. PLoS One 12, e0178548 (2017).
Gwiazdowski, R. A., Foottit, R. G., Maw, H. E. L. & Hebert, P. D. The Hemiptera (Insecta) of Canada: constructing a reference library of DNA barcodes. PLoS One 10, e0125635 (2015).
Funk, D. J. & Omland, K. E. Species-level paraphyly and polyphyly. Annu. Rev. Ecol. Evol. Syst. 34, 397–423 (2003).
Acknowledgements
We would like to express our gratitude to the Canadian Centre for DNA Barcoding (CCDB) for their technical assistance and support for molecular analysis, as well as the Barcode of Life Data System (BOLD) team at the Centre for Biodiversity Genomics for their support on data management and storage. We would also like to thank Jandir Cruz Santos for critically analysing nomenclature and providing helpful comments on the manuscript. The study was carried out within the framework of the European Cooperation in Science and Technology (COST) Action (FA1404-COREMI) ‘Improving current understanding and research for sustainable control of the poultry red mite Dermanyssus gallinae’.
Author information
Authors and Affiliations
Contributions
M.R.Y., L.R., Y.G. and E.P. devised the study; M.A., L.B., D.H., J.F.LB., S.G. and E.G.R. collected the specimens and prepared the mites for molecular analysis and museum archiving; M.L.M. and E.U. identified the specimens; E.P. managed and coordinated the activities of the study; M.R.Y. wrote the manuscript and analysed the data; all authors contributed to discussion of ideas and manuscript revisions.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Young, M.R., Moraza, M.L., Ueckermann, E. et al. Linking morphological and molecular taxonomy for the identification of poultry house, soil, and nest dwelling mites in the Western Palearctic. Sci Rep 9, 5784 (2019). https://doi.org/10.1038/s41598-019-41958-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-41958-9
This article is cited by
-
DNA barcodes enable higher taxonomic assignments in the Acari
Scientific Reports (2021)
-
A simple PCR-based method for the rapid and accurate identification of spider mites (Tetranychidae) on cassava
Scientific Reports (2020)
-
Taxonomic integrative and phylogenetic identification of the first recorded Triatoma rubrofasciata in Zhangzhou, Fujian Province and Maoming, Guangdong Province, China
Infectious Diseases of Poverty (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
