
Abstract
Gaucher disease is caused by mutations in GBA1 encoding acid β-glucosidase (GCase). Saposin C enhances GCase activity and protects GCase from intracellular proteolysis. Structure simulations indicated that the mutant GCases, N370S (0 S), V394L (4L) and D409V(9V)/H(9H), had altered function. To investigate the in vivo function of Gba1 mutants, mouse models were generated by backcrossing the above homozygous mutant GCase mice into Saposin C deficient (C*) mice. Without saposin C, the mutant GCase activities in the resultant mouse tissues were reduced by ~50% compared with those in the presence of Saposin C. In contrast to 9H and 4L mice that have normal histology and life span, the 9H;C* and 4L;C* mice had shorter life spans. 9H;C* mice developed significant visceral glucosylceramide (GC) and glucosylsphingosine (GS) accumulation (GC»GS) and storage macrophages, but lesser GC in the brain, compared to 4L;C* mice that presents with a severe neuronopathic phenotype and accumulated GC and GS primarily in the brain. Unlike 9V mice that developed normally for over a year, 9V;C* pups had a lethal skin defect as did 0S;C* mice resembled that of 0S mice. These variant Gaucher disease mouse models presented a mutation specific phenotype and underscored the in vivo role of Saposin C in the modulation of Gaucher disease.
Similar content being viewed by others


Introduction
Acid β-glucosidase (GCase), encoded by GBA1, is the lysosomal hydrolase that hydrolyzes glucosylceramide (GC) and glucosylsphingosine (GS) to ceramide and sphingosine, respectively1. Disruptive GBA1 mutations are causal to Gaucher disease by leading to insufficient GCase function and resultant GC and GS accumulation1. Three types of Gaucher disease are clinically defined, based on age at disease onset and organ involvement. Type 1 is the non-neuronopathic variant with highly variable visceral disease2. Type 1 patients also have an increased life-time risk of developing Parkinson disease variants indicating that mutations in GBA1 are genetic risk factors3. Types 2 and 3 have early onset of primary, but variable, central nervous system (CNS) degeneration and are distinguished phenotypically by the rapidity of CNS disease progression in type 2 during the first year of life4. Several hundred GBA1 mutations have been identified in affected patients2,5. Genotype/phenotype correlations show that the presence of homozygosity or heteroallelism of N370S predicts absence of early onset progressive CNS disease, and L444P homozygosity predisposes to variable primary CNS disease6. However, the relationships of other GBA1 mutations and disease phenotypes are poorly understood.
In humans, the presence of the N370S allele in Gaucher disease patients is associated with type 1 and highly variable visceral involvement2,7,8. The D409H alleles have significant frequency and homozygotes (designated here as 9H) manifest early onset of variable visceral and the CNS involvement9. 9H patients also uniquely manifest calcific aortic root and valvular disease10. The V394L allele in humans has been reported only in the heteroallele state and is associated with type 1 or types 2 and 3 depending on the heteroallele11. In mice, Gba1 mutations have been created to mimic those found in human patients12. In comparison to humans, N370S homozygosity (designated here as 0S) in mice leads to death within 24 hours due to a defect in the skin permeability barrier12. This is likely due to the lesser hydrolytic efficiency of the N370S enzyme toward the longer chain fatty acid acyl moieties on GC in murine skin vs. human13,14,15. Mice homozygous for V394L (designated here as 4L) and 9H have defective GCase activity and survive up to 2 years with relatively mild visceral abnormalities12,16. The mild phenotype in such murine models limits our understanding of in vivo effect of the GBA1/Gba1 mutations.
Human GCase protein structures have been solved by X-ray crystallography and only one mutant GCase structure, N370S, has been characterized by X-ray and biochemical analyses13,17,18. The approach of studying protein - structure-function relationship has relied on structural modeling and dynamic simulation based on the crystal structure information19,20. A computation tool (Swiss-Pdb Viewer) can be applied to simulate GBA1 mutations and their dynamic alterations, side chain interactions and force field energy changes at the atomic resolution for better understanding mutant GCases protein function.
Saposin C is a lysosomal protein that functions in maximizing enzymatic activity of GCase and in protecting GCase against intracellular proteolysis21,22,23. Mutations in the Saposin C region of the prosaposin gene (PSAP) produce variant forms of Gaucher disease24,25. Specific Saposin C deficient mice (C−/−, designated here as C*) were generated by a knock-in point mutation within the Saposin C domain of the Prosaposin locus (Psap), preserving Saposin A, B, and D, but leading to undetectable Saposin C protein and reductions of GCase activity and protein, and a slowly progressive CNS phenotype developing after 8–12 months26. Mice with Saposin C deficiency (C*) and homozygosity for 4L (combined model designated 4L;C*) have greater reductions in 4L GCase levels and develop a severe CNS disease phenotype compared to C* mice26,27.
To study the pathogenic effect of Gba1 mutations and gain insights into the in vivo effects of GCase and Saposin C, additional Gba1 mutant mouse models (0S, 9H, and 9V) combined with C* (designated as “Gba1 mutation”;C*,. e.g, 0S;C*) were created and analyzed for biochemical, histopathologic, and phenotypic abnormalities. Compared to the models of 4L or 9H homozygotes combined with a hypomorphic prosaposin transgene that expresses subnormal levels of mouse prosaposin and four saposins22, these models allow the study of Saposin C’s specific effects with GCase mutants. These studies reveal mutation-dependent and tissues-specific phenotypes in Gba1 mutant mice that are deficient in Saposin C and also highlight the critical role of Saposin C, or its potential variants, in GCase function.
Results
Simulation analysis of structural effects of GCase mutations
To understand differential effects of mutations on GCase conformation and function, human crystal structures13,17,18 were used to simulate the effects of D409H, D409V, V394L and N370S on GCase by analyzing side chain interactions and force field energy changes. In force field energy computing, negative energy values mean favorable energy environment, whereas positive values indicate unfavorable energy environment for a given amino acid (Supplementary Table 1).
D409 (wild type, WT) at pH 7.2 and pH 5.5 maintained the same side chain interactions with 7 surrounding amino acids (Fig. 1, Supplementary Table 1). The force field energy for D409 changed from −17.148 KJ/mole at pH 7.2 to −12.592 KJ/mole at pH 5.5 (Supplementary Table 1). Histidine (H) has a polar amino side chain and can either be protonated or deprotonated. D409H showed a gain of an additional side chain interactions with I406 at pH7.2 and involved 5 additional interacting amino acids at pH 5.5 (Supplementary Table 1). Force field energy analyses for D409H showed conversion to an unfavorable energy environment (positive), 17.879 KJ/mole at pH7.2 and 10.184 KJ/mole at pH 5.5, suggesting this mutation would produce a significant conformational change and altered side chain interactions in neutral and acidic environments. The D409V contains a non-polar amino acid, valine, which lost all WT interactions with surrounding amino acid side chains (Fig. 1, Supplementary Table 1). The force field energy value for D409V was positive, i.e., unfavorable, 56.649 KJ/mole at pH 7.2, and more unfavorable, 72.925 KJ/mole at pH 5.5, relative to WT and D409H. These results indicate that D409V has greater unfavorable energy environment than D409H that could result in their differential in vivo hydrolytic properties. D409 is within the GCase binding motif (399DSPIIVDITKD409) for LIMP-2 (lysosomal integral membrane protein 2), the internal trafficking chaperone of GCase for lysosomal targeting28,29. Mutation at this position (D409V, D409H) leads to an unstable enzymes that predisposed them to protease degradation compared to WT.

Modeling of mutation effect on GCase structure. (A) GCase structure showing side chain interaction with D409. (B) Enlarged side chain interaction region. Red clouds show electronic force field at position 409. WT D409 at pH7.2 or pH 5.5 maintain the same side chain interactions with 7 surrounding amino acids showing green/yellow at pH 7.2 and white/blue at pH5.5. Carbons on amino acid are labeled as red dots. D409H gains additional side chain interactions at pH7.2 and turns more dramatic alterations in its conformation at pH 5.5. D409V interact with surrounding amino acids. This mutation at 409 changes D to V (Valine), a non-polar amino acid side chain, which lost all WT interactions with surrounding amino acid side chains. Human PDB crystal structures 2F61, pH7.2, 2.5 Å and 3GXI, pH 5.5, 1.84 Å from Swiss PDB Viewer (DeepView, SPDBV,Version 4.10) program were used for modelling. Amino acids involved side chain interaction are listed in Supplementary Table 1.
V394 is located on Domain 1 of GCase at the anti-parallel β-sheet, which is close to the active site pocket (a loop formation) opening13,30. The amino acids involved in the side chain interaction in V394L mutant are different from WT (Supplementary Table 1). The force field energy calculated at the WT, i.e., V394, is dramatically increased from 11.884 to 2627.135 KJ/mole (220 fold) for V394L mutant at pH 7.2 (Supplementary Table 1). At pH 5.5, the force field energy for V394L mutant changes from 12.841 to 28645.000 KJ/mole (2230 fold) compared to WT, especially in the non-bound energy calculation, suggesting this mutation affects GCase both in structure as well as accessibility for side chain interaction leading to reduced enzymatic activity.
The side chain interaction at N370S was analyzed based on its crystal structures at pH 7.1 (3KEH) and acidic pH 5.4 (3KEO)18. Located at Domain III, the role of N370 in the catalytic cycle is significant, probably associated with local conformational effects at or near the active site15. Here, N370S at pH 5.4 showed slight conformation changes compared to WT (Supplementary Table 1). The WT N370 has a negative energy force (−193 to −199 KJ/mole), which may allow the water solvent to maximize its entropy, lowering the total free energy at this region toward catalytic function. Once it was mutated into N370S, the energy force shifts to higher entropic stage (−16 and −26 KJ/mole) and sequentially alters the local conformational (side chains) interaction that could contribute to reduced catalytic activity.
The simulation analyses suggest that each mutant leads to different alteration on side chain interaction, which may underlie the phenotypic variation.
Mouse models of homozygotes for Gba1 mutations
The mouse models having the GCase mutations, 9H, 9V, 0S and 4L, were generated previously to study their in vivo effects12. Homozygotes for 9V, 9H and 4L in mice had reduced tissue GCase activity (Table 1). However, these mutant mice at about 1 year of age do not accumulate significant levels of substrates, do not develop severe CNS and visceral phenotypes, and have normal life spans (Table 1)12,16. Mice homozygous for 0S die within the first 24–48 hrs of birth, due to skin permeability defects12. Thus, these mutants have limitations in studying the mutations’ in vivo effects.
Double homozygotes for Gba1 mutations and Saposin C deficiency
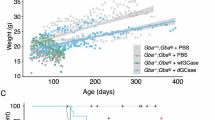
To understand differential in vivo effect of these mutations, mice were created with homozygosity for Gba1 mutations in combination with Saposin C deficiency. Saposin C has activity optimization and protective functions on GCase23. Deficiency of Saposin C leads to reduced GCase activity that could potentiate the disease phenotype in Gba1 mutant mice26,27. Four combined Gba1 homozygous mutation mice with Saposin C deficiency (Gba1 mutation;C*) were generated (Table 1). The 9V;C* and 0S;C* mice died within 24 hours after birth due primarily to skin permeability abnormalities. 4L;C* mice showed primary CNS deficits and had short life span (~7 weeks); phenotypic and pathologic findings have been published and summarized in Table 1 27,31. The 9H;C* mice developed a neurological phenotype resembled that of WT;C* mice26, but with earlier (~3 months) vs. ~8 months (WT;C*) onset and with shorter lifespan (Table 1)26. The 9H;C* behavioral phenotype included hind-limb clasping during tail hanging by 3 months and the development of kyphotic posturing at 12 months of age (Fig. 2A). The 9H;C* mice also showed mild hind limb paresis and gait ataxia. Compared to the 7 weeks survival of 4L;C* mice, 9H;C* mice survived to about 13 months of age27.

CNS pathology in 9H;C* mice. (A) Phenotype. 9H;C* mice showed hind-limb clasping during tail hanging at 3 months of age (left panel) and kyphotic posturing at 11 months of age (Right panel). As a control, C+/− mouse did not show hind-limb clasping. (B) CNS pathology in 9H;C* mice compared to WT;C* and 9H;C+/− control mice at 12 months of age. (Top panels) Loss of Purkinje cells (H&E, arrows) was evident in 9H;C* and WT;C* cerebellum and was accompanied with activated microglial cells positive for anti CD68 antibody (CD68, brown) staining. (Middle panels) Dorsal root ganglion in 9H;C* mice contained foamy storage materials in cells (H&E, arrows) and had CD68 positive cells (brown). (Lower panels) Dorsal horn of spinal cord in 9H;C* mice had axonal spheroids (H&E, arrows) and CD68 positive cells (brown). WT;C* mice had fewer foamy cells, axonal spheroids and CD68 positive cells than 9H;C* mice. As a control, 9H;C+/− mice tissues showed normal histology. (C) CD68 positive signals (brown) distributed differently in 9H;C* and 4L;C* brains. CD68 signals were restricted in caudate putamen (cp), thalamus (th) and cerebellum (cb) regions (arrows) in 9H;C* brain and distributed in most regions in 4L;C* brain.
CNS and visceral histopathology of 9H;C* and 4L;C* mice
9H;C* mice developed severe brain and spinal cord histopathology, similar to, but earlier than, the WT;C* mice, major losses of Purkinje cells in cerebellum, inclusions in dorsal root ganglia, and axonal degeneration in spinal cord by 6 months of age (Fig. 2B)26. However, these were about 6 months earlier than the appearance of the corresponding lesions in WT;C* mice26. Proinflammatory reactions in 9H;C* mice were shown by positive CD68 staining of activated macrophages (Fig. 2B and C). In comparison to 4L;C* at 45 days of age in which the CD68 signals were distributed throughout the entire brain, the CD68 signals in 9H;C* brains were restricted in thalamus, basal ganglia and dentate nucleus of cerebellum in 9H;C* brain at 1 year of age (Fig. 2C). Proinflammation was observed in spinal cord in both 9H;C* and 4L;C* models (Fig. 2B)27.
Visceral involvement was also present in the 9H;C* mice. Engorged macrophages were observed in liver, lung and spleen (Fig. 3). In contrast to 4L;C* livers and lungs that had WT level CD68 signals and no storage cells (Fig. 3B), massive CD68 positive storage cells were in 9H;C* liver, lung and spleen (Fig. 3B and C). Electron micrographs of storage cells showed typical tubular structure to the accumulated materials which resembled of the storage materials in human GD Kupffer cells and other macrophages (Fig. 3A-B)4. In lung, there were many dense aggregates and membrane like materials in interstitial or alveolar macrophages (Fig. 3A–D).

Visceral pathology of 9H;C* mice. (A) H&E stained 9H;C* liver and lung at 13 months of age showed storage cells (arrows) in the liver (A) and lung (C). Ultrastructural studies demonstrated the storage cells form multi nucleic cluster in the liver (B). The storage materials had tubule form (B insert). The membrane inclusions were in the lung storage cells (D). (B) Anti-CD68 antibody (brown) stained liver and lung. 12-month WT mouse liver (A) and lung (B) showed background level of CD68 signals. 9H;C* liver (C) and lung (D) at 12 months of age had engorged CD68 positive macrophages (arrows). 4L;C* liver (E) and lung (F) at 45 days of age did not have storage cells. Scale bar = 100 µm for all images. (C) Compared to age-matched WT spleen stained by H&E (A) and anti-CD68 (B), 9H;C* spleen at 12 months of age had storage cells (arrows) by H&E (C) and CD68 positive cells (D).
Skin histopathology of 9V;C* and 0S;C* mice
Similar to 0S and Gba1−/− (i.e., GCase null) pups, 9V;C* and 0S;C* mice died within 24 hours of birth12. 9V;C* and 0S;C* pups had ichthyotic skin with wrinkly appearance, compared to smooth skin in WT and WT;C* pups at 1 day of age (Fig. 4A). By H&E staining, skin from WT and WT;C* pups had normal stratum corneum showing a basket weave appearance (Fig. 4B), whereas this layer was compact in 9V;C* and 0S;C* skin, which was very similar to that in Gba1−/− and 0S skin epidermis (Fig. 4B). Ultrastructural analyses of stratum corneum from 9V;C* and 0S;C* mice showed loosely packed layers and irregular lamella structure (Fig. 4C). In comparison, the corresponding WT pup skin had a lamellar structure. Histology of 9V;C* and 0S;C* visceral organs and brain appeared normal, e.g., no storage cells were found in liver, spleen and lung of 9V;C* and 0S;C* pups.

Skin of 9V;C* and 0S;C* mice. (A) 9V;C*, 0S;C* and 0S pups had ichthyotic skin compared to smooth skin of WT and WT;C* pups. (B) H&E stained skin epidermal sections from 1-day old pups. Normal stratum corneum (SC) in WT (A) and WT;C* (B) skin had basket weave appearance. SC was compact in 9V;C* (C), 0S;C* (D), Gba1−/− (E) and 0S (F) skin epidermis. (C) Ultrastructural studies of stratum corneum in 1-day old pup skin. Normal lamellar structure (arrow) in WT skin. 9V;C* and 0S;C* had loosely packed layers and irregular lamella structure (arrow).
GCase activity deficiency in combined Gba1 mutation and Saposin C deficient mice
GCase activity in the tissues of the Gba1 mutation;C* mice were compared to those from WT and WT;C* mice. Consistent with previous studies, GCase activity was reduced by ~40% in the organs of WT;C* mice (Fig. 5)26. In 9H;C* mice, GCase activity in liver, lung, spleen and cerebrum were about or less than 5% of WT level, but at similar levels as that in 9H tissues, although at these very low activity levels are difficult to compare directly (Fig. 5A). 9H GCase is a very unstable protein13. The similarity of GCase activity in 9H;C* and 9H tissues suggest that may not reflect potential differences detectible in vivo, i.e., the very low in vivo levels are not reflected by the in vitro activity assessments. Compared to 9V mice, 9V;C* had apparently reduced GCase activity in liver, lung and brain (Fig. 5B). 0S;C* mice also showed similarly decreased activity in liver, lung and brain compared to 0S/0S tissues (Fig. 5B). Reduced GCase activity by ~50% was reported previously in 4L;C* compared to 4L tissues27. These results showed that Saposin C deficiency in 9V, 0S and 4L mice leads to reduction of mutant GCase activity.

GCase activity. (A) GCase activity in 9H;C* mice tissues were reduced compared to WT and WT;C* mice. Compared to 9H mice, GCase activity in 9H;C* tissues were not changed in the lung, spleen and cerebrum, but slightly elevated in the liver. (B) 9V;C* and 0S;C* mice had reduced GCase activity in the liver, lung and brain compared to 9V and 0S tissues, respectively. Student’s t-test (n = 3–6 mice).
Analyses of substrate levels in the mice tissues
GC levels in 9V;C* and 0S;C* tissues were compared to WT and WT;C* mice at 1 day of age. 9V;C* and 0S;C* liver and lung had significantly increased GC compared to WT (Fig. 6). 9V;C* brain showed a 1.5-fold increase in GC above WT level (Fig. 6A). GC levels in 0S;C* brain were comparable to the WT level (Fig. 6A). GS levels in 9V;C* liver, lung and brain and 0S;C* lung were detectable and slightly above WT level (Table 2).

Tissue GC and GS analysis by LC/MS. (A) 9V;C* and 0S;C* mice had GC accumulation in liver and lung. 9V;C* had GC accumulation and 0S;C* mice had WT GC level in brain. (B) 9H;C* visceral and brain tissues showed GC accumulation increased with age. (C) GC levels were increased in 4L;C* brain and lung compared to WT mice. (D) Epidermal GC and GS levels were significantly increased in 9V;C*, 9V, 0S;C* and 0S mice at 1 day of age compared to WT. Epidermal GC and GS levels in 9V;C* were higher than 9V. (E) Total ceramides were significantly increased in 0S, 0S;C*, 9V;C* and WT;C* mice epidermis compared to WT. Epidermal ceramide levels in 9Vmice were not significantly different from WT. Student’s t-test (n = 3–6 mice).
In 9H;C* mice GC levels were massively increased in liver and moderately elevated in lung, spleen and brain (Fig. 6B). The GC levels in 9H;C* visceral tissues increased with age (Fig. 6B). In contrast to 4L;C* mice that had higher GC accumulation in brain than viscera (Fig. 6C and Table 1)27, 9H;C* had a greater GC content in visceral tissues than brain. GS levels in 9H;C* mice were also higher in both visceral tissues and brain (Table 2).
GC and GS in skin epidermis of 1-day old 9V;C* and 0S;C* pups were compared to age-matched WT, WT;C*, 9V and 0S mice. 9V;C* epidermal GC and GS were significantly increased compared to WT, WT;C* or 9V (Fig. 6D). GC and GS accumulated in 0S;C* epidermis and levels were significantly higher compared with WT and WT;C* mice (Fig. 6D). GC and GS levels were comparable in 0S;C* and 0S epidermis, but higher in 9V;C* than 9V (Fig. 6D). The accumulated epidermal GC species detected ranged from GC16:0 to GC30:0 with the major accumulated species being GC16:0 and GC26:0 (Supplementary Fig. 1A). Ceramide levels in 0S epidermis were 2-fold above WT levels, however, ceramide levels in 9V;C*, 0S;C* and WT;C* were increased less than 2-fold from WT level, but not increased in 9V epidermis (Fig. 6E). Ceramide species profile revealed that C16-OH, C24 and C26-1 are the major species in mouse epidermis. Various ceramide species were increased in those mutants compared to WT mice epidermis (Supplementary Fig. 1B).
In summary, simulation analysis suggest the conformational alterations in 9H, 9V, 4L and 0S mutant GCase results in reduced enzymatic activity and stability. Although the homozygous Gba1 mutations in mice develop relatively mild phenotypes, deficiency of Saposin C in Gba1 mutants potentiate the disease and reveal mutation-dependent phenotype variation.
Discussion
The findings from our studies suggest GCase structure altered by specific mutations exhibits differential phenotypes in Gaucher disease. Enzymatic properties of the mutant 9H, 9V, 4L and 0S have been characterized in human recombinant proteins and mouse fibroblasts13. Using non-natural substrates in vitro, 9H and 9V GCases have ~5% of WT GCase activity and are unstable within cells, whereas 0S and 4L mutants are stable, but have reduced catalytic activity to ~12–14% of WT levels. 0S and 4L mutations have similar enhanced activation by the phosphatidylserine (PS) induced conformational change, whereas Saposin C’s activation effects on these GCases was similar to WT levels13. The in vivo outcome of these mutants were explored in the mice with homozygosity for a specific Gba1 mutation together with Saposin C deficiency. Compared to point mutated Gba1 mutant mice, 9H, 9V, 0S and 4L, backcrossing of C* mice with those Gba1 mutants produced the 9H;C*, 4L;C*, 9V;C* and 0S;C* mice with additionally reduced mutant enzyme activity and increased substrates levels. Saposin C deficiency did not change the phenotype in 0S due to early death from the skin permeability abnormality in 0S mice12. In 9H;C*, 9V;C* and 4L;C* mice, the deficiency of Saposin C potentiated the disease progression in the variant phenotypes.
The distinct phenotypes from 9V;C* (1-day survival) and 9H;C* (13 months survival) mice suggest that D409 is a critical amino acid affecting catalytic function of GCase. Aspartic acid (D) has an acidic/negatively charged side chain, whereas histidine (H) has a basic/positively charge side chain, and valine (V) is a non-polar amino acid. GCase has three domains resolved by X-ray crystallization. Domain 1 (residues1–27 and 383–414), where D409 resides, is predicted to be important in enzyme folding and stability, domain 2 (residues 30–75 and 431–497) has an immunoglobulin-like structure, and domain 3 (residues 76–381 and 416–430) forms TIM barrel-helix6 and helix7 consisting of catalytic site13,30. LIMP2 is an intracellular receptor for lysosomal targeting of GCase28,32. The binding motif of GCase for LIMP2 is contained in the span covered by amino acids 399 to 409 in Domain III of GCase28,29. The mutant GCases, D409V or H, disrupts lysosomal targeting of these GCases leading to their proteolysis. The crystal structure of mouse GCase is presently not available, but is predicted to be very similar to the human WT enzyme. Human and mouse GCases are highly homologous, have 85% amino acid identity, normal and mutant GCases, e.g., 4L and 9V and 9H, have similar respective properties13. These similarities provide the basis for modeling of the human GCase structures at neutral and acidic pH to simulate mutation effects in vitro (human) and in vivo (mouse) of a given amino acid and to gain insight into GCase’s function. Analysis of side chain interaction and its force field energy indicates that D409 has side chain interaction with N19 (within 6 Å), a critical glycosylation site in GCase function13,33. The 9V mutation abolished nearly all side chain interactions in Domain 1 and the favorable energy environment, which leads to greater conformational changes at pH 5.5. In comparison, 9H has a significant conformational change and altered side chain interaction in an acidic environment. 9V and 9H mice have similar mild phenotypes up to 12 months12, but the interaction with Saposin C deficiency brings out the differential properties of the GCase mutant enzyme alone without the protective effects of Saposin C. This simulation result supports the differential phenotype of two mutants with severe, early death of 9V;C* and mild, chronic form of 9H;C*.
The V394 is located in Domain 1 close to the active site pocket. The modeling of V394L implicates an altered conformation change of this GCase leading to its functional effects on catalysis. The force field energy at V394 is dramatically increased when Valine was replaced by Leucine (V394L) at pH 7.2 and pH 5.5, indicating significant alterations of the side chain geometries with resultant negative effects on active site function. N370 is located in Domain 3 on the helical turn near loop1 and contributes to the hydrogen bonding network to stabilize the orientation of this loop. The residue Y313 in loop 1 (residues 311–319) plays a key role in moving this loop to open the active site for binding15. At pH 7.2, the N370 side chain forms a stable interaction with L310 (Table 1) and at pH 5.5, N370 forms an even more stable interaction with W312 and D315 (Table 1, red) to stabilize the Y313 (a chelated-like interaction). Mutation of 370 from N to S alters the original with L310 to W312 and forms two new interactions with V376 and G377. This is a significant alteration of loop 1 (amino residues 311–319) structure. The serine side chain of N370S at acidic pH 5.4 cannot form a chelated-like interaction with W312 and D315 and sways the contact with W312 and A320. This has been suggested to affect substrate access to the active site15,34. The simulation analyses of V394L and N370S support the structural alteration change on mutated GCase induced enzymatic function deficit. The abnormal skin permeability in 0S and 0S;C* mice prevented the investigation of disease development on this mutant. Whereas 4L mice have nearly normal phenotype and life span12, the effect of 4L on disease phenotype was revealed in 4L;C* mice in the absence of Saposin C.
The mouse models developed here not only underscore the Gba1 mutation on disease development but also provide insight into the in vivo role of Saposin C for mutant GCase function. Saposin C exists as a dimer and is required by GCase for optimal activity and protein stabilization against proteolysis21,23. Mutated Saposin Cs, which have been cleaved from prosaposin, i.e., the mature form, are unstable and rapidly degraded35. Mutations in Saposin C lead to a rare form of Gaucher-like disease presenting neuronopathic and non-neuronopathic symptoms24,36,37,38. The mouse models of Saposin C deficiency develop slow progression of neurological phenotypes, but no substrate and storage cells in visceral organs26,39. In Saposin C deficient mice, ~40% reductions of WT or mutant GCase proteins and activities are due to increased GCase proteolysis in the lysosome23,26,27, i.e., the loss of Saposin C’s protective effects. The exact mechanism and mode of interaction of GCase and Saposin C has not been fully defined. Saposin C has membrane lipid binding properties and forms a protein complex with GCase at the lipid bilayers, as demonstrated by in vitro experiments40,41. Saposin C plays role in lipid presentation by CD1b, the molecule responsible for lipid-antigen presentation to T-cells in immune response42,43. Saposin C’s membrane interactions are required for providing GCase accessibility to its substrates, GC and GS embedded in the membrane44,45. Studies of V394L with PS or Saposin C reveal a surface-accessible loop structure containing V394 (394–414) sensitive to PS and Saposin C activation13. Together with D409 in the same loop, this region exhibits sensitivity to conformational changes altering protein stability and activity as well as activator interactions (e.g. PS and Saposin C)28. How Saposin C deficiency differentially affects 9H or 9V is not known, partially due to their instability during purification, which inhibits direct modeling studies. However, 3D-Docking models of GCase and Saposin C predicts interactions with domain 2 and helix 6 of domain 3 on GCase46. Both D409 and V394 are not within those domains. Recent structural study showed Saposin A binds its cognate enzyme galactocylceramidase and form a heterotetramer complex47. Further studies of the structural complex of Saposin C and GCase will be needed to resolve the mechanism and mode of interaction.
Anionic phospholipids-containing membranes are essential for Saposin C’s function40. Phospholipid composition could influence Saposins C’s action and affect its interaction with GCase, consequently mutated GCase may have defect in response PS or Saposin C’s activation13. By reducing anionic phospholipids to 20% of total lipids, Saposin C promotes binding and activation of normal GCase, but loses its effects on N370S GCase48. The different phenotype of 9H;C*, 4L;C* or 9V;C* mice could be influenced by phospholipids compositions that affect the interaction of Saposin C and mutant GCase in specific cells/organs.
The different phenotyps of these mutant mice could be explained by changes in substrate specificity of mutant GCase. GCase has two substrates: GC and GS. GCs are a group of lipids containing glucose and ceramide with fatty acids of various chain length16,49. GS is deacyl form of glucosylceramide resulting from acid ceramidase hydrolysis50,51. Degradation of GC requires three components, GCase, Saposin C and phospholipids52,53. It is evident that there is mutation-specific quantitative differences in GC species and GS accumulations that influences tissue/regional expression of Gaucher disease phenotypes in these mouse models16. 4L;C* mice had major GC18:0 degradation defects in the brain, whereas the analogous mice with 9H;C* led to all GC species accumulating massively in visceral tissues16. GS was poorly degraded in brain by 4L and 9H GCases, but not by 9V and 0S GCase. Such differences are anticipated from the basic kinetic properties of the variant GCases, and the ratio of the rate constants for the cleavage of GC and GS54. Saposin C could also influence the substrate preferences for various GCase variants in vivo.
Defective GCase activity in the hydrolysis of GC to ceramide may affect maturation or change in lipid membranes that form the normal epidermal barrier55. In both Gaucher disease type 2 patients and Gba1 knock out mice, epidermal abnormalities are associated with the accumulation of GC56,57,58. The 9V;C* and 0S;C* pups showed compact stratum corneum structure and irregular layers of lamellar body. Significantly increased epidermal GC and GS likely account for the epidermal abnormalities in those mice skins. Unexpectedly, the ceramide level was not significantly reduced in those mice although GC was increased, which is in contrast to previous report of reduced ceramide in Gaucher disease type 2 patient and in knock out mice56,57,58. In those studies, total GC and ceramides were determined by thin layer chromatography56,57,58. In current study, epidermal glycosphingolipids were quantitated by LC/MS. With the available ceramide standards, the detectable longest fatty acid chain length ceramide was C18:1/24:0, which may omit long chain and complex ceramide that contribute to major portion of ceramides in epidermis59. With species < GC24, accumulated GCs did not lead to reduction of same chain length of ceramide. In addition, this study demonstrated that deficiency of Saposins C alone did not cause GC and GS accumulation and abnormal stratum corneum in epidermis which is dissimilar to deficiency of prosaposin that leads to aberrant lamellar membrane structure and GC accumulation60.
In summary, 9H;C*, 9V;C*, 0S;C* and 4L;C* mice provide additional in vivo models to study the tissue specific pathogenic effect of GBA1 mutations, e.g. 9V;C* and 0S;C* for skin permeability, 4L;C* for neuronopathic phenotype27 and 9H;C* for chronic form of both viscera and brain pathology in Gaucher disease. Valvular disease found in human patients10 was not observed grossly in 9H12 or 9H/C* mice which presents a limitation for studying valvular disease using these models. Biochemical and pathological data from this study support a functional interaction of GCase and Saposin C. Without Saposin C, the Gba1 mutant mice developed mutation-dependent and tissues-specific phenotypes. The results of these studies provide insights to GCase mutations and their correlation with tissue specific variation in substrate accumulation and disease phenotype, which lays the groundwork for comparative human studies in exploring genotype and phenotype correlations in Gaucher disease.
Methods
Mouse models and tissues collection
Gba1 mutant mice with 9H, 9V, and 0S homozygosity were generated as described12. Saposin C deficient mice (WT;C*) were created by knock-in of a point mutation on Saposin C domain of Psap26. The combined Gba1 mutation and Saposin C deficiency mice (designated as “Gba1 mutation”;C*) were generated by cross-breeding of C* mice with specific Gba1 mutant mice26. The resultant doubly homozygous mice are designated 9H;C*, 9V;C*, and 0S;C*. Generation of 4L;C* mice was described previously27. Saposin C heterozygous mice (C+/−) and 9H mice with C+/− (9H;C+/−) did not show abnormal histology or a behavioral phenotype and were used as controls. The strain backgrounds of the various mutant mice and WT mice were C57BL/129. The mice were maintained in microisolators in accordance with institutional guidelines under IACUC approval at Cincinnati Children’s Hospital Research Foundation. The tissues were collected from adult mice after transcardial perfusion with saline and from pups without perfusion. The collected tissues were stored at −80 °C for enzyme and lipid analyses or fixed in fixative for histology studies.
Histopathological analyses
Mouse tissues were fixed in 10% formalin and embedded in paraffin. The tissues were sectioned and stained with Hematoxylin and Eosin (H&E) and analyzed under light microscopy. Karnovsky’s fixative was used for ultrastructural studies. For immunohistochemistry, frozen tissue sections fixed with 4% paraformaldehyde were incubated with rat anti-mouse CD68 monoclonal antibody (Serotec, Oxford, UK) at 1/200 dilution in PBS with 5% BSA overnight at 4 °C as described22. Detection was performed using ABC Vectastain and Alkaline Phosphatase Kit II (Black) according to the manufacturer’s instruction. The slides were counterstained with Hematoxylin.
Enzyme activity
Tissues were homogenized in 1% Na taurocholate and 1% Triton X-100, with 0.25% each in final assay mixtures. GCase activities were determined fluorometrically with 4- methylumberlliferyl-β-D-glucopyranoside (4MU-Glucose) (Biosynth AG, Switzerland) in the presence and absence of the GCase irreversible inhibitor, 1 mM Conduritol B epoxide (Millipore. CA)61. WT GCase activities in control tissues were run in parallel62. Protein concentrations were determined using BCA Protein Assay Reagent (Pierce, Rockford, IL).
Lipid analyses
Tissue glycosphingolipids were extracted in methanol/chloroform/water (2:1:0.7) as described previously63 and subjected to alkaline methanolysis and desalting on Sephadex G-25 fine columns. The extracted samples were taken up in methanol containing an internal standard. GC and GS analyses were carried out by ESI-LC-MS/MS using a Waters Quattro Micro API triple quadrupole mass spectrometer (Milford, MA) interfaced with an Acquity UPLC system16. Quantification of GCs with various chain lengths was achieved by interpolation of calibration curve for the natural GCs with the most closely related fatty acid chain length. The quantification of GS was based on the curve using GS-d5 (Avanti) as internal standard. Linear responses for GCs and GS were in the range of 25 pg–10 ng.
Epidermis was collected from newborn pup abdomen skin. The skin samples (~1 cm2) were incubated with 1 mL of 0.25% Trypsin at 4 °C for 18 hours. Epidermis was separated from tissues and stored at −80 °C for lipid extraction59. Glycosphingolipids in the epidermis were extracted with 2 mL of chloroform/methanol/water (30:60:8), sonicated for 15 min at 50 °C, and centrifuged for 5 min at 1000 × g. The extraction was repeated 3 times. The combined extracts were dried under N2 followed by dissolving in 5 mL of chloroform/methanol/water (2:1:0.15) and subjected to alkaline methanolysis as described above. The extracted epidermal glycosphingolipids from 1 mg epidermis were taken up in methanol containing an internal standard for quantitation of GC and GS as above16. Ceramides in epidermis were quantitated by LC/MS64. Glycosphingolipids levels were normalized by wet tissues weights.
Simulation analysis of GCase mutation
Human GCase crystal structures, pH7.2 (2F61, 2.5 Å)13 and pH 5.5 (3GXI, 1.84 Å)17, were used to model the D409 and V394 wild type (WT) GCases and their respective mutant forms, D409H, D409V and V394L. Human GCase crystal structures, pH7.1 (3KEH, 2.5 Å) and pH 5.4 (3KEO, 1.84 Å), were applied for modeling N370 WT and mutant N370S18. Swiss PDB Viewer (DeepView,SPDBV,Version 4.10) was applied for structure modeling analysis and GROMOS 96 was used for force field energy computations65. The distance parameter for computing interactions of target mutation site was set to 6 Å and force field energy changes within introduced mutation were computed. All amino acids in the side chains that interact with the mutated amino acid at this position (e.g. D409H, V394L and N370S) within 6 Å were listed in Supplementary Table 1. The side chain interactions of mutant were compared to WT GCase structure at each position. Differences on the amino acids involved in the interaction in mutant compared to WT are highlighted in red. The parameters for force field energy computation analysis at given position include the energy in bonds, angles, torsion, improper, non-bonded, electrostatic and constraint energy (K joule/mole or KJ/mole). Negative energy values represent favorable energy environment whereas positive values represent unfavorable energy environment for a given amino acid. The pH environment effects on side chain interactions (acidic, pH 5.5 versus neutral, pH 7.2) were also computed.
Statistical analysis
The data were analyzed by Student’s t-test or One-way ANOVA test with Dunnett posttest using GraphPad Prism.
References
Grabowski, G. A., Petsko, G. A. & Kolodny, E. H. In The Online Metabolic and Molecular Bases of Inherited Diseases (eds Valle, D. et al.) Ch. 146, (The McGraw-Hill Companies, Inc., 2010).
Grabowski, G. A. et al. In The Metabolic and Molecular Bases of Inherited Diseases (eds Scriver, C. R. et al.) (McGraw-Hill, 2006).
Petrucci, S., Consoli, F. & Valente, E. M. Parkinson Disease Genetics: A “Continuum” From Mendelian to Multifactorial Inheritance. Curr Mol Med (2014).
Beutler, E. & Grabowski, G. A. In The Metabolic and Molecular Basis of Inherited Disease Vol. III (eds Scriver, C. R., Beaudet, A. L., Sly, W. S., & Valle, D.) 3635–3668 (McGraw-Hill, 2001).
Hruska, K. S., LaMarca, M. E., Scott, C. R. & Sidransky, E. Gaucher disease: mutation and polymorphism spectrum in the glucocerebrosidase gene (GBA). Hum Mutat 29, 567–583, https://doi.org/10.1002/humu.20676 (2008).
Grabowski, G. A., Zimran, A. & Ida, H. Gaucher disease types 1 and 3: Phenotypic characterization of large populations from the ICGG Gaucher Registry. Am J Hematol 90(Suppl 1), S12–18, https://doi.org/10.1002/ajh.24063 (2015).
Tsuji, A., Omura, K. & Suzuki, Y. Intracellular transport of acid alpha-glucosidase in human fibroblasts: evidence for involvement of phosphomannosyl receptor-independent system. J Biochem (Tokyo) 104, 276–278 (1988).
Charrow, J. et al. The Gaucher registry: demographics and disease characteristics of 1698 patients with Gaucher disease. Arch Intern Med 160(2835–2843), ioi90854 (2000).
Eyal, N., Wilder, S. & Horowitz, M. Prevalent and rare mutations among Gaucher patients. Gene 96, 277–283 (1990).
Pasmanik-Chor, M. et al. The glucocerebrosidase D409H mutation in Gaucher disease. Biochem Mol Med 59, 125–133 (1996).
Theophilus, B., Latham, T., Grabowski, G. A. & Smith, F. I. Gaucher disease: molecular heterogeneity and phenotype-genotype correlations. Am J Hum Genet 45, 212–225 (1989).
Xu, Y. H., Quinn, B., Witte, D. & Grabowski, G. A. Viable mouse models of acid beta-glucosidase deficiency: the defect in Gaucher disease. Am J Pathol 163, 2093–2101 (2003).
Liou, B. et al. Analyses of variant acid beta-glucosidases: effects of Gaucher disease mutations. J Biol Chem 281, 4242–4253, https://doi.org/10.1074/jbc.M511110200 (2006).
Smith, L., Mullin, S. & Schapira, A. H. V. Insights into the structural biology of Gaucher disease. Experimental neurology 298, 180–190, https://doi.org/10.1016/j.expneurol.2017.09.010 (2017).
Liou, B. & Grabowski, G. A. Participation of asparagine 370 and glutamine 235 in the catalysis by acid beta-glucosidase: the enzyme deficient in Gaucher disease. Mol Genet Metab 97, 65–74, https://doi.org/10.1016/j.ymgme.2009.01.006 (2009).
Sun, Y. et al. Substrate compositional variation with tissue/region and Gba1 mutations in mouse models–implications for Gaucher disease. PLoS One 8, e57560, https://doi.org/10.1371/journal.pone.0057560 (2013).
Lieberman, R. L., D’Aquino, J. A., Ringe, D. & Petsko, G. A. Effects of pH and iminosugar pharmacological chaperones on lysosomal glycosidase structure and stability. Biochemistry 48, 4816–4827, https://doi.org/10.1021/bi9002265 (2009).
Wei, R. R. et al. X-ray and biochemical analysis of N370S mutant human acid beta-glucosidase. J Biol Chem 286, 299–308, https://doi.org/10.1074/jbc.M110.150433 (2011).
Guex, N., Peitsch, M. C. & Schwede, T. Automated comparative protein structure modeling with SWISS-MODEL and Swiss-PdbViewer: a historical perspective. Electrophoresis 30(Suppl 1), S162–173, https://doi.org/10.1002/elps.200900140 (2009).
Johansson, M. U., Zoete, V., Michielin, O. & Guex, N. Defining and searching for structural motifs using DeepView/Swiss-PdbViewer. BMC bioinformatics 13, 173, https://doi.org/10.1186/1471-2105-13-173 (2012).
Sandhoff, K., Kolter, T. & Van Echten-Deckert, G. Sphingolipid metabolism. Sphingoid analogs, sphingolipid activator proteins, and the pathology of the cell. Ann N Y Acad Sci 845, 139–151 (1998).
Sun, Y., Quinn, B., Witte, D. P. & Grabowski, G. A. Gaucher disease mouse models: point mutations at the acid {beta}-glucosidase locus combined with low-level prosaposin expression lead to disease variants. J Lipid Res 46, 2102–2113 (2005).
Sun, Y., Qi, X. & Grabowski, G. A. Saposin C is required for normal resistance of acid beta-glucosidase to proteolytic degradation. J Biol Chem 278, 31918–31923 (2003).
Christomanou, H., Chabas, A., Pampols, T. & Guardiola, A. Activator protein deficient Gaucher’s disease. A second patient with the newly identified lipid storage disorder. Klinische Wochenschrift 67, 999–1003 (1989).
Kang, L. et al. A rare form of Gaucher disease resulting from saposin C deficiency. Blood Cells Mol Dis, https://doi.org/10.1016/j.bcmd.2017.04.001 (2017).
Sun, Y. et al. Specific saposin C deficiency: CNS impairment and acid beta-glucosidase effects in the mouse. Hum Mol Genet 19, 634–647, https://doi.org/10.1093/hmg/ddp531 (2010).
Sun, Y. et al. Neuronopathic Gaucher disease in the mouse: viable combined selective saposin C deficiency and mutant glucocerebrosidase (V394L) mice with glucosylsphingosine and glucosylceramide accumulation and progressive neurological deficits. Hum Mol Genet 19, 1088–1097, https://doi.org/10.1093/hmg/ddp580 (2010).
Liou, B., Haffey, W. D., Greis, K. D. & Grabowski, G. A. The LIMP-2/SCARB2 binding motif on acid beta-glucosidase: basic and applied implications for Gaucher disease and associated neurodegenerative diseases. J Biol Chem 289, 30063–30074, https://doi.org/10.1074/jbc.M114.593616 (2014).
Zunke, F. et al. Characterization of the complex formed by beta-glucocerebrosidase and the lysosomal integral membrane protein type-2. Proc Natl Acad Sci USA 113, 3791–3796, https://doi.org/10.1073/pnas.1514005113 (2016).
Dvir, H. et al. X-ray structure of human acid-beta-glucosidase, the defective enzyme in Gaucher disease. EMBO Rep 4, 704–709, https://doi.org/10.1038/sj.embor.embor873 (2003).
Dasgupta, N. et al. Neuronopathic Gaucher disease: dysregulated mRNAs and miRNAs in brain pathogenesis and effects of pharmacologic chaperone treatment in a mouse model. Hum Mol Genet, https://doi.org/10.1093/hmg/ddv404 (2015).
Reczek, D. et al. LIMP-2 is a receptor for lysosomal mannose-6-phosphate-independent targeting of beta-glucocerebrosidase. Cell 131, 770–783, https://doi.org/10.1016/j.cell.2007.10.018 (2007).
Berg-Fussman, A., Grace, M. E., Ioannou, Y. & Grabowski, G. A. Human acid beta-glucosidase. N-glycosylation site occupancy and the effect of glycosylation on enzymatic activity. J Biol Chem 268, 14861–14866 (1993).
Offman, M. N. et al. Comparison of a molecular dynamics model with the X-ray structure of the N370S acid-beta-glucosidase mutant that causes Gaucher disease. Protein Eng Des Sel 24, 773–775, https://doi.org/10.1093/protein/gzr032 (2011).
Motta, M. et al. Gaucher disease due to saposin C deficiency is an inherited lysosomal disease caused by rapidly degraded mutant proteins. Hum Mol Genet 23, 5814–5826, https://doi.org/10.1093/hmg/ddu299 (2014).
Kang, L. et al. A rare form of Gaucher disease resulting from saposin C deficiency. Blood Cells Mol Dis 68, 60–65, https://doi.org/10.1016/j.bcmd.2017.04.001 (2018).
Tamargo, R. J., Velayati, A., Goldin, E. & Sidransky, E. The role of saposin C in Gaucher disease. Mol Genet Metab 106, 257–263, https://doi.org/10.1016/j.ymgme.2012.04.024 (2012).
Tylki-Szymanska, A. et al. Non-neuronopathic Gaucher disease due to saposin C deficiency. Clin Genet 72, 538–542, https://doi.org/10.1111/j.1399-0004.2007.00899.x (2007).
Yoneshige, A., Suzuki, K., Suzuki, K. & Matsuda, J. A mutation in the saposin C domain of the sphingolipid activator protein (Prosaposin) gene causes neurodegenerative disease in mice. J Neurosci Res 88, 2118–2134, https://doi.org/10.1002/jnr.22371 (2010).
Wang, Y., Grabowski, G. A. & Qi, X. Phospholipid vesicle fusion induced by saposin C. Arch Biochem Biophys 415, 43–53 (2003).
Alattia, J. R., Shaw, J. E., Yip, C. M. & Prive, G. G. Molecular imaging of membrane interfaces reveals mode of beta-glucosidase activation by saposin C. Proc Natl Acad Sci USA 104, 17394–17399, https://doi.org/10.1073/pnas.0704998104 (2007).
Leon, L. et al. Saposins utilize two strategies for lipid transfer and CD1 antigen presentation. Proc Natl Acad Sci USA 109, 4357–4364, https://doi.org/10.1073/pnas.1200764109 (2012).
Winau, F. et al. Saposin C is required for lipid presentation by human CD1b. Nature immunology 5, 169–174, https://doi.org/10.1038/ni1035 (2004).
Kolter, T. & Sandhoff, K. Lysosomal degradation of membrane lipids. FEBS Lett 584, 1700–1712, https://doi.org/10.1016/j.febslet.2009.10.021 (2010).
Qi, X. & Grabowski, G. A. Differential membrane interactions of saposins A and C: implications for the functional specificity. J Biol Chem 276, 27010–27017, https://doi.org/10.1074/jbc.M101075200 (2001).
Atrian, S. et al. An evolutionary and structure-based docking model for glucocerebrosidase-saposin C and glucocerebrosidase-substrate interactions - relevance for Gaucher disease. Proteins 70, 882–891, https://doi.org/10.1002/prot.21554 (2008).
Hill, C. H. et al. The mechanism of glycosphingolipid degradation revealed by a GALC-SapA complex structure. Nature communications 9, 151, https://doi.org/10.1038/s41467-017-02361-y (2018).
Salvioli, R. et al. The N370S (Asn370– > Ser) mutation affects the capacity of glucosylceramidase to interact with anionic phospholipid-containing membranes and saposin C. Biochem J 390, 95–103, https://doi.org/10.1042/BJ20050325 (2005).
Kuske, T. T. & Rosenberg, A. Quantity and fatty acyl composition of the glycosphingolipids of Gaucher spleen. J Lab Clin Med 80, 523–529 (1972).
Yamaguchi, Y., Sasagasako, N., Goto, I. & Kobayashi, T. The synthetic pathway for glucosylsphingosine in cultured fibroblasts. J Biochem 116, 704–710 (1994).
Ferraz, M. J. et al. Lysosomal glycosphingolipid catabolism by acid ceramidase: formation of glycosphingoid bases during deficiency of glycosidases. FEBS Lett 590, 716–725, https://doi.org/10.1002/1873-3468.12104 (2016).
Berent, S. L. & Radin, N. S. Mechanism of activation of glucocerebrosidase by co-beta-glucosidase (glucosidase activator protein). Biochim Biophys Acta 664, 572–582 (1981).
Vaccaro, A. M. et al. Function of saposin C in the reconstitution of glucosylceramidase by phosphatidylserine liposomes. FEBS Lett 336, 159–162 (1993).
Osiecki-Newman, K. et al. Human acid beta-glucosidase: inhibition studies using glucose analogues and pH variation to characterize the normal and Gaucher disease glycon binding sites. Enzyme 40, 173–188 (1988).
Holleran, W. M. et al. Permeability barrier requirements regulate epidermal beta-glucocerebrosidase. J Lipid Res 35, 905–912 (1994).
Doering, T., Proia, R. L. & Sandhoff, K. Accumulation of protein-bound epidermal glucosylceramides in beta-glucocerebrosidase deficient type 2 Gaucher mice. FEBS Lett 447, 167–170 (1999).
Holleran, W. M. et al. Consequences of beta-glucocerebrosidase deficiency in epidermis. Ultrastructure and permeability barrier alterations in Gaucher disease. J Clin Invest 93, 1756–1764, https://doi.org/10.1172/JCI117160 (1994).
Sidransky, E. et al. Epidermal abnormalities may distinguish type 2 from type 1 and type 3 of Gaucher disease. Pediatr Res 39, 134–141, https://doi.org/10.1203/00006450-199604001-00814 (1996).
Jennemann, R. et al. Integrity and barrier function of the epidermis critically depend on glucosylceramide synthesis. J Biol Chem 282, 3083–3094, https://doi.org/10.1074/jbc.M610304200 (2007).
Doering, T. et al. Sphingolipid activator proteins are required for epidermal permeability barrier formation. J Biol Chem 274, 11038–11045 (1999).
Xu, Y. H. et al. Turnover and distribution of intravenously administered mannose-terminated human acid β-glucosidase in murine and human tissues. Pediatr. Res. 39, 313–322 (1996).
Grace, M. E., Newman, K. M., Scheinker, V., Berg-Fussman, A. & Grabowski, G. A. Analysis of human acid β-glucosidase by site-directed mutagenesis and heterologous expression. J. Biol. Chem. 269, 2283–2291 (1994).
Sun, Y. et al. Ex Vivo and in Vivo Effects of Isofagomine on Acid beta-Glucosidase Variants and Substrate Levels in Gaucher Disease. Journal of Biological Chemistry 287, 4275–4287, https://doi.org/10.1074/jbc.M111.280016 (2012).
Sun, Y. et al. Tissue-specific effects of saposin A and saposin B on glycosphingolipid degradation in mutant mice. Hum Mol Genet 22, 2435–2450, https://doi.org/10.1093/hmg/ddt096 (2013).
van Gunsteren, W. F. et al. Tironi. Biomolecular Simulation: The GROMOS96 Manual and User Guide. 1–1042 (Vdf Hochschulverlag AG an der ETH Zürich, 1996).
Acknowledgements
The authors thank Jaclyn Brandewie, Becky C. Coyle, Lisa McMillin and Georgianne Ciraolo for their technical assistance. This work is supported by NIH awards to GAG (R01DK36681) and in part to YS (R01NS086134 and R21NS095047).
Author information
Authors and Affiliations
Contributions
All authors contributed to the manuscript. Benjamin Liou performed most of biochemical experiments and data analysis, and wrote manuscript. Wujuan Zhang performed LC/MS lipids analyses. Venette Fannin performed mice experiments and lipids preparation. Brian Quinn performed lipids preparation. Huimin Ran generated mouse models and performed histology experiments. Kui Xu generated mouse models and performed skin experiments. Kenneth D.R. Setchell participated LC/MS lipids analyses and manuscript editing. David Witte participated in the ultrastructure analyses. Gregory A. Grabowski designed study and participated manuscript editing. Ying Sun designed study, performed experiments and data analysis, wrote manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Liou, B., Zhang, W., Fannin, V. et al. Combination of acid β-glucosidase mutation and Saposin C deficiency in mice reveals Gba1 mutation dependent and tissue-specific disease phenotype. Sci Rep 9, 5571 (2019). https://doi.org/10.1038/s41598-019-41914-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-41914-7
This article is cited by
-
GAU-PED study for early diagnosis of Gaucher disease in children with splenomegaly and cytopenia
Orphanet Journal of Rare Diseases (2023)
-
Sphingolipid lysosomal storage diseases: from bench to bedside
Lipids in Health and Disease (2021)
-
Ocular phenotypes in a mouse model of impaired glucocerebrosidase activity
Scientific Reports (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
