
Abstract
The intra-renal dopamine (DA) system is highly expressed in the proximal tubule and contributes to Na+ and blood pressure homeostasis, as well as to the development of nephropathy. In the kidney, the enzyme DOPA Decarboxylase (DDC) originating from the circulation. We used a twin/family study design, followed by polymorphism association analysis at DDC locus to elucidate heritable influences on renal DA production. Dense single nucleotide polymorphism (SNP) genotyping across the DDC locus on chromosome 7p12 was analyzed by re-sequencing guided by trait-associated genetic markers to discover the responsible genetic variation. We also characterized kinetics of the expressed DDC mutant enzyme. Systematic polymorphism screening across the 15-Exon DDC locus revealed a single coding variant in Exon-14 that was associated with DA excretion and multiple other renal traits indicating pleiotropy. When expressed and characterized in eukaryotic cells, the 462Gln variant displayed lower Vmax (maximal rate of product formation by an enzyme) (21.3 versus 44.9 nmol/min/mg) and lower Km (substrate concentration at which half-maximal product formation is achieved by an enzyme.)(36.2 versus 46.8 μM) than the wild-type (Arg462) allele. The highly heritable DA excretion trait is substantially influenced by a previously uncharacterized common coding variant (Arg462Gln) at the DDC gene that affects multiple renal tubular and glomerular traits, and predicts accelerated functional decline in chronic kidney disease.
Similar content being viewed by others


Introduction
Dopamine (DA) in the central and peripheral neural systems has an established role in motor and behavior control. The kidney also possesses a dopaminergic system that seems to be independent from neural DA systems. In fact intra-renal production of DA is not regulated by renal sympathetic nerve activity as evidenced by renal denervation1. Instead, DA is formed locally in proximal tubule epithelial cells2 from its circulatory precursor levodopa (L-DOPA) after filtration at the glomerulus. L-DOPA is transported into tubular cells, where it is decarboxylated to DA by the enzyme DOPA Decarboxylase (DDC), which is regulated by dietary sodium (Na+)3,4. DA then exits these cells across apical and basolateral surfaces to exert paracrine actions via G-protein coupled DA receptors, predominantly dopamine receptor1(DRD1) across the nephron, signaling largely through Gs to adenylyl cyclase5. The renal dopaminergic system is thus a physiological regulator of Na+ excretion through inhibition of tubular Na+ reabsorption.
Dopaminergic actions in the kidney are not limited to maintaining Na+ homeostasis. DA may increase glomerular filtration rate (GFR) by post-glomerular (efferent) arteriolar constriction6 and modulates renin expression7 as well as angiotensin II, also controlling Na+ excretion and blood pressure (BP)8,9.
Not surprisingly, defects in renal DA production or receptor signaling are linked to development of hypertension10,11,12 In animal models intra-renal DA production is linked to development of hypertension and decreased longevity13, as well as angiotensin II-mediated renal injury14 and progression of nephropathy15.
We used a classical twin design approach to ask whether renal DA synthesis and excretion are heritable, and we performed a genetic association analysis to determine specific effects of the DDC locus on the urinary DA trait. Finally, we have performed a functional analysis of 462Gln DDC variant.
Methods
All protocols were approved by the appropriate committees of the University of California – San Diego, Human Research Protection Program (HRPP), and each subject gave written informed consent prior to participation.
Subjects and clinical characterization
Twin and sibling subjects
The University of California, San Diego (UCSD) twin/sibling study has previously been described16. Twin and sibling participants were recruited from southern California by access to a population birth record-based twin registry17, as well as by newspaper advertisement as described previously18. Subjects included both dizygotic (DZ) and monozygotic (MZ) twin pairs. Zygosity of twins was confirmed genetically by microsatellite and SNP markers18. Clinical characterization was conducted as previously described18. BP status (high vs. normal) was defined by history (medical record or self-report), presence of antihypertensive medications, and measurement of seated BP by arm cuff [hypertension: either/or ≥140/≥90 mmHg systolic BP (SBP)/diastolic BP (DBP), or both]. None of the subjects had a history of cardiovascular disease or renal failure, and plasma creatinine concentrations were ≤1.5 mg/dL. Estimated glomerular filtration rate (eGFR) was estimated by the MDRD (Modification of Diet in Renal Disease) algorithm.
Twin/sibling physiological phenotyping
Brachial cuff BPs with SBP/DBP measured as K1/K4, as well as heart rate, were obtained non-invasively in seated subjects in triplicate using an oscillometric device (DynaPulse; PulseMetric Inc., Vista, CA) as described16. Triplicate values (within ±10% of the mean) were averaged. We and others previously validated DynaPulse measurements against more invasive devices16,19.
Twin/sibling biochemical phenotyping
Subjects were instructed to fast during the 6 h preceding the evaluation. Fasting blood and freshly voided urine samples were collected from each subject. Plasma and urine samples were quickly frozen to −70 °C, in preparation for later catecholamine assay at the same time. A sensitive radioenzymatic assay was used to measure plasma and urine catecholamines [dopamine (DA), norepinephrine (NE), and epinephrine (EPI)] through the catechol-O-methylation process20. The radioenzymatic assay for catecholamines involved transfer of a 3H label to catecholamines from S-adenosylmethionine during O-methylation, mediated by the enzyme catechol-O-methyltransferase (COMT). Prior to O-methylation, plasma catecholamines were extracted into dilute acetic acid to remove COMT inhibitors in plasma. Assay sensitivities (lower limits of detection) were ~10 pg for DA and NE, and ~6 pg for EPI. Inter-assay coefficients of variation were ~10% for urine DA, ~20% for plasma DA, ~10% for NE and ~13% for EPI. Plasma and urine creatinine were measured by autoanalyzer (Beckman-Coulter; Brea, CA), and urine catecholamine results were normalized to creatinine in the same sample.
Genotyping of the DDC locus
We obtained the exon/intron structure for human DDC, as well as putative sequence, at http://genome.ucsc.edu, focusing on the longer kidney/liver (~107 kbp) isoform (RefSeq NM_001082971.1).
Genomic deoxyribonucleic acid (DNA) from twin and sibling pairs was isolated from blood leukocytes with Qiagen columns, after proteinase K digestion, as previously described18. SNP genotyping across the DDC locus was accomplished using the Illumina 610-Quad genotyping array www.Illumina.com. DDC LD blocks are displayed in Supplementary Information Fig. 1.
Systematic polymorphism characterization across the DDC locus
After initial positive DDC-on-DA excretion association we searched for functional variants across the DDC locus by re-sequencing exons, untranslated regions (UTRs), and promoter, in 12 subjects who carried the trait-associated (minor) allele in intron-8 tagging SNP rs11575358 (G > A), versus 11 subjects who did not carry this allele. A public draft of the human21 genome sequence was obtained from the UCSC Genome Bioinformatics website http://genome.ucsc.edu and used as a scaffold for primer design and sequence alignment. Positions were numbered with respect to the messenger ribonucleic acid (mRNA) cap (transcriptional initiation) site in national Center for Biotechnology Reference Sequences (NCBI RefSeq) source clone NM_001082971.1 (kidney and liver splicing isoform), using polymerase chain reaction (PCR) primers designed by Primer322 to span each of the 15 exons, and 50–100 bp of flanking intronic sequences, as well as 965 bp of the proximal promoter (upstream of the cap site). Primers are shown in Supplementary Table 1. PCR, amplicon purification and analysis of target sequences were performed according to the protocol described previously23. Sequence was determined on an ABI-3730XL dideoxy capillary sequencer. Polymorphism and heterozygosity were visualized from the ABI tracings using Codon Code Aligner http://www.codoncode.com/aligner.
Genotyping of coding variant Arg462Gln (rs11575542, G/A) in DDC exon-14
An amplified fragment length polymorphism (AFLP) analysis was also used to genotype the Arg462Gln at the DDC locus, with forward primer 5′-ggcttcttcctgatgtacgg-3′, and reverse primer 5′-aataaggaagagaaggccgg-3′. Following PCR (30 plus cycles) and digestion with restriction enzyme StuI (1 unit, 2 hours), 2% agarose gel electrophoresis separated restriction fragments. Genotyping yielded a single 482-bp fragment for G/G (Arg/Arg, major allele) homozygotes; two shorter fragments (257 and 225 bp) for A/A homozygotes (Gln/Gln); while G/A heterozygotes (Arg/Gln) displayed 3 fragments (482, 257, and 225 bp) (Supplemental Fig. 2).
Replication of marker (DDC)-on-trait (DA) association
MariMRSnes
US Marines (all young healthy men) from the Marine Resiliency Study (MRS) http://marineresilience.org/24 were evaluated. Of 2600 Marines assessed at pre-deployment for MRS on bases in Southern California, 361 were included in this analysis. Participants were healthy males of self-identified European (white, non-Hispanic) ancestry, ranging in age from 18–37 years (mean 21.7 ± 2.5 years). Biological samples including freshly voided urine, nucleated cells from whole blood (for genomic DNA) and EDTA plasma by venipuncture were collected for further analysis.
Caregivers
The study population included caregivers from The Alzheimer Caregiver Coping Study25, conducted at UCSD. To be eligible for the study, participants were required to be at least 55 years of age, married, and providing in-home care for their spouses. Participants were excluded if they were diagnosed with a serious medical condition, had hypertension greater than 200/120 mmHg, or if they were taking anti-coagulant medication (exclusion criteria due to other data collected for this prospective study).
Statistical analyses
Heritability (h2) and genetic covariance
Heritability (h2) estimates of physiological and biochemical traits were obtained through twin pair (MZ or DZ) variance components in Sequential Oligogenic Linkage Analysis Routines (SOLAR)26 available at http://solar.txbiomedgenetics.org. Heritability is estimated by the equation: h2 = VG/VP, where VG is additive genetic variance and VP is total phenotypic variance. Here a normal distribution in twin pairs is assumed in order to maximize the likelihood with a mean dependent on traits of interest. Genetic covariance of DA excretion with other correlated heritable traits was estimated as ρG in SOLAR, while environmental covariance was estimated as parameter ρE.
SNP-on-phenotype effects across the DDC locus were initially tested in MERLIN v1.1.2 http://www.sph.umich.edu/csg/abecasis/merlin/ with an additive model, to explicitly account for family structure. Descriptive (mean ± SEM) and inferential statistics were computed by generalized estimating equations (GEEs) to account for correlated observations within families, using SPSS (IBM Corporation; Armonk, NY) predictive analytics software. GEE also tested for allele and diploid genotype effects on specified traits. The SNP was defined as a 3-level variable representing the three possible genotypes (homozygous variant, heterozygous, and homozygous wild-type), initially testing an additive effect in the model. Analyses were adjusted for age and sex. In addition to additive models, after inspection of distribution of the observations, heterozygotes and minor (or major) allele homozygotes were grouped together, as needed to test dominant/recessive models.
Marker-on-trait association display
Results across the DDC locus, centered on intronic tagging-SNP peak associations (e.g., rs11575340) as well as results centering on functional variant Arg462Gln (rs11575542) were displayed by local SNAP (SNP Annotation and Proxy Search) plot as described at http://www.broadinstitute.org/mpg/snap/ldplot.php.
Grouped analysis were carried out testing fixed-effect (i.e., diploid genotype) models in STATA (v12, Stata Corp, College Station, TX), after individual study regression analyses in SPSS (v17), incorporating individual data to derive significance as well as pooled genotype effect size (beta, or slope/allele) and its SE (standard error).
Bioinformatics
To determine conservation of sequence at the functional coding genetic variant (Arg462Gln) and local homology, we performed sequence searches using BLASTP http://blast.ncbi.nlm.nih.gov, followed by multiple sequence alignments with CLUSTAL Omega (1.1.0) http://www.ebi.ac.uk/Tools/msa/clustalo/.
Human DDC enzyme (EC 4.1.1.28) coding genetic variant Arg462Gln (rs11575542): Mutagenesis, eukaryotic expression, and characterization of enzymatic kinetics
Site-directed mutagenesis
The full-length human DDC cDNA DNA clone, was obtained from a human pheochromocytoma cDNA library, and code for a 480 amino acid protein with a predicted molecular mass of ~53.9 kDa. The cDNA was inserted (in the correct 5′ → 3′ orientation) into a eukaryotic (pSV40 promoter) expression plasmid, as previously described13. The wild-type (Arg462) version was used as a template for site-directed mutagenesis (QuikChange, Agilent) to create the 462Gln variant, which was verified by dideoxy-sequencing.
COS cell transfection and electrophoresis
COS cells were transfected with the pSV40 DDC expression plasmids with a modified calcium-phosphate procedure as described13, followed by cell lysis, SDS-PAGE electrophoresis, protein transfer to polyvinylidene difluoride membranes (Bio-Rad, Hercules, CA), and either protein staining (coomassie blue) or detection by immunoblotting with a rabbit anti-human DDC polyclonal antibody, to visualize the previously described Mr ~ 50 kDa immunoreactive band. We also detected beta-actin with an anti-beta-actin monoclonal antibody (Sigma A5441, St. Louis, MO) to normalize the expression level of DDC protein in COS cells, with protein loads of 25, 50, and 100 µg protein/lane (in triplicate).
Expressed human DDC enzyme (EC 4.1.1.28) kinetics: Vmax and Km
COS-cell-expressed DDC (Arg462 versus Gln462) gave rise to enzymatic activity that catalyzed the decarboxylation of L-3,4-dihydroxyphenylalanine (L-DOPA) to DA, at pH 6.5 in the presence of cofactor pyridoxal phosphate (10 µM). Cell lysates were prepared and enzymatic activity (including Km (substrate concentration at which half-maximal product formation is achieved by an enzyme) as a function of substrate concentration, and Vmax (maximal rate of product formation by an enzyme) at saturating substrate concentration). Km and Vmax values were calculated using a Hanes-Woolf plot. Vmax results were normalized to DDC protein expression, determined by immunoblot DDC/beta-actin ratio (in triplicate).
After transfection of COS cells with DDC-expression plasmid, we performed a Western blotting to estimate the expression levels of wild type (WT) or variant DDC protein. The expression level of variant DDC was 83% less than that of WT DDC (as shows in the results), probably caused by the differences in transfection efficiency and the copy number of expression plasmids in the cells. We assayed the DDC activity in the same lysates of transfected COS cells, and determined Km and Vmax values of each lysate. Then, the Vmax value of variant DDC was divided by 0.83 to normalize with the expression level of WT DDC.
Results
Renal DA excretion: Heritability (h2) and genetic covariance
Twin pairs variance component analyses indicated that catecholamine secretion and renal excretion traits were all substantially heritable (Table 1, Fig. 1A), with DA excretion heritability (h2 = VG/VP) = 62.7 ± 5.2% of trait variance (p = 5.96E-16). Notably, the renal and plasma DA traits did not significantly correlate, nor did they display shared heritability (genetic covariance), reinforcing the separate control of the two systems.

Renal DA excretion: Effects of heredity and the DDC gene. (A) Heritability (h2) of catecholamine secretion and urinary excretion. Results for plasma catecholamine concentrations (pg/ml) and urine catecholamine excretion (ng/gm creatinine) emerge from variance components analyses by SOLAR in MZ versus DZ twin pairs. h2 is expressed as % of trait variance attributable to gene action, ±SEM. VG: genetic variance; VP: total phenotypic variance. (B) Human DOPA decarboxylase (DDC): Exon/intron and inter-species sequence conservation. Polymorphism discovery across the human DDC locus: re-sequencing strategy. There are 15 exons at human DDC. Eighteen polymerase chain reaction amplicons spanned the promoter and coding regions. Amplicons with double arrows were read in both directions. The SNP Arg462Gln (rs11575542) characterized in this paper is located in exon-14. (C) Natural genetic variation at DOPA decarboxylase (DDC, AADC) on chromosome 7p12: Prediction of renal DA production in twin pairs. Local region of the Manhattan plot displayed by SNAP (SNP Annotation and Proxy Search) plot http://www.broadinstitute.org/mpg/snap/ldplot.php. The peak association (p = 1.35E-06; see diamond) was at DDC intron-5 tagging SNP rs11575340. The DA secretion trait-associated coding variant (characterized in this paper) is at DDC Arg462Gln (exon-14, rs11575542). Marker-on-marker LD is shown as R2 (with respect to rs11575340), on a red color scale.
Renal DA excretion: Cis-QTL at the DDC locus
During dense SNP genotyping across the DDC locus (Fig. 1B), we observed a broad DDC marker-on-DA trait association peak in twin/sibling pairs (Fig. 1C) spanning the ~107 kbp DDC locus, but confined to a ~275 kbp linkage disequilibrium (LD) block at the locus, as defined by the local recombination pattern (cM/Mb tracing). A peak tagging SNP (rs11575340, within intron-5) was highly associated with urine DA excretion levels (p = 1.135E-06). By contrast, the plasma DA trait did not display association with the DDC locus (all p > 0.05). Other DDC SNPs “tagging” the urinary DA trait at high significance (p < 1.0E-05) were rs11575358 (intron-8), rs11575522 (intron-12), and exonic SNP Arg462Gln (rs11575542, exon-14). The association of DDC with DA excretion was confirmed with additional independent groups (Twins/siblings, Marines, and caregivers) using tagging SNPs rs11575358 and rs11575522. Grouped analyses indicate significant associations for each marker SNP rs11575358 (p = 1.0E-04 OR: 0,879), and rs11575522 (p = 1.0E-04 OR: 0,890) (Table 2).
The trait-associated DDC region on chromosome 7p12, was also plotted in CEU subjects at www.HapMap.org as pair-wise LD parameter R2 across the ∼107 kbp locus (Supplemental Fig. 1), indicating a substantial LD block spanning the locus.
Trait aggregation with DA excretion: Stratification by urine DA quantiles
We next asked whether other traits were associated with DA excretion. Table 3 provides a description of the twin/sibling cohort, divided into two groups (quantiles, upper and lower) stratified around the DA excretion median value. Elevated urine DA was substantially more common in women than men (p = 1.5E-07), a trend borne out by direct comparison of excretion in women versus men (173.1 ± 3.68 versus 134.6 ± 5.49 ng/gm, p = 1.0E-5). Participants with higher DA excretion showed significant (p < 0.05) elevations in several other traits, including urine catecholamines (NE and EPI), excretion of K+, Na+, and Cl−, as well as the fractional excretion of Na+ (FENa+) (though not fractional excretion of Li+. (FELi+)), and eGFR.
However, subjects with increased DA excretion did not display changes in circulating catecholamines (including plasma DA) or components of the renin-angiotensin-aldosterone system), BP or heart rate (HR).
Visual display of trait aggregation for excreted DA (p = 1.0E-06) with NE (p = 1.0E-06) and EPI, (p = 1.0E-06), is given in Supplemental Fig. 3a. Supplemental Figure 3b displays the significant correspondence of urine DA with urine Na+ excretion (p = 1.0E-06) and FENa+: increasing levels of excreted Na+ led to higher FENa+ (p = 0.019). Supplemental Figure 3c displays urine DA and eGFR coordinately increasing (p = 9.0E-03). The effect on glomerular function is reflected in the excretion of other electrolytes: K+ (p = 0.019) and Cl− (p = 1.0E-06) (Table 3).
Systematic polymorphism discovery across the DDC locus
In 23 unrelated individuals (n = 46 chromosomes) stratified by presence/absence of DDC rs11575358 trait-associated (“risk”, A, minor) allele (11 with, 12 without), the promoter and all 15 exons and adjacent intronic regions were re-sequenced. The 17 amplicons and their sequencing directions are represented in Fig. 1B. As shown in Table 4 a total of 17 polymorphisms were detected. Of these, two common variants were discovered in the promoter: a common SNP (rs56233242, G-899T) and an Insertion/Deletion (Ins/Del) variant (rs76759613, G-554/−). We identified 2 non-synonymous (amino acid replacement) SNPs, one previously known and located in exon-2 (E2-206; G/A, Val60Met) and a previously uncharacterized SNP located in exon-14 (rs11575542, E14-143; G/A, Arg462Gln). We also found a second novel SNP in intron-3 (I3-21; C/T) and a common five-base Ins/Del in intron-9 (rs59827423, I9-107; CAGGG/−). The other 10 SNPs detected were common and intronic.
We selected 23 subjects based on genotype, according to trait-associated intron-8 tagging SNP rs11575358 (G > A): 11 cases (carrying the DA-trait-associated A-allele, one A/A homozygote and 10 G/A heterozygotes) and 12 controls (G/G homozygotes).
Inter-species amino acid sequence alignments and conservation are presented in Supplemental Table 2. Results reveal that Arg 462 is completely in old world primates, though not in new world primates (squirrel monkey, marmoset). 462Gln is found in several other mammalian and vertebrate species: mole rat, guinea pig, and ricefish. The local region around codon 462 is within a carboxy-terminal alpha-helix (helix-12, amino acids 457–477) on X-ray crystallography of human DDC.
Pleiotropic effects of DDC genetic variant Arg462Gln on multiple human traits
With diploid genotypic ratios of Arg/Arg:Arg/Gln:Gln/Gln at 429:23:1, Arg462Gln (rs11575542) was in Hardy Weinberg equilibrium (HWE) (Chi2 = 1.32, p = 0.25), with a minor allele frequency of ~2.76%. Its effects on physiological and biochemical traits were evaluated in twins/siblings (Table 5), revealing pleiotropic effects of Arg462Gln on multiple traits.
Notable effects were observed in the renal excretion traits. Carriers of the 462Gln (minor) allele (heterozygous [n = 23] or homozygous [n = 1]) showed increased DA and EPI excretion. Directionally coordinate effects on eGFR and urine DA are depicted in Fig. 2A. Figure 2B displays parallel effects on albumin excretion and FELi+, an index of proximal tubular avidity for Na+. Thus 462Gln-augmented renal DA synthesis and excretion may give rise to a cascade of events in the kidney: local DA may trigger both natriuresis and afferent arteriolar vasodilation (or efferent constriction), and the ensuing glomerular hyperfiltration increases albumin excretion.

Association of DDC genetic variant Arg462Gln (rs11575542) with DA excretion and renal function. Results are shown as mean ± SEM in twins/siblings. Numbers are in parentheses (n) indicate the n for the observation. Significant differences (p < 0.05) are in bold. (A) Pleiotropic effects on urine DA excretion, p = 3.68E-05, and eGFR, p = 0.032. Minor allele homozygotes Gln/Gln (A/A) and heterozygous Arg/Gln (G/A) subjects displayed increased eGFR and urine DA excretion compared to homozygous major allele Arg/Arg (G/G) subjects. (B) Pleiotropic effects on FELi+ (p = 0.008) and urine albumin excretion (p = 0.033). Minor allele homozygotes (Gln/Gln, A/A) and heterozygotes (Arg/Gln, G/A) displayed increased FELi+, as well as urine albumin excretion, compared to major allele homozygotes (Arg/Arg, G/G).
Notably, DDC variant Arg462Gln did not affect plasma catecholamine (including plasma DA) concentrations, the renin-angiotensin system (plasma renin and plasma aldosterone), c-reactive protein (CRP) levels, or systemic hemodynamic traits (BP and HR) (Table 5).
Human DDC variant enzyme kinetics
Expression
Figure 3A shows immunoblot results of human DDC protein expression in COS cells: wild-type (Arg462) and variant (Gln462), with beta-actin control normalization. Figure 3B shows the results of the quantifying the expression of DDC in transfected cos cells in controls (column 1), wild type (Arg462) and variant (462Gln), with a higher expression in wild type cells.

Human DDC enzyme genetic variants. (A) Expression of human DDC protein in COS cells: Detection by anti-AADC immunoblot, normalized with beta-actin. Protein standard size ladders bracket the experimental results. C (control), W (wild-type, Arg462), and V (variant, 462Gln). Blots were initially examined at protein loads of 25, 50, and 100 µg protein/lane; here, triplicate determinations for 50 µg protein loads are shown, along with size standards. (B) Quantifying expression of DDC expressed in COS cells: Wild-type (Arg462) versus variant (462Gln). DDC and beta-actin immunoreactive bands (at 50 µg protein/lane, in triplicate) were scanned for optical density, and mean ± one SD values are shown. Expression of 462Gln was ~83% of that for Arg462. (C) Kinetic analyses with L-DOPA substrate. Graph shows a Hanes-Woolf plot of [S]/v as a function of [S], where [S] is the concentration of substrate [L-DOPA] and v is the reaction velocity (nmol/min/mg) at that [S]. The 462Gln variant displays a substantially lower Vmax (at 19.4 ± 0.2 versus wild-type 29.8 ± 0.3 nmol/min/mg protein; 35% reduction at p < 0.0001), though similar Km (at 31.0 ± 5.3 vs wild-type 31.6 ± 4.6 μM; p = 0.94). Replicates: at [S] = 0.5, n = 2; others, n = 3. Results are shown as mean ± one SD. Since typical L-DOPA substrate concentrations in humans are far below the Km value (at L-DOPA of ~1.08 nM [2.13 ng/ml] in plasma and ~100 nM [1.22 µg/hr] in urine, then the DDC system is likely to be catalyzing DA synthesis over a relatively linear range in vivo.
Kinetics
We analyzed the kinetic parameters comparing wild-type and variant enzymatic action using L-DOPA as substrate (Fig. 3C), using a Hanes-Woolf plot. The 462Gln variant displayed a substantially lower Vmax (at 19.4 ± 0.2 versus wild-type 29.8 ± 0.3 nmol/min/mg protein; 35% reduction at p < 0.0001), though similar Km (at 31.0 ± 5.3 versus wild-type 31.6 ± 4.6 μM).
Discussion
The independent renal DA system highly expressed in proximal tubule subserves both tubular and glomerular functions. Intra-renal DA also influences the renin-angiotensin system and thereby controls progression of renal damage in kidney pathologies14,15.
The role of common genetic variation in control of endogenous dopaminergic activity is previously unexplored. In the twin study, we first determined that DA excretion is a highly heritable trait (at 62.7 ± 5.2% of trait variance, p = 5.96E-16), although it was genetically uncorrelated with plasma DA. We then characterize a functional non-synonymous variant (Arg462Gln) in the DDC coding region that seems to control common heritable variation in DA secretion and its downstream renal consequences in an apparently healthy population. Replication was performed with a grouped analysis of three independent cohorts that validated the findings (Table 2).
The paradox of elevated DA excretion in DDC deficiency
We observed a DDC genetic variant (Arg462Gln) wherein the minor allele (462Gln) seemed to increase renal DA production but reduce enzymatic DDC activity. Mendelian human autosomal recessive DDC deficiency is characterized by predominantly neurological manifestations such as developmental delay, prominent motor abnormalities, oculogyric crises and autonomic features27,28. Available treatment options (DA agonists, vitamin B6 and monoamine oxidase inhibitors) are only variably effective29. However, many such DDC-deficient patients exhibit normal or even elevated levels of urinary DA excretion in the presence of a less active DDC with normal levels of plasma DA30. Such findings have been referred to the “paradox of hyperdopaminuria in DDC deficiency”. Clues to the origin of this paradox involve the presence of a separate/local renal DA system synthesizing the transmitter in proximal tubular epithelial cells of the kidney cortex31. This local kidney system displays high efficiency in transforming blood-derived L-DOPA to renal DA. As a consequence of systemic DDC deficiency in the brain and elsewhere, more substrate (L-DOPA) and cofactors may be delivered to DDC in the kidney cortex32. Other theories propose alternative substrates for renal DA formation such as 3-methoxy-tyrosine33, other metabolic pathways such as tyramine hydroxylation by renal CYP2D6 to form DA34 or even tyrosinase as an alternative pathway for DOPA and DA synthesis even in the absence of tyrosine hydroxylase35.
Selective kidney DDC loss-of-function in experimental animals: Renal functional consequences
The DDC 462Gln allele elevate DA eGFR and albumin excretion in healthy individuals (Table 5, Fig. 2B). These results suggest that glomerular hyperfiltration may be a risk factor for accelerated renal decline in 462Gln carriers36. By contrast, after selective loss of intra-renal DA production by targeted ablation of the DDC gene in the proximal tubule, transgenic mice display protection from renal injury induced by angiotensin II14 or during diabetic nephropathy15.
Why might humans and mice display such apparent differences in renal effects of intra-renal DA deficiency? First of all, in the mouse, DDC ablation could be created precisely in the proximal tubule by targeting the knockout to cells expressing the Cre recombinase driven by a proximal tubule-specific (γ-GT) promoter37, while humans express DDC from the same gene in both CNS and kidney. Secondly, our results as well as other human studies on the “paradox of hyperdopaminuria in DDC deficiency” indicate that a relatively inactive DDC enzyme can actually result in elevated renal DA excretion32 by several proposed mechanisms. Thus, the human and mouse results may ultimately be compatible.
Advantages and limitations of this study
Here we were able to mobilize several complementary techniques to understand genetic control of renal dopamine excretion: twin pair variance components to estimate heritability, extensive human phenotyping, dense SNP genotyping, validation by grouped analysis of three independent groups, systematic polymorphism discovery across the DDC locus, functional testing of a mutant enzyme, and extension into a disease population. The non-synonymous amino acid substitution (Arg462Gln) occurred in a carboxy-terminal alpha-helix far from the DDC active site38; however, the Arg → Gln substitution yields a substantial change in amino acid properties specified by codon 462 (cationic → neutral), and its position far from the active site is consistent with the unchanged Km (i.e., no change in substrate affinity) in the face of diminished Vmax (i.e., essentially non-competitive or allosteric inhibition).
Nonetheless, questions remain. DDC and DA production are widespread, occurring not just in the kidney, but also in liver and in the nervous system. In the intact human organism (as opposed to a more manipulable experimental animal) it may be difficult to understand precisely the roles of such different sites of origin of the amine. Indeed, there may be other origins of DA, such as tyramine hydroxylation by renal CYP2D634 or even tysosinase39. Since there is only a single DDC gene on chromosome 7p12, subserving expression in both brain and kidney (albeit with alternative initial exons), and since DDC is in the biosynthetic pathway for serotonin as well as dopamine, it is likely that our results also have implications for neuropsychiatric disease; however, in this report we confined our investigation to renal traits.
Conclusions and perspectives
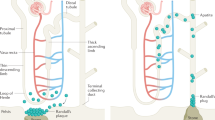
We conclude that DDC exon-14 non-synonymous variant Arg462Gln is functional and this variant has clinical implications as follows, and it alters enzyme kinetic parameters, Vmax Km, resulting in changes in urine DA excretion. The change in DA secretion influences renal function, modifying renal tubular avidity for Na+ and Li+, with consequent changes in eGFR and albumin excretion, all this changes eventuating in accelerated renal decline in patients with CKD (Fig. 4). These findings augment our understanding of the kidney dopaminergic system, and shed new light on the link between DA production and renal dysfunction in humans, a topic previously investigated largely in animal models.

Hypothetical schema for effects of DDC genetic variation on catecholamine and renal traits. The genetic variation of human CDD and its effects on AD and renal traits: a schematic hypothesis. This framework correlates the experimental results from the genetic variation of the gene that codes for CDD, the change in the activity of the CDD enzyme and how it influences the first DA excretion (biochemical trait), then affects the excretion of electrolytes and GFR (traits). physiological), the excretion of albumin in urine and, finally, determines the progression of CKD (disease trait).
Change history
14 February 2020
An amendment to this paper has been published and can be accessed via a link at the top of the paper.
References
Wang, Z. Q., Siragy, H. M., Felder, R. A. & Carey, R. M. Intrarenal dopamine production and distribution in the rat. Physiological control of sodium excretion. Hypertension 29, 228–34 (1997).
Hayashi, M., Yamaji, Y., Kitajima, W. & Saruta, T. Aromatic L-amino acid decarboxylase activity along the rat nephron. Am J Physiol 258, F28–33 (1990).
Seri, I. et al. Influence of Na+ intake on dopamine-induced inhibition of renal cortical Na(+)-K(+)-ATPase. Am J Physiol 258, F52–60 (1990).
Hayashi, M., Yamaji, Y., Kitajima, W. & Saruta, T. Effects of high salt intake on dopamine production in rat kidney. Am J Physiol 260, E675–9 (1991).
Jose, P. A., Eisner, G. M. & Felder, R. A. Renal dopamine receptors in health and hypertension. Pharmacol Ther 80, 149–82 (1998).
Luippold, G., Schneider, S., Vallon, V., Osswald, H. & Muhlbauer, B. Postglomerular vasoconstriction induced by dopamine D(3) receptor activation in anesthetized rats. Am J Physiol Renal Physiol 278, F570–5 (2000).
Zhang, M. Z. et al. Intrarenal dopaminergic system regulates renin expression. Hypertension 53, 564–70 (2000).
Gildea, J. J. Dopamine and angiotensin as renal counterregulatory systems controlling sodium balance. Curr Opin Nephrol Hypertens 18, 28–32 (2009).
Carey, R. M. The intrarenal renin-angiotensin and dopaminergic systems: control of renal sodium excretion and blood pressure. Hypertension 61, 673–80 (2013).
Chugh, G., Lokhandwala, M. F. & Asghar, M. Altered functioning of both renal dopamine D1 and angiotensin II type 1 receptors causes hypertension in old rats. Hypertension 59, 1029–36 (2012).
Hussain, T., Kansra, V. & Lokhandwala, M. F. Renal dopamine receptor signaling mechanisms in spontaneously hypertensive and Fischer 344 old rats. Clin Exp Hypertens 21, 25–36 (2009).
Jose, P. A., Eisner, G. M., Drago, J., Carey, R. M. & Felder, R. A. Dopamine receptor signaling defects in spontaneous hypertension. Am J Hypertens 9, 400–5 (1996).
Zhang, M. Z. et al. Intrarenal dopamine deficiency leads to hypertension and decreased longevity in mice. J Clin Invest 121, 2845–54 (2011).
Yang, S. et al. Intrarenal dopamine modulates progressive angiotensin II-mediated renal injury. Am J Physiol Renal Physiol 302, F742–9 (2012).
Zhang, M. Z. et al. Intrarenal dopamine inhibits progression of diabetic nephropathy. Diabetes 61, 2575–84 (2012).
Zhang, L. et al. Functional allelic heterogeneity and pleiotropy of a repeat polymorphism in tyrosine hydroxylase: prediction of catecholamines and response to stress in twins. Physiol Genomics 19, 277–91 (2012).
Davis, J. T. et al. Autonomic and hemodynamic origins of pre-hypertension: central role of heredity. J Am Coll Cardiol 59, 2206–16 (2012).
Cockburn, M., Hamilton, A., Zadnick, J., Cozen, W. & Mack, T. M. The occurrence of chronic disease and other conditions in a large population-based cohort of native Californian twins. Twin Res 5, 460–7 (2002).
Brinton, T. J. et al. Arterial compliance by cuff sphygmomanometer. Application to hypertension and early changes in subjects at genetic risk. Hypertension 28, 599–603 (1996).
Ziegler, M. G., Kennedy, B. & Elayan, H. A sensitive radioenzymatic assay for epinephrine forming enzymes. Life Sci 43, 2117–22 (1998).
Giardina, G. et al. Open conformation of human DOPA decarboxylase reveals the mechanism of PLP addition to Group II decarboxylases. Proc Natl Acad Sci USA 108, 20514–9 (2011).
Rozen, S. & Skaletsky, H. Primer3 on the WWW for general users and for biologist programmers. Methods Mol Biol 132, 365–86 (2000).
Wen, J. D. & Gray, D. M. Selection of genomic sequences that bind tightly to Ff gene 5 protein: primer-free genomic SELEX. Nucleic Acids Res 32, e182 (2004).
Baker, D. G. et al. Predictors of risk and resilience for posttraumatic stress disorder among ground combat Marines: methods of the Marine Resiliency Study. Prev Chronic Dis 9, E97 (2012).
Mausbach, B. T. et al. A 5-year longitudinal study of the relationships between stress, coping, and immune cell beta(2)-adrenergic receptor sensitivity. Psychiatry Res 160, 247–55 (2008).
Brun, L. et al. Clinical and biochemical features of aromatic L-amino acid decarboxylase deficiency. Neurology 75, 64–71 (2010).
Hyland, K., Surtees, R. A., Rodeck, C. & Clayton, P. T. Aromatic L-amino acid decarboxylase deficiency: clinical features, diagnosis, and treatment of a new inborn error of neurotransmitter amine synthesis. Neurology 42, 1980–8 (1992).
Allen, G. F., Land, J. M. & Heales, S. J. A new perspective on the treatment of aromatic L-amino acid decarboxylase deficiency. Mol Genet Metab 97, 6–14 (2009).
Abeling, N. G. et al. Pathobiochemical implications of hyperdopaminuria in patients with aromatic L-amino acid decarboxylase deficiency. J Inherit Metab Dis 23, 325–8 (2000).
Soares-da-Silva, P. & Fernandes, M. H. Synthesis and metabolism of dopamine in the kidney. Effects of sodium chloride, monoamine oxidase inhibitors and alpha-human atrial natriuretic peptide. Am J Hypertens 3, 7S–10S (1990).
Wassenberg, T. et al. The paradox of hyperdopaminuria in aromatic L-amino Acid deficiency explained. JIMD Rep 4, 39–45 (2012).
Ibarra, F. R. et al. Demethylation of 3-O-methyldopa in the kidney: a possible source for dopamine in urine. Am J Physiol 270, F862–8 (1996).
Wassenberg, T. et al. Urinary dopamine in aromatic L-amino acid decarboxylase deficiency: the unsolved paradox. Mol Genet Metab 101, 349–56 (2010).
Ruderman, A. I. Prerequisties for effective radiotherapy for lung cancer. Cancer Treat Rep 60, 1475–7 (1976).
Brenner, B. M. Hemodynamically mediated glomerular injury and the progressive nature of kidney disease. Kidney Int 23, 647–55 (1983).
Qi, W. N. et al. Reperfusion injury in skeletal muscle is reduced in inducible nitric oxide synthase knockout mice. J Appl Physiol 97, 1323–8 (2004).
Ichinose, H., Kurosawa, Y., Titani, K., Fujita, K. & Nagatsu, T. Isolation and characterization of a cDNA clone encoding human aromatic L-amino acid decarboxylase. Biochem Biophys Res Commun 164, 1024–30, decarboxylases. Proc Natl Acad Sci USA 108, 20514–9 (1989).
Rios, M. et al. Catecholamine synthesis is mediated by tyrosinase in the absence of tyrosine hydroxylase. J Neurosci 19, 3519–26 (1999).
Acknowledgements
We acknowledge the National Institutes of Health (DK094894, HL58120, DK079337, 5UL1RR031980, 5P60MD000220), the Department of Veterans Affairs HSR&D, Marine Corps and Navy BUMED, and Fundación Alfonso Martín Escudero (FAME) for providing support to this research. Special mention to Dr. Daniel T. O’Connor, he designed the research and supervised all the steps. Dan passed away, but every day he continues teaching us. National Institutes of Health (R01-DK094894, P30-DK079337, R01-HL58120, UL1-TR000100, P60-MD000220), Department of Veterans Affairs, Fundación Alfonso Martín Escudero (FAME).
Author information
Authors and Affiliations
Contributions
J.P.M.G., M.S.L., M.G.Z., D.G.B., M.M., D.T.O. and H.I.: designed the project. J.P.M., C.M.H., K.Z., S.V., N.B., M.M. and A.J.S.: Performed the genetic experiments. H.K. and H.I.: performed the enzyme kinetics experiments. J.P.M.G., K.Z. and C.M.H. wrote the main manuscript text. J.P.M.G., D.T.O., C.M.N., D.L.-J., R.G.S. and D.G.B., reviewed the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Miramontes-Gonzalez, J.P., Hightower, C.M., Zhang, K. et al. A new common functional coding variant at the DDC gene change renal enzyme activity and modify renal dopamine function. Sci Rep 9, 5055 (2019). https://doi.org/10.1038/s41598-019-41504-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-41504-7
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
