
Abstract
The effects of biogeographical separation and parent material differences in soil bacterial structure and diversity in offshore islands remain poorly understood. In the current study, we used next-generation sequencing to characterize the differences in soil bacterial communities in five offshore subtropical granite islands (Matsu Islets, MI) of mainland China and two offshore tropical andesite islands (Orchid [OI] and Green Islands [GI]) of Taiwan. The soils of OI and GI were more acidic and had higher organic carbon and total nitrogen content than MI soils. The bacterial communities were dominated by Acidobacteria and Proteobacteria but had different relative abundance because soils were derived from different parent material and because of geographic distance. Non-metric multi-dimensional scaling revealed that the communities formed different clusters among different parent material and geographically distributed soils. The alpha-diversity in bacterial communities was higher in tropical than subtropical soils. Mantel test and redundancy analysis indicated that bacterial diversity and compositions of OI and GI soils, respectively, were positively correlated with soil pH, organic carbon, total nitrogen, microbial biomass carbon and nitrogen. These results suggest that variations in soil properties of offshore islands could result from differences in soil parent material. Distinct soils derived from different parent material and geographic distance could in turn alter the bacterial communities.
Similar content being viewed by others


Introduction
Soil bacteria are essential parts of soil ecosystems and are involved in mineralizing organic matter, biogeochemical cycling of carbon and nitrogen and many other soil processes1,2,3. Their distribution can be altered by soil properties4,5, plant species6, litter quality and root exudates7,8,9 as well as temperature and precipitation in different climatic conditions10,11. For instance, bacterial phylotype richness and composition, including the relative abundance of Acidobacteria, Actinobacteria, and Bacteroidetes, is significantly correlated with soil pH4. Bacterial communities cluster separately among four different tropical tree species6. Recently, studies of microbial biogeography have focused on the microbial communities across spatial distance12,13. Other than revealing a causal relation between microbes and their environments, such studies could also provide insights into possible environmental drivers of change in microbial communities14.
Our previous study using clone libraries revealed similar soil bacterial communities in two isolated islands, but climate conditions and soil characteristics affected the bacterial community composition between offshore islands and inland soils of Taiwan15. However, this approach with a low number of sequences provided little resolution for estimating diversity and structure of bacterial communities. The work could also be site-specific, providing less information about bacterial communities across biomes and regions.
The current study investigated the island soil bacterial diversity and structure by biogeographical separation and parent material differences based on high-throughput sequencing. The offshore, subtropical granite islands of China, Matsu Islets (MI), were under long-term military control and restricted industrial activities. Some ecological studies were conducted in this region16,17,18; however, no study has focused on the soil microbial diversity in these islets. We included two more tropical andesite islands of Taiwan, Orchid Island (OI) and Green Island (GI), for comparison. For more detailed and comprehensive information on soil bacterial biogeography, we examined such broad and diverse regions across larger spatial and climatic scales. Our first objective was to elucidate the bacterial diversity and structure of these offshore islands with different soil properties. The second objective was to compare the structure of bacterial communities in soils derived from different parent material and with geographical isolation among these islands. This research could provide important information for understanding how soil bacterial communities respond to environmental factors on these islands.
Results
Soil properties
Basic properties of soil samples from the surveyed islands are listed in Table 1. MI soils were more acidic and had less organic carbon (org. C) and total nitrogen (TN) content than other soils. Both microbial biomass C (MBC) and N (MBN) were significantly reduced in MI soils (P < 0.05). OI and GI are characterized by higher pH, org. C, TN, MBC and MBN as compared with MI soils. Microbial biomass phosphorus (MBP) content was significantly higher in GI soils than other island soils (P < 0.05) (Table 1). Soils in MI were classified as Typic Haplustult according to the US Soil Taxonomy19, whereas soils on GI and OI are Typic Paleudult, which reflects a relative discrepancy in climate between MI and GI or OI.
Bacterial community composition
The major phyla obtained from Miseq libraries (a total of 84,000 sequences for GI and each island of MI; 168,000 sequences for OI) were Acidobacteria (24–34%) and Proteobacteria (27–38%) (Fig. 1). Actinobacteria was the third most abundant phylum and comprised only 3% to 7% of the communities. Other phyla, such as Bacteroidetes, Gemmatimonadetes and Verrucomicrobia, each accounted for less than 8% of the communities (Fig. 1). Among Acidobacteria, groups (Gp) 6, 1, and 2 were the most abundant, and the relative abundance of Gp6 was greater than 6%, except in Dongyin (DY) communities (Table 2). Within Proteobacteria, Alphaproteobacteria was the most abundant class (18–39%), followed by Gammaproteobacteria and Betaproteobacteria. Among communities, Phenylobacterium (1.1%) of Caulobacterales and Bradyrhizobium (0.5%) of Rhizobiales were the two most abundant genera of Alphaproteobacteria. Burkholderia (0.9%) was the most abundant genus of Betaproteobacteria, and its relative abundance was higher in granite than andesite island communities (Table 2).

Relative abundance of bacterial phyla among soil bacterial communities for all offshore islands. Site abbreviations are in Table 1.
The relative abundance among communities showed some variation, despite a similar abundant phyla. Within MI communities, Acidobacteria comprised more than 30% of sequences, except in DY soils. However, the proportion of Acidobacteria in OI and GI soils was only about 25%. The relative abundance of Proteobacteria revealed a similar trend — more than 32% of MI communities, and only about 26% to 30% of OI and GI communities. Within Proteobacteria, the relative abundance of Alphaproteobacteria and Gamaproteobacteria was higher in MI than OI and GI soil communities. The phylum Verrucomicrobia accounted for 6% to 11% of OI and GI communities but less than 5% of all MI soils.
K-shuff analysis of the soil bacterial communities also revealed differences among these offshore islands (Table S1). The non-metric multidimensional scaling (NMDS) plot derived from Ckf of K-shuff analysis indicated that the community structures among MIs did not significantly differ from each other but significantly differed from OI and GI structures (Fig. 2, Table S1).

Shannon diversity index of soil bacterial communities for all offshore islands. Operational taxonomic units (OTUs) were calculated at the 3% evolutionary distance. Site abbreviations are in Table 1.
Bacterial community diversity
Besides the distinctive community composition, the bacterial alpha-diversity among these communities (a total of 84,000 sequences for GI and each island of MI; 168,000 sequences for OI) also differed. Calculated at an evolutionary distance <0.03 (about 97% sequence similarity), the Shannon diversity index revealed that the bacterial diversity was similar among the individual islands of MI (Fig. 3). The OI community was more diverse than the GI community and the diversity was higher for OI and GI than MI communities (Fig. 3). The results of Chao 1 estimator and rarefaction curve analyses supported these results (Fig. S1). β-diversity, analyzed by community comparison of the NMDS plot indicated the different clusters formed between subtropical granite and tropical andesite islands (Fig. 4). NMDS plot analysis also revealed differences between OI and GI communities, thereby suggesting that the different soil properties affect bacterial communities on these two andesite islands. Taken together, MI communities showed close distribution, whereas OI and GI communities formed two separate clusters. These results reflect the close geographic distribution and soil parent material of MI and the distant geographic isolation and soils derived from different soil parent materials among MI, OI and GI soils.

Non-metric multidimensional scaling plot derived from Ckf of K-shuff analysis. Site abbreviations are in Table 1.

Non-metric multidimensional scaling plot of soil communities for all offshore islands. Site abbreviations are in Table 1.
Relation between bacterial communities and soil properties
The community composition of MI soils was significantly altered by the soil properties of TN, MBC and MBN. In contrast, only soil pH was correlated with bacterial diversity (Table S2). Similarly, soil org. C, MBC, MBN and MBP were significantly correlated with OI and GI community composition, but their bacterial diversity was correlated with all soil properties, except for soil acidity (Table S2). Soil pH, org. C, TN, MBC, MBN and MBP significantly altered the composition and alpha-diversity of bacterial communities on the Mantel test (Table S2). Redundancy analysis (RDA) was further used to elucidate the relation between bacterial composition and soil properties. The bacterial composition was positively associated with soil properties for OI and GI soils (Fig. 5). The distribution of some of the most abundant taxa, Acidobacteria, Actinobacteria, Alphaproteobacteria and Gammapreoteobacteria, was negatively correlated with these soil characteristics, except MBP. The distribution of other phylogenetic groups, including Betaproteobacteria, Deltaproteobacteria and Verrucomicrobia, was positively correlated with soil pH, org. C, TN, MBC and MBN. In addition, on combining the replicates of each site for RDA, the results further confirmed the different clusters between MI, OI and GI soils and the correlations between communities and soil properties (Fig. S2).

Redundancy analysis of bacterial communities in all offshore islands. Site abbreviations are in Table 1. Abbreviations in figure: Org. C, organic carbon; TN, total nitrogen; MBC, microbial biomass carbon; MBN, microbial biomass nitrogen; MBP, microbial biomass phosphorus; Acido: Acidobacteria; Actino, Actinobacteria; α-Proteo, Alphaproteobacteria; β-Proteo, Betaproteobacteria; γ-Proteo, Gammaproteobacteria; δ-Proteo, Deltaproteobacteria; Ver, Verrucomicrobia.
Discussion
The soil bacterial communities of offshore, isolated islands examined in this study were all dominated by Acidobacteria and Proteobacteria, but the relative abundance differed. The bacterial diversity also varied among MI, OI and GI soil communities. OI and GI have higher annual temperature and precipitation than MIs. Higher temperature and precipitation could shape the bacterial communities20 and result in greater bacterial diversity in tropical soils. In addition, higher org. C and total nitrogen content in tropical island soils could provide sufficient nutrient availability to affect bacterial community composition21.
The soil parent material could be another key factor affecting soil bacterial communities among MI, OI and GI soils. Soil parent material could provide a basic nutritional environment for the development of microbial communities22. One study showed that soil bacterial structure is determined by the nature of the parent material, and the community still maintained characteristics of the original populations even after a long time23. The soil mineralogy of soils derived from different parent materials, granite and andesite, is distinct and strongly controls the soil C processes in temperate forest soils24. It also affects the accumulation of soil microbially derived residues25, which suggests that soil microbial communities differ under different parent materials. A study found variable reactions of different bacterial phylotypes, and operational taxonomic units (OTUs) affiliated with Acidobacteria, Actinobacteria, and Alpha- and Betaproteobacteria mainly responded to granite soils26. Moreover, the soils had more neutral pH values on OI and GI than MI. The increased abundance of Acidobacteria in MI soils and the negative correlation between Acidobacteria distribution and soil pH based on RDA agree with previous studies27,28. These results support the importance of soil pH affecting soil communities across spatial scales5 and in local regions. The soil community variability among these offshore islands could have resulted from the quite different soil parent materials and were determined from the early stage of soil formation. The non-random distribution in soil bacteria could reflect the correlation with key environmental variables across different spatial scales29,30.
The variation in bacterial communities among these island soils could also reveal the effect of geographic distance. The distance from MI to OI and GI is >400 km. Some studies have revealed a significant effect of distance at intermediate scales of 10 to 3,000 km on soil community composition31,32, and environmental differences also had effects on the communities at this scale31. In the present study, the geographic distances among island soils were at the intermediate scale, so both environmental differences and geographic distances could alter the soil communities. In addition, the same cluster formed by the communities of MI indicated that the local environmental properties could be more essential factors affecting bacterial communities.
Acidobacteria Gp6 was the most abundant genus-level group of Acidobacteria, followed by Gp1, 2, 3 and 4. The relative abundance of Acidobacteria Gp4 and 6 was higher in OI and GI communities, whereas that of Acidobacteria Gp1, 2, 3 was higher in MI soils. The abundance of Acidobacteria Gp1, 2 and 3 is negatively related to soil pH, and that of Acidobacteria Gp4 and 6 is positively related to soil pH33. The differences in abundance among these Acidobacteria subdivisions could reflect the responses of community composition to the soil properties. It also indicates the regulation of soil pH for this phylum and its individual subdivisions.
Alphaproteobacteria was the most abundant class of the Proteobacteria in these island soils, although relative abundances differed among communities. Many sequences are affiliated with the genus Bradyrhizobium, order Rhizobiales, which include species that could perform nitrogen fixation, organic matter decomposition, and plant growth promotion34,35. This group also includes a number of nonsymbiotic bacteria with varied biochemical functions such as denitrification36 and photosynthesis37. Their versatile metabolism ability suggests their importance in nutrient availability to the soil ecosystems, especially in GI communities. Among Betaproteobacteria, the genus Burkholderia was abundant and is found in rhizosphere soil communities that perform nitrogen fixation and plant growth promotion38. On the basis of their physiological characteristics, they could be involved in parts of the carbon and nitrogen biogeochemical cycles in these offshore island soil ecosystems. In addition, Burkholderia was more abundant in MI than OI and GI soil communities, perhaps because MI soils have low nitrogen content and the genus is able to fix nitrogen under these conditions.
The relative abundance of Verrucomicrobia in OI soils was especially high among these communities. This phylum has a wide distribution across a range of biomes39. A recent study indicated that their distribution was significantly affected by soil pH, total carbon and nitrogen, and carbon/nitrogen ratio40. The high soil pH and total carbon and nitrogen content in OI and GI soil may explain why Verrucomicrobia was abundant on these islands and not MI. Moreover, this phylum includes members involved in polysaccharide degradation41 and nitrogen fixation42, so they may have essential roles in the tropical soil ecosystem. It was also an abundant phylum in the soil community analyzed with the clone library method15. Members of this phylum could be further isolated to elucidate their roles in soils of these tropical islands.
The genus Gemmatimonas of Gemmatimonadetes was one of the abundant genera in offshore island soil communities. This group comprises about 2.2% of soil communities analyzed by high-throughput sequencing43. Gemmatimonas was relatively more abundant in MI than OI and GI soil communities. The finding could reflect the adaption to low soil moisture of this group43 because of the reduced annual precipitation in MI. Moreover, an obligate aerobic Gemmatimonas strain had a unique regulatory mechanism of N2O reduction44, which indicates the potential role of this genus in nitrogen cycling in this ecosystem, especially in MI soils.
In conclusion, bacterial diversity was higher in tropical andesite than subtropical granite island soil communities. The major phyla were similar, but their relative abundance among communities differed. The subtropical granite and tropical andesite island soil communities formed different clusters and had distinct relations with soil properties, including soil pH, org. C, TN, MBC, MBN, and MBP. These results suggest that different parent material and geographic distance affected soil properties and altered the bacterial composition and diversity among these offshore island soils. In the local region, the environmental properties could be the more essential factors affecting bacterial communities, whereas environmental differences and geographic distances could both alter the soil communities across a large scale.
Materials and Methods
Site description and soil sampling
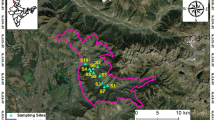
This study was conducted on offshore islands, including MI of subtropical granite and OI and GI of tropical andesite (Fig. 6). The map was prepared on the basis of State Information System database, Taiwan (https://ngis.wra.gov.tw/NgisGIS/). MIs are in the East China Sea and located 10–50 km off southeastern China, about 210 km northwest of Taiwan, facing the Taiwan Strait. Five islands in this region were used for sampling: Beigan (BG) (26°22′N, 119°99′E), Nangan (NG) (26°15′N, 119°93′E), Dongju (DJ) (25°95′N, 119°97′E), Shiju (SJ) (25°97′N, 119°94′E), and DY (26°36′N, 120°49′E) islands. BG and NG, DJ and SJ are close to one another; DY is more distant from other four islands. These islands have a subtropical marine climate with mean precipitation about 1,000 mm and annual average temperature 18.6 °C. The soil parent material is granite and the environments have remained little disturbed since broadleaved forests were established about 5 decades ago. OI (22°01′N, 121°34′E) is in the Pacific Ocean and located at about 60 km off the southeastern coast of Taiwan and facing the Pacific Ocean. It is a tropical island with mean precipitation >3000 mm and annual average temperature 22.6 °C. The vegetation is natural and consists of a little-disturbed secondary broadleaved forest. GI (22°39 N, 121°29) is also in the Pacific Ocean and located about 30 km off the eastern coast of Taiwan, facing the Pacific Ocean. A secondary broadleaved forest is the main vegetation. The mean temperature is 23.5 °C and mean precipitation about 2,500 mm. The lands chosen as sampling locations for this study are owned by the Forestry Bureau and accessible to the public. Research proposals were approved and granted by the Ministry of Science and Technology, Taiwan. Field and laboratory studies did not involve any animal husbandry, nor any protected or endangered biological species. The surface soil (0–10 cm) of each island was sampled in four replicates (eight replicates for OI because of a much larger area) of 50 × 50-m plots established 50-m apart along transect lines. After removing the surface litter, three subsamples at each plot were collected by using a soil auger, 8 cm in diameter and 10 cm deep, and pooled. The soil samples were sieved with a 2-mm sieve to remove the debris of litter and gravel and one part was kept at 4 °C before biochemical and molecular assays. Other samples were freeze-dried and stored at −20 °C for DNA extraction.

Sampling sites at offshore islands.
Soil analysis
Soil pH was determined in a 1:1 soil/water slurry by using a portable Jenco 6009 pH/mV meter45. Soil org. C and TN were examined by the combustion method with an NCS elemental analyzer (Model NA1500 Fisons, Italy). Soil MBC and MBN were assayed by using the chloroform fumigation extraction method as described46. In brief, soil samples were equally separated into two sets. One set was fumigated with chloroform for 24 hr and extracted with 0.5 M potassium sulphate (K2SO4). The other set was directly extracted with 0.5 M K2SO4. MBC and MBN were calculated as the differences between the fumigated and unfumigated K2SO4-extractable C and N by multiplying coefficients (KEC = 2.22 and KEN = 4.95) [(fumigated C (N) − unfumigated C (N)) × coefficients]47,48.
DNA extraction, PCR amplification, and high-throughput sequencing
Total soil DNA was extracted from 0.3 g soil samples with the PowerSoil DNA extraction kit (MoBio Laboratories, Carlsbad, CA) as directed by the manufacturer’s instructions. PCR amplification was performed according to the Illumina 16S Metagenomic Sequencing Library preparation guide (Illumina) with slight modification, targeting the V4 region (primer 515f and 806r) of the bacterial 16S rRNA gene with index sequences and adapters. PCR involved initial denaturation at 95 °C for 2 min, followed by 30 cycles of denaturation at 95 °C for 20 s, annealing at 55 °C for 15 s, elongation at 72 °C for 5 min, and a final elongation at 72 °C for 10 min. Paired-end sequencing of the PCR amplicons involved the MiSeq 250-bp paired sequencing system (Illumina) according to the manufacturer’s instructions.
Sequence data analyses
Illumina sequencing data were pair-assembled using PEAR49. The data were then processed with the Ribosomal Data Project (RDP) pipeline (http://pyro.cme.msu.edu; RDP release 11.5; release date: 2016.09.30). The sequences that did not have Ns and were >200 bp long, were free of chimeras by using Uchime50, and had quality scores >25 were used for further analysis. Taxonomic information was analyzed by using the Naïve Bayesian rRNA classifier51 of the RDP pipeline with a confidence cutoff of 80%. Data at the genus level were also examined and the Miseq 250-bp paired sequencing system could be used to analyze communities at this level with high accuracy52. The RDP pipeline was also used to calculate the Shannon diversity index53 based on the Complete Linkage Clustering data for OTUs with an evolutionary distance of 0.03. To avoid measurement differences for estimating the alpha and beta diversity from sample size, the sequences for each replicate were normalized to 21,000 sequences with random selection from the larger samples for community comparisons. K-shuff analysis was used for community comparisons54. NMDS community comparisons and relations between bacterial composition, diversity and soil properties were assessed by using PRIMER v655. RDA involved the vegan package in R v.3.2.1 to determine the relation between bacterial composition and soil properties. All sequences were deposited in MG-RAST (Metagenome Rapid Annotation using Subsystems Technology)56 with accession numbers 4812289.3, 4812290.3 and 4812291.3.
References
Bardgett, R. D., Freeman, C. & Ostle, N. J. Microbial contributions to climate change through carbon cycle feedbacks. The ISME journal 2, 805–814, https://doi.org/10.1038/ismej.2008.58 (2008).
Burton, J., Chen, C., Xu, Z. & Ghadiri, H. Soil microbial biomass, activity and community composition in adjacent native and plantation forests of subtropical Australia. Journal of Soils and Sediments 10, 1267–1277, https://doi.org/10.1007/s11368-010-0238-y (2010).
Chatterjee, A., Vance, G. F., Pendall, E. & Stahl, P. D. Timber harvesting alters soil carbon mineralization and microbial community structure in coniferous forests. Soil Biology and Biochemistry 40, 1901–1907, https://doi.org/10.1016/j.soilbio.2008.03.018 (2008).
Ushio, M., Wagai, R., Balser, T. C. & Kitayama, K. Variations in the soil microbial community composition of a tropical montane forest ecosystem: Does tree species matter? Soil Biology and Biochemistry 40, 2699–2702, https://doi.org/10.1016/j.soilbio.2008.06.023 (2008).
Lauber, C. L., Hamady, M., Knight, R. & Fierer, N. Pyrosequencing-based assessment of soil pH as a predictor of soil bacterial community structure at the continental scale. Applied and Environmental Microbiology 75, 5111–5120, https://doi.org/10.1128/aem.00335-09 (2009).
Oh, Y. M. et al. Distinctive bacterial communities in the rhizoplane of four tropical tree species. Microbial Ecology 64, 1018–1027, https://doi.org/10.1007/s00248-012-0082-2 (2012).
Quideau, S. A., Chadwick, O. A., Benesi, A., Graham, R. C. & Anderson, M. A. A direct link between forest vegetation type and soil organic matter composition. Geoderma 104, 41–60, https://doi.org/10.1016/S0016-7061(01)00055-6 (2001).
Grayston, S. J. & Prescott, C. E. Microbial communities in forest floors under four tree species in coastal British Columbia. Soil Biology and Biochemistry 37, 1157–1167, https://doi.org/10.1016/j.soilbio.2004.11.014 (2005).
Xu, Z. et al. Soil carbon and nutrient pools, microbial properties and gross nitrogen transformations in adjacent natural forest and hoop pine plantations of subtropical Australia. Journal of Soils and Sediments 8, 99–105, https://doi.org/10.1065/jss2008.02.276 (2008).
Sowerby, A. et al. Microbial community changes in heathland soil communities along a geographical gradient: interaction with climate change manipulations. Soil Biology and Biochemistry 37, 1805–1813, https://doi.org/10.1016/j.soilbio.2005.02.023 (2005).
Nielsen, U. N., Osler, G. H. R., Campbell, C. D., Burslem, D. F. R. P. & van der Wal, R. The influence of vegetation type, soil properties and precipitation on the composition of soil mite and microbial communities at the landscape scale. Journal of Biogeography 37, 1317–1328, https://doi.org/10.1111/j.1365-2699.2010.02281.x (2010).
Martiny, J. B. H. et al. Microbial biogeography: putting microorganisms on the map. Nature Reviews Microbiology 4, 102–112, https://doi.org/10.1038/nrmicro1341 (2006).
Fierer, N. & Jackson, R. B. The diversity and biogeography of soil bacterial communities. Proc. Natl. Acad. Sci. USA 103, 626–631, https://doi.org/10.1073/pnas.0507535103 (2006).
Green, J. & Bohannan, B. J. M. Spatial scaling of microbial biodiversity. Trends in Ecology & Evolution 21, 501–507, https://doi.org/10.1016/j.tree.2006.06.012 (2006).
Lin, Y.-T., Whitman, W. B., Coleman, D. C. & Chiu, C.-Y. Comparison of soil bacterial communities between coastal and inland forests in a subtropical area. Applied Soil Ecology 60, 49–55, https://doi.org/10.1016/j.apsoil.2012.03.001 (2012).
Shih, H. T., Chen, G. X. & Wang, L. M. A new species of freshwater crab (Decapoda: Brachyura: Potamidae) from Dongyin Island, Matsu, Taiwan, defined by morphological and molecular characters, with notes on its biogeography. Journal of Natural History 39, 2901–2911, https://doi.org/10.1080/00222930500214010 (2005).
Shen, H. P., Chang, C. H. & Chih, W. J. Earthworms from Matsu, Taiwan with descriptions of new species of the genera Amynthas (Oligochaeta: Megascolecidae) and Drawida (Oligochaeta: Moniligastridae). Zootaxa 3973, 425–450, https://doi.org/10.11646/zootaxa.3973.3.2 (2015).
Ho, C.-C., Shih, H.-T. & Chen, W.-H. Eight phytoseiid mites from the Matsu Islands. Plant Protection Bulletin 45, 143–154, https://doi.org/10.6342/NTU.2011.01873 (2003).
Soil Survey Staff. Keys to soil taxonomy (12th ed.). Washington, DC: USDA‐Natural Resources Conservation Service (2014).
Romanowicz, K. J., Freedman, Z. B., Upchurch, R. A., Argiroff, W. A. & Zak, D. R. Active microorganisms in forest soils differ from the total community yet are shaped by the same environmental factors: the influence of pH and soil moisture. FEMS Microbiology Ecology 92, fiw149, https://doi.org/10.1093/femsec/fiw149 (2016).
Hofmann, K., Lamprecht, A., Pauli, H. & Illmer, P. Distribution of prokaryotic abundance and microbial nutrient cycling across a high-alpine altitudinal gradient in the Austrian Central Alps is affected by vegetation, temperature, and soil nutrients. Microbial Ecology 72, 704–716, https://doi.org/10.1007/s00248-016-0803-z (2016).
Ulrich, A. & Becker, R. Soil parent material is a key determinant of the bacterial community structure in arable soils. FEMS Microbiology Ecology 56, 430–443, https://doi.org/10.1111/j.1574-6941.2006.00085.x (2006).
Sheng, R. et al. Bacterial succession in paddy soils derived from different parent materials. Journal of Soils and Sediments 15, 982–992, https://doi.org/10.1007/s11368-014-1058-2 (2015).
Rasmussen, C., Southard, R. J. & Horwath, W. R. Mineral control of organic carbon mineralization in a range of temperate conifer forest soils. Global Change Biology 12, 834–847, https://doi.org/10.1111/j.1365-2486.2006.01132.x (2006).
Ding, X., Zhang, B., Lü, X., Wang, J. & Horwath, W. R. Parent material and conifer biome influence microbial residue accumulation in forest soils. Soil Biology and Biochemistry 107, 1–9, https://doi.org/10.1016/j.soilbio.2016.12.020 (2017).
Zumsteg, A., Bååth, E., Stierli, B., Zeyer, J. & Frey, B. Bacterial and fungal community responses to reciprocal soil transfer along a temperature and soil moisture gradient in a glacier forefield. Soil Biology and Biochemistry 61, 121–132, https://doi.org/10.1016/j.soilbio.2013.02.017 (2013).
Xiong, J. et al. Geographic distance and pH drive bacterial distribution in alkaline lake sediments across Tibetan Plateau. Environmental Microbiology 14, 2457–2466, https://doi.org/10.1111/j.1462-2920.2012.02799.x (2012).
Chu, H. et al. Soil bacterial diversity in the Arctic is not fundamentally different from that found in other biomes. Environmental Microbiology 12, 2998–3006, https://doi.org/10.1111/j.1462-2920.2010.02277.x (2010).
Griffiths, R. I. et al. The bacterial biogeography of British soils. Environmental Microbiology 13, 1642–1654, https://doi.org/10.1111/j.1462-2920.2011.02480.x (2011).
Nemergut, D. R. et al. Global patterns in the biogeography of bacterial taxa. Environmental Microbiology 13, 135–144, https://doi.org/10.1111/j.1462-2920.2010.02315.x (2011).
Yannarell, A. C. & Triplett, E. W. Geographic and environmental sources of variation in lake bacterial community composition. Applied and Environmental Microbiology 71, 227–239, https://doi.org/10.1128/aem.71.1.227-239.2005 (2005).
Reche, I., Pulido-Villena, E., Morales-Baquero, R. & Casamayor, E. O. Does ecosystem size determine aquatic bacterial richness? Ecology 86, 1715–1722, https://doi.org/10.1890/04-1587 (2005).
Jones, R. T. et al. A comprehensive survey of soil acidobacterial diversity using pyrosequencing and clone library analyses. ISME J 3, 442–453, https://doi.org/10.1038/ismej.2008.127 (2009).
Zhang, L. & Xu, Z. Assessing bacterial diversity in soil. Journal of Soils and Sediments 8, 379–388, https://doi.org/10.1007/s11368-008-0043-z (2008).
Yarwood, S. A., Myrold, D. D. & Högberg, M. N. Termination of belowground C allocation by trees alters soil fungal and bacterial communities in a boreal forest. FEMS Microbiology Ecology 70, 151–162, https://doi.org/10.1111/j.1574-6941.2009.00733.x (2009).
Sameshima-Saito, R., Chiba, K. & Minamisawa, K. Correlation of denitrifying capability with the existence of nap, nir, nor and nos genes in diverse strains of soybean Bradyrhizobia. Microbes and Environments 21, 174–184, https://doi.org/10.1264/jsme2.21.174 (2006).
Giraud, E. et al. Legumes Symbioses: Absence of Nod genes in photosynthetic Bradyrhizobia. Science 316, 1307–1312, https://doi.org/10.1126/science.1139548 (2007).
Coenye, T. & Vandamme, P. Diversity and significance of Burkholderia species occupying diverse ecological niches. Environmental Microbiology 5, 719–729, https://doi.org/10.1046/j.1462-2920.2003.00471.x (2003).
Bergmann, G. T. et al. The under-recognized dominance of Verrucomicrobia in soil bacterial communities. Soil Biology and Biochemistry 43, 1450–1455, https://doi.org/10.1016/j.soilbio.2011.03.012 (2011).
Shen, C., Ge, Y., Yang, T. & Chu, H. Verrucomicrobial elevational distribution was strongly influenced by soil pH and carbon/nitrogen ratio. Journal of Soils and Sediments 17, 2449–2456, https://doi.org/10.1007/s11368-017-1680-x (2017).
Martinez-Garcia, M. et al. Capturing single cell genomes of active polysaccharide degraders: an unexpected contribution of Verrucomicrobia. PLOS ONE 7, e35314, https://doi.org/10.1371/journal.pone.0035314 (2012).
Khadem, A. F., Pol, A., Jetten, M. S. M. & Op den Camp, H. J. M. Nitrogen fixation by the verrucomicrobial methanotroph ‘Methylacidiphilum fumariolicum’ SolV. Microbiology 156, 1052–1059, https://doi.org/10.1099/mic.0.036061-0 (2010).
DeBruyn, J. M., Nixon, L. T., Fawaz, M. N., Johnson, A. M. & Radosevich, M. Global biogeography and quantitative seasonal dynamics of Gemmatimonadetes in soil. Applied and Environmental Microbiology 77, 629–6300, https://doi.org/10.1128/AEM.05005-11 (2011).
Park, D., Kim, H. & Yoon, S. Nitrous oxide reduction by an obligate aerobic bacterium, Gemmatimonas aurantiaca strain T‐27. Applied and Environmental Microbiology 83, e00502–e00517, https://doi.org/10.1128/aem.00502-17 (2017).
McLean, E. O. Soil pH and lime requirement. Page, A. L. (Ed.), Methods of Soil Analysis, Part 2 Chemical and Microbiological Properties, ASA, Madison, WI., pp. 199–224 (1982).
Witt, C., Gaunt, J. L., Galicia, C. C., Ottow, J. C. G. & Neue, H.-U. A rapid chloroform-fumigation extraction method for measuring soil microbial biomass carbon and nitrogen in flooded rice soils. Biology and Fertility of Soils 30, 510–519, https://doi.org/10.1007/s003740050030 (2000).
Inubushi, K., Brookes, P. C. & Jenkinson, D. S. Soil microbial biomass C, N and ninhydrin-N in aerobic and anaerobic soils measured by the fumigation-extraction method. Soil Biology &. Biochemistry 23, 737–741, https://doi.org/10.1016/0038-0717(91)90143-8 (1991).
Wu, J., Joergensen, R. G., Pommerening, B., Chaussod, R. & Brookes, P. C. Measurement of soil microbial biomass C by fumigation extraction - An automated procedure. Soil Biology &. Biochemistry 22, 1167–1169, https://doi.org/10.1016/0038-0717(90)90046-3 (1990).
Zhang, J., Kobert, K., Flouri, T. & Stamatakis, A. PEAR: a fast and accurate Illumina Paired-End reAd mergeR. Bioinformatics 30, 614–620, https://doi.org/10.1093/bioinformatics/btt593 (2014).
Edgar, R. C., Haas, B. J., Clemente, J. C., Quince, C. & Knight, R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27, 2194–2200, https://doi.org/10.1093/bioinformatics/btr381 (2011).
Wang, Q., Garrity, G. M., Tiedje, J. M. & Cole, J. R. Naïve Bayesian Classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Applied and Environmental Microbiology 73, 5261–5267, https://doi.org/10.1128/aem.00062-07 (2007).
Jeon, Y. S., Park, S. C., Lim, J., Chun, J. & Kim, B. S. Improved pipeline for reducing erroneous identification by 16S rRNA sequences using the Illumina MiSeq platform. Journal of Microbiology 53, 60–69, https://doi.org/10.1007/s12275-015-4601-y (2015).
Shannon, C. E. & Weaver, W. The mathematical theory of communication. University of Illinois Press, Urbana (1963).
Jangid, K. et al. K-shuff: A novel algorithm for characterizing structural and compositional diversity in gene libraries. PLOS ONE 11, e0167634, https://doi.org/10.1371/journal.pone.0167634 (2016).
Clarke, K. R. & Gorley, R. N. Primer v6: user manual/tutorials. Primer-E Ltd, Plymouth (2006).
Meyer, F. et al. The metagenomics RAST server- a public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC Bioinformatics 9, 386, https://doi.org/10.1186/1471-2105-9-386 (2008).
Acknowledgements
The authors thank the Ministry of Science and Technology of Taiwan, Republic of China, for financially supporting this research (MOST 105–2313-B-001-002 and MOST 106-2303-B-001-002). The authors are also grateful to Ms Pei-Yi Yu and Ms Yun-chien Hsu from the Biodiversity Research Center, Academia Sinica, Taipei, Taiwan, for soil analyses.
Author information
Authors and Affiliations
Contributions
Y.T.L. and Y.F.L. performed statistical analyses and built statistical models. I.J.T. and E.H.C. helped in analysing and interpreting data. S.H.J. helped conduct the soil survey and collect soil samples. Y.J.L. helped with data curation. C.Y.C. originally formulated the idea and developed the methodology. Y.T.L. wrote the original draft. I.J.T. and C.Y.C. edited and reviewed the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lin, YT., Lin, YF., Tsai, I.J. et al. Structure and Diversity of Soil Bacterial Communities in Offshore Islands. Sci Rep 9, 4689 (2019). https://doi.org/10.1038/s41598-019-41170-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-41170-9
This article is cited by
-
Responses of Nutrients and Bacterial Communities to Temperature and Nitrogen Addition in Rhizosphere Soil for Malus sieversii Seedlings
Journal of Soil Science and Plant Nutrition (2024)
-
Bacterial Communities and Diversity of Western Ghats Soil: A Study of a Biodiversity Hotspot
Current Microbiology (2023)
-
Metallophores production by bacteria isolated from heavy metal-contaminated soil and sediment at Lerma–Chapala Basin
Archives of Microbiology (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
