
Abstract
The trade of bumble bees started in the early nineties for pollinator-dependent greenhouse plants. Nowadays, its rearing and transport have received public attention, since managed bees can transfer pathogens to wild bee populations. Therefore, guaranteeing pathogen-free bumble bees is fundamental. The major protein source used in rearing facilities is honey bee-collected pollen. This can carry pathogens, however to date, solid data on the risk of this food source to the health of bumble bees is lacking. Here we performed a large pathogen screening of non-irradiated honey bee-collected pollen to discover particles infective to Bombus terrestris. We identified seven parasites (Apicystis bombi, Ascosphaera apis, Crithidia mellificae, Nosema ceranae, Paenibacillus larvae and two parasites resembling Nosema thomsoni and Microsporidium sp. Oise) and four viruses (CBPV, DWV, IAPV and SBV) in 17 pollen batches from two major European pollen source regions (Spain and Romania). Ascosphaera apis was capable of infecting bumble bees; the larvae showed similar symptoms to chalkbrood disease reported in honey bees. Bumble bee breeding facilities need to be cautious about the potential presence of this disease, which was originally reported in honey bees. Thorough diagnostic and control methods are needed, as risk of spillover to wild bee species is possible.
Similar content being viewed by others

Introduction
Bee domestication began with the honey bee, attractive for its production capacities of honey, wax and propolis. Nowadays, various bee species are managed, and their pollination services are exploited to form a ‘new’ pollination industry1. A good example is bumble bee management and trade, which started in the early nineties within enclosed facilities. The bumble bee market was initially focused on providing pollination services in greenhouses and has expanded to open-field pollination of bumble bee-visited crops2. Currently, the international bumble bee market is still expanding2,3, with pollination activities in more than sixty countries.
The health status of managed bumble bees is crucial in international trade. First, several viral and non-viral pathogens can reduce fitness, resulting in shorter life span, reduced learning ability4,5,6 and likely reduced pollination efficiency5. Second, commercial bees may act as a reservoir of pathogens and can be responsible for the transmission of pathogens from commercial bees to wild pollinators7,8,9. Such transmission is a specific case of pathogen spillover, more generally defined as a transmission driven by a species acting as a pathogen reservoir. Bee transport can facilitate the presence of such reservoirs influencing natural host-parasite dynamics, and therefore influence host populations within natural ecosystems10. With regard to spillover, it has been argued that it can have devastating effects on endemic bee fauna7,11. Furthermore, other stressors harm bee populations (e.g. land-use and pesticide), which can act in synergy with parasite stress12. For example, Nosema bombi prevalence in declining bumble bees (Bombus occidentalis, B. pensylvanicus, B. affinis and B. terricola) in North America is correlated with fungicide presence12.
Pathogen-free bumble bee management is essential in order to limit spillover dynamics or introductions of new parasitic species or genetic strains. Although the rearing of bumble bees occurs indoors with no direct contact with the outdoor environment, there are still two main routes of pathogen influx into the breeding facilities. The first potential transmission route is the bumble bee itself, as a certain genetic stock is needed to reach the desired production capacities. Queens first undergo quarantine measures and new offspring can be tested to obtain pathogen-free nests and daughter queens. Thus, pathogen-free facilities are a matter of thorough diagnostics and time/cost investment efforts. A second potential influx route is the honey bee-collected pollen (pollen pellets on the corbicula of honey bees)5,13,14, which is used as the main protein source of the bumble bee’s diet. A recent study on two pollen batches showed that honey bee-collected pollen was a probable source of infective Apicystis bombi oocysts5. This is surprising since A. bombi was mainly known as a bumble bee parasite. Then again, very little is known about the natural host range of many bee parasites14 and the occurrence in honey bees had been reported sporadically15. These results led us to speculate that honey bee-collected pollen could serve as potential source for other parasites and viruses. Indeed, many pathogens originally reported in honey bees were later also found in bumble bees in Europe and North America4,6,16,17,18,19.
Sanitary control measures on honey bee-collected pollen are an important aspect of good bumble bee rearing practices. Yet no thorough prevalence study of parasites and viruses in honey bee-collected pollen for commercial purpose has been performed to date. The aim of this study was to perform screening for wide variety of parasites and viruses on pollen used for bumble bee rearing (N = 17 different pollen batches). The use of broad-range PCR diagnostics, where possible, allowed us to screen for potential undiscovered threats. Finally, we performed a realistic oral infection test on a subset of parasite contaminated pollen batches to investigate the infectivity risk.
Results
Pathogen diversity in honey bee-collected pollen
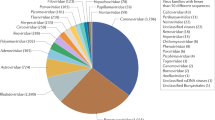
We screened a total of 17 different pollen batches for the presence of parasites and viruses; the batches originated from 4 different suppliers located in Romania and Spain. In total, 7 parasites and 4 viruses were detected (see Fig. 1). The most prevalent pathogen within all honeybee-collected pollen samples was Crithidia spp. (70.6%), Sacbrood virus (SBV) (58.8%), A. bombi (52.9%) and Ascosphaera apis (47.1%) followed by the Microsporidia Nosema ceranae (23.5%), Nosema thomsoni (17.6%), Microsporidium sp. Oise (11.7%) and the viruses Deformed wing virus (DWV) (11.7%) and Israeli acute paralysis virus (IAPV) and Chronic bee paralysis virus (CBPV) with 5.8%. Sequences of N. ceranae, N. thomsoni and Microsporidium sp. Oise had 99% identity with sequences from GenBank accession numbers AB860144.1, EU219086.1 and HM566197.1, respectively. All the parasites were retrieved in both regions (Romania and Spain); while Paenibacillus larvae was found only in one pollen batch from Romania. Furthermore, a minimum of 1 parasite and a maximum of 4 parasites was found per pollen batch tested. Concerning the viruses, SBV was highly present in the batches from Romania and this exclusively, while CBPV, IAPV and DWV were found only in suppliers from Spain (Fig. 1). Nosema apis, N. bombi and the viruses Kashmir bee virus (KBV), Acute bee paralysis virus (ABPV) and Black queen cell virus (BQCV) were not detected in any pollen sample.

The cumulative prevalence in percentage of all the parasites retrieved in 17 pollen batches from Spain and Romania. Bars in yellow indicate pathogens identified in samples from Spain and in blue, the ones from Romania.
Infection risk of pollen to larvae and adults of bumble bee
In total we tested 7 different parasites A. bombi, A. apis, Crithidia spp., N. ceranae, N. thomsoni, Microsporidium sp. Oise and P. larvae in the infection assay. We only detected an active infection for A. apis. The known honey bee parasite A. apis was particularly prominent in the gut of bumble bee larvae. It was detected in three out of the four microcolonies that were supplied with pollen containing the parasite (Table 1). The other parasites A. bombi, Crithidia spp., N. ceranae, N. thomsoni, Microsporidium sp. Oise and P. larvae were not detected in any bumble bee tissue, although they were present in the pollen.
Infectivity of A. apis in bumble bee larvae
Microscopic identification and morphological description
We detected larvae presenting symptoms still inside the cocoon in three out of the four microcolonies supplied with pollen with A. apis. One of these dead larvae was found in one contaminated colony that was negative for the infection test with living larvae. Two larvae were blackened and mummified showing common symptoms of chalkbrood disease as described within the host Apis20,21(Fig. 2) and one infected exhibiting an unusual symptom, distinctly creamy in appearance (Fig. 2C,F). In addition, we detected vegetative and reproductive stages of Ascosphaera fungi in these larvae (Figs 2 and 3). The measurements of the fungus were equivalent to this species22,23,24 presenting spore cysts with a diameter of 39.3–52.0 μm (average = 44.9 ± 3.8 μm), a spore ball with 12 μm in diameter and ascospores with lengths of 1.9–3.4 μm (average = 2.6 ± 0.3 μm).

Different health conditions of Bombus terrestris larvae at 4th instar. (A) Healthy bumble bee larva. (B–F) Bumble bee larvae showing symptoms of chalkbrood disease after ingestion of honey bee-collected pollen with Ascosphaera apis. (B) Unremoved larva contaminated with A. apis in closed cocoon by co-worker in colony. (F) A close up of an opened creamy larva infected with A. apis. Scale bars = 50 mm.

Stages of Ascosphaera apis infecting bumble bee larvae. (A) Close-up of an opened spore cyst. (B) A spore ball when released from a cleistotheca. (C) Bacilliform ascospores. Scale bars = 10 µm.
Phylogenetic analysis
All ITS sequences of Ascosphaera fungi found in honey bee-collected pollen, alive larvae, and symptomatic dead larvae matched 100% identity (336–345 bp fragments length) with ITS sequences from voucher strains of A. apis from Genbank database (NCBI). The phylogenetic analysis placed all sequences of A. apis found in pollen and larvae from this study together with existed database sequences (Fig. 4).

Phylogeny of the internal transcribed spacer region for selected Ascosphaera species. ITS sequences from this study were concatenated to strains of A. apis (ATCC MYA-4450, Genbank accession #FJ172292; ATCC MYA-4451, Genbank accession #FJ172293) together to another ITS from the genus Ascosphaera, Genbank accession #U68313.1 (see Anderson et al.22). Phylogenetic analysis performed using the maximum likelihood algorithm. Analyses was executed using 1000 bootstrap replicates.
Ascosphaera apis quantification
The load of A. apis was significantly different among all the sample groups (honey bee-collected pollen, living larvae and dead larvae manifesting symptoms). The number of genome copies of the fungus was higher in dead larvae exhibiting symptoms compared to living larvae and pollen samples (P < 0.0001, Fig. 5).

Specific qPCR measurement of 18S rRNA gene from Ascosphaera apis. Bars indicate the relative quantity (normalized to 1) of Ascosphaera apis transcripts in honey bee-collected pollen, alive larvae and symptomatic dead larvae. Error bars denote ± s.e.m. from six (n = 6), nine (n = 9) and three (n = 3) independent samples, respectively. Error flags are SE and the size are in accordance to the different scales of the y-axis. Statistical significance calculated using the independent samples t-test, ***P-value < 0.001.
Discussion
The presence of pathogens in bumble bee breeding facilities receives attention mainly because of the potential disease transmission risk towards wild bees3,5,8,14,25. Although a main aspect, only a few studies looked at the pathogen influx routes into these sites. Honey bee-collected pollen can be identified as a potential introduction route of pathogens. However, to date, little knowledge is present to understand how substantial this problem is. This understanding is not only important in terms of regulation, for instance on the use of unsterilized pollen and its origin, but also provides valuable information for the sector in terms of sanitary controls within the facility. In this study, we found that each of the 17 pollen batches contained at least one parasite or virus (at max. 5). This highlights a widespread potential problem for bee breeding facilities. It is essential to assess the impact of each of these pathogens on bumble bees and more importantly assess the impact of pathogens able to infect multiple hosts (e.g. honey bees and bumble bees).
The clearest result is the infection of A. apis in the larvae of Bombus terrestris. ITS sequencing revealed that the Ascosphaera fungi detected in honey bee-collected pollen and in infected bumble bee larvae were identical with each other and with the A. apis sequences (#FJ172292, #FJ172293 and #U68313) deposited at GenBank. We did not only identify the presence of this parasite, but we could also demonstrate infection. We detected chalkbrood-like symptoms in A. apis positive larvae, which also contained higher parasite load. The massive growth of A. apis and the occurrence of symptoms seems to be causal, but it can also be triggered by co-occurring parasites. Since we only have three observations of symptoms, we cannot perform co-occurrence analysis and infer such exact causality. These results are important since the use of many reared bumble bee species in biological pollination worldwide poses a risk to wild pollinators if parasites are shared. For this reason, it is important to know the host preference and virulence of A. apis. To date, the established infection based on symptoms, fungus sporulation or via microscopy of this fungus, refers to larvae of the European honey bee, Apis mellifera26, the Asian honey bee, Apis cerana27, the carpenter bee Xylocopa californica arizonensis Cresson28 and Xylocopa augusti21. There is no definitive evidence that A. apis can infect wild bumble bee larvae, and anecdotal reports of the parasite are mainly associated with the rearing of bumble bees in artificial conditions. For instance, Ascosphaera spp. (no species identification) was isolated from a dead bumble bee larva reared in the laboratory29, where the authors speculated that honey bee-collected pollen could be the infection source. Recently, isolates of A. apis were detected in wild North American bumble bee queens of Bombus griseocollis, B. nevadensis and B. vosnesenskii24, again after bringing them into the lab, and thus there was no identification if the captured queens already had the infection or were infected by feeding on contaminated pollen. PCR diagnostics have identified Ascosphaera in many bee species, and so it was concluded that the fungus was very common in the environment of pollinators, yet mainly in the context of being vectored by non-host pollinators17. Here, we show a true infection in managed B. terrestris. We noticed sign of chalkbrood in A. apis-infected bumble bees which is a common symptom that was originally reported in the larvae of honey bees30. Bumble bee breeders should be aware of this fact, and chalkbrood symptomology and the presence of this parasite must be verified through implemented sanitary measures. It is important to highlight that the diseased larvae were not thrown out of their brood cell, and thus they were only spotted after opening the brood cell manually. Therefore, the eradication of this parasite from breeding facilities is important to prevent spillover toward wild bumble bees, as the managed bees could act as a parasite reservoir for A. apis.
We identified another pathogen namely P. larvae, that is typically associated with honey bees, but that is not known to infect bumble bees. This pathogen might represent a threat since it is likely to cause infection in breeding facilities. Thus, its presence in pollen samples is still alarming, and we consider the introduction of P. larvae as a potential danger. This bacterium is the etiological agent of the American Foulbrood (AFB), the most important disease of honey bees worldwide31. Due to its high virulence, honey bee hives need to be eradicated when diagnosed with this quarantine organism. We only had one positive sample to test potential transmission towards commercial bees through infection experiment and they remained negative. To the best of our knowledge, no wild bumble bees have been identified as harboring P. larvae. Yet, artificial infection with bacterial cultures is needed to perform a meaningful risk evaluation. We underline the importance of these common honey bee pathogens in the pollen for further specific study, as the breeding process of bumble bees with honey bee-collected pollen facilitates an artificial contact with a potential new host. The rearing process could facilitate spillover across species, which would be less likely to occur in natural conditions. Although possible, this latter point remains speculative.
The detection of microsporidium sequences related to Microsporidium sp. Oise and particularly N. thomsoni also presents an interesting finding. Nosema thomsoni is associated with moths32,33 and the ladybird Harmonia axyridis, and the parasite association with the latter is thought to threaten native ladybird species in Europe34. The 18S sequencing reveled matches (99%) with N. thomsoni from moths, with sequences within the N. thomsoni clade of Nosema species found in Asian bumble bees35, and with the sequence found in the solitary bee Andrena vaga in Belgium36 and recently in Andrena haemorrhoa37. We did not observe infectivity of any microsporidium or with N. ceranae-contaminated pollen. This could be attributed to a number of reasons: (1) the infectious potential can be lost due to the freezing process that the pollen undergoes for instance N. ceranae is known to be sensitive to cold temperature38, (2) the inoculation loads were not high enough, and/or (3) these parasites are not able to infect bumble bees. Nosema thomsoni can be regarded as a multihost parasite, and its presence in honey bee-collected pollen is more likely an environmental contamination of other pollinators visiting the flower before the pollen was collected by the honey bee. In this case, similar contact with this parasite also occurs in nature. Furthermore, the recent discovery of different Nosema species in bumble bees with use of molecular diagnostics cannot always be linked with true infections, as remarked by Brown39.
Based on the results, it is clear how problematic the influx of non-sterilized honey bee-collected pollen into facilities is, as the pollen from every supplier was contaminated with at least 3 pathogens. Although, many parasites of bees and insects found here did not establish infection, it does not suggest an invulnerability for those parasites. This first documentation of A. apis in B. terrestris larvae via feeding pollen is alarming and endorses the need of measures to reduce or prevent the entry of pathogens in commercial establishments. Pollen trade and transportation are therefore also important to be considered in the context of potential parasite spillover from commercial to wild bumble bees. We are aware that some bumble bee breeders advertise the use of gamma-irradiated pollen, which is a good practice. Nonetheless, to our knowledge, no legislation is existing yet on the sterility and sanitary control measures to import pollen or use it for insect breeding in general. We encourage further studies on sterilization measures and their efficacy for bee pathogens and potential trade-offs towards nutritional value.
Materials and Methods
Prevalence screening and diagnostics
Honey bee-collected pollen and Nucleic acids extraction
We assessed pollen intended to feed bumble bees in breeding facilities in Europe. In total 17 frozen pollen batches from four different suppliers in Spain and Romania were tested for viral and non-viral pathogens of bees. Sampling was carried out and obtained from May until July of 2016. Twenty pollen pellets from each pollen were pooled (approximately 0.3 g) in a 2 ml-microcentrifuge tube (Eppendorf). We added 800 µl of RLT buffer supplemented with β-mercapto ethanol (100/1; v/v) (RNeasy Mini Kit; Qiagen, Venlo, the Netherlands). After, the samples were homogenized for 2 min at 300 rpm and for 2 min at 200 rpm with 0.1 mm-diameter zirconia beads and two 5 mm diameter-metal beads in a Qiagen TissueLyser. The mixture was centrifuged for 2 min at 20000 g. For DNA extraction 200 µl of the supernatant was mixed with 400 µl of Lysis buffer G and 40 µl of Proteinase K, and incubated for 1 h at 52 °C with shaking (400 rpm). Further extraction was done according to the manufacturer’s protocol (Invisorb Spin Tissue Mini Kit, Protocol 1; Stratec, Berlin, Germany), DNA was stored at −20 °C until further use. RNA extractions were done starting with 200 µl of supernatant and 200 µl of 70% ethanol was added. Further extraction was done according to the manufacturer’s protocol (RNeasy Mini Kit; Qiagen), RNA was stored at −80 °C until further use.
cDNA synthesis
To screen for RNA viruses, RNA concentration and purity were determined using a NanoDrop ND-1000 spectrophotometer. The cDNA was synthesized using 500 ng of RNA and random hexamers and SuperScript II Reverse Transcriptase (Life Technologies; Merelbeke, Belgium) according to the manufacturer’s instructions. The cDNA was stored at 20 °C, until further use.
Parasite screening and molecular identification
PCR with broad range primers (if possible) was used to detect a wide variety of parasites: Protozoan parasites are screened with Neogregarine primers to detect A. bombi and Leishmaniinae primers to detect Crithidia spp. and Lotmaria passim (technical details are described in Meeus et al.40); microsporidia was screened with the universal primers of Fernández et al.41. For Nosema species identification, we designed external primers for nested PCR (Supplementary Table 1) and performed Sanger sequencing on the amplicon obtained after PCR with the internal primers of Weiss & Vossbrinck42. The Fungus Ascosphaera spp. was screened with genus-broad primers described by James & Skinner43 and the bacteria P. larvae with specific primers of Dobbelaere et al.44. Positive and negative controls were added in all PCR reactions except for P. larvae. The viruses IAPV, DWV, KBV and ABPV were checked by multiplex PCR as described in Meeus et al.45 and confirmed by simplex virus-specific primers as described in Sguazza et al.46. SBV, CBPV and BQCV were checked by simplex virus-specific primers as described in Sguazza et al.46.
PCR products were visualized on a 1% agarose gel by staining it in ethidium bromide and submitted to UV illumination for checking the DNA amplification. All positive pathogens retrieved on agarose gel were sent for sequencing (by LGC genomics, Berlin, Germany) in the forward direction to confirm the species. The BlastN against the nucleotide collection was performed to check the percentage match for all sequences. For known honey bee and bumble bee parasites we provide the species names, while for the others, we provide the tentative species names and the Genbank accession number of the matching sequence. The Leishmaniinae positive had very faint bands and the quality of the electropherograms was not high enough to guarantee optimal species differentiation. For primers and PCR conditions (see Supplementary Table 1).
Infection experiments
Experimental design
Seeing the high diversity of parasites found in the honey bee-collected pollen and knowing that often high viral particle dosages are needed to establish oral infection6,47, we decided to focus on the study of the infection potential of parasites. In order to check for the infection potential of parasites within honey bee-collected pollen, we used a subset of pollen batches used for the prevalence study described above. Batches containing various parasites were selected to increase parasite repeats within the common microcolonies, when possible. All parasites present in the pollen were used in the infection experiment. A pollen ball (approximately 30 g) was provided to micro-colonies (i.e. small colony of 5 newly emerged workers of which one female will become dominant and start laying eggs). Bumble bees were provided by the company Biobest (Westerlo, Belgium). In total, we tested 8 micro-colonies and 7 parasites. We had one micro-colony control with 15 kGy-radiated pollen; the other micro-colonies received pollen naturally contaminated with at least one of the following parasites A. bombi (n = 4 colonies), Crithidia spp. (n = 5), N. ceranae (n = 2), N. thomsoni (n = 1), Microsporidium sp. Oise (n = 2), P. larvae (n = 1) and A. apis (n = 4). Pollen balls were made by mixing pollen pellets with sugar syrup at 40% (w/v). All micro-colonies were kept with sugar syrup (Biogluc) ad libitum under standardized laboratory conditions at 30 °C and continuous darkness48.
Infection status of bumble bees feeding on parasite-contaminated pollen
The infection status of 2 adults and 3 larvae (4th stage) per colony was determined on fifteen and twenty days after colony initiation, respectively. Larvae and adults were dissected in order to obtain the gut and internal organs (mainly fat body). The sampling of body parts was to ensure that we could identify true infections for those parasites infecting inside the hemocoel. Each individual tissue was then put separately in 2 ml-Eppendorf tube with 800 µl of RLT buffer supplemented with β-mercapto ethanol (100/1; v/v) (RNeasy Mini Kit; Qiagen) and stored at −80 °C. Before extraction, samples were thawed in an incubator at 37 °C for 10 min with shaking (300 rpm). After incubation, samples were centrifuged during 2 min at 2000 g. The samples were homogenized for 2 min at 300 rpm and for 2 min at 200 rpm with 0.1 mm-diameter zirconia beads and two 5 mm-diameter metal beads in a Qiagen TissueLyser. The mixture was centrifuged for 2 min at 20000 g. DNA extraction on 200 µl of the supernatant and PCRs are performed as described previously for the pollen above.
Infectivity of Ascosphaera apis in bumble bee larvae
In order to check if A. apis was replicating in larvae, we checked them by microscopy and qPCR.
Microscopic identification and morphological measurements
Three larvae potentially manifesting symptoms from sealed cocoons were screened for vegetative or reproductive stages of A. apis. Each larva was sampled from different micro-colonies fed with pollen contaminated with A. apis. The larvae exhibiting symptoms were slide mounted for microscopy in distilled water using Wild Heerbrugg Switzerland M20 microscope. Pictures were taken by using Leica DFC295 microscope camera and Leica Application Suite version 3. Measurements of the parasite’s morphology were performed using ImageJ 1.48 software (US National Institutes of Health, http://imagej.nih.gov/ij/download.html).
Molecular identification and phylogenetic analysis
Symptomatic larvae were submitted to DNA extraction as described before using (RNeasy Mini Kit; Qiagen). PCR was performed by using the primers as described by James & Skinner43. PCR products were submitted to a 1% agarose gel and stained with ethidium bromide for checking the DNA amplification and intensification. All ITS sequences of Ascosphaera fungi found in dead larvae presenting symptoms, as well as from honey bee-collected pollen and living larvae samples were BLAST against the database of the website of the National Center for Biotechnology Information (NCBI) (https://blast.ncbi.nlm.nih.gov/Blast.cgi) to confirm species identity. Sequence data were checked for errors, manually edited and compared to reference sequence of A. apis from the American Type Culture Collection. These sequences including different Ascosphaera species as deposited in Genbank database, were aligned (By Muscle) and subjected to a phylogenetic analysis processed with MEGA 7 software.
Pathogen quantification
The quantification of A. apis was checked by qPCR analysis among three different sources: all the contaminated pollen batch samples with the fungus (n = 6), infected living larvae (n = 9) and symptomatic dead larvae (n = 3). Ascosphaera apis quantification was performed by using a CFX96 Real-Time PCR Detection system (Bio-Rad, Hercules, CA, USA), performing each reaction in triplicate. The total reaction volume of 20 µL contained 10 µL of GoTaqr qPCR Master Mix, (Promega, Madison, WI, USA), 1 µL (10 µM) Forward and 1 µL (10 µM) Reverse primer targeting the 18S rRNA gene (see Supplementary Table 1) and 8 µL of diluted DNA. Nuclease free water was used as a negative control (NTC). The standard curve was obtained from the highest infected sampled and was serially diluted (E = 93.2%; R2 = 0.995).
Statistical analysis
The pathogen prevalence was determined per locality (country) and plotted using the software SigmaPlot 13.0. The difference in pathogen load among pollen, living larvae and dead larvae exhibiting symptoms was subjected to an Independent samples t-test (P < 0.05), using the software SPSS v.25 and graphically designed on SigmaPlot 13.0.
Data Availability
The datasets generated and analyzed in this study are available from the corresponding author on reasonable request.
References
VanEngelsdorp, D. & Meixner, M. D. A historical review of managed honey bee populations in Europe and the United States and the factors that may affect them. J. Invertebr. Pathol. 103, S80–S95 (2010).
Velthuis, H. H. W. & Van Doorn, A. A century of advances in bumblebee domestication and the economic and environmental aspects of its commercialization for pollination. Apidologie 37, 421–451 (2006).
Sachman-Ruiz, B., Narváez-Padilla, V. & Reynaud, E. Commercial Bombus impatiens as reservoirs of emerging infectious diseases in central México. Biol. Invasions 17, 2043–2053 (2015).
Fürst, M. A., McMahon, D. P., Osborne, J. L., Paxton, R. J. & Brown, M. J. F. Disease associations between honeybees and bumblebees as a threat to wild pollinators. Nature 506, 364–366 (2014).
Graystock, P. et al. Hygienic food to reduce pathogen risk to bumblebees. J. Invertebr. Pathol. 136, 68–73 (2016).
Meeus, I., de Miranda, J. R., de Graaf, D. C., Wäckers, F. & Smagghe, G. Effect of oral infection with Kashmir bee virus and Israeli acute paralysis virus on bumblebee (Bombus terrestris) reproductive success. J. Invertebr. Pathol. 121, 64–69 (2014).
Meeus, I., Brown, M. J. F., De Graaf, D. C. & Smagghe, G. Effects of invasive parasites on bumble bee declines. Conserv. Biol. 25, 662–671 (2011).
Graystock, P. et al. The Trojan hives: pollinator pathogens, imported and distributed in bumblebee colonies. J. Appl. Ecol. 50, 1207–1215 (2013).
Murray, T. E., Coffey, M. F., Kehoe, E. & Horgan, F. G. Pathogen prevalence in commercially reared bumble bees and evidence of spillover in conspecific populations. Biol. Conservation 159, 269–276 (2013).
Meeus, I., Pisman, M., Smagghe, G. & Piot, N. Interaction effects of different drivers of wild bee decline and their influence on host-pathogen dynamics. Curr. Opin. Insect Sci. 26, 136–141 (2018).
Kent, C. F. et al. Conservation genomics of the declining North American bumblebee Bombus terricola reveals inbreeding and selection on immune genes. Front. Genet. 9, 316 (2018).
McArt, S. H., Urbanowicz, C., McCoshum, S., Irwin, R. E. & Adler, L. S. Landscape predictors of pathogen prevalence and range contractions in US bumblebees. Proc. R. Soc. B 284, 20172181 (2017).
Singh, R. et al. RNA viruses in hymenopteran pollinators: evidence of inter-taxa virus transmission via pollen and potential impact on non-Apis hymenopteran species. PLoS One 5, e14357 (2010).
Goulson, D. & Hughes, W. O. H. Mitigating the anthropogenic spread of bee parasites to protect wild pollinators. Biol. Conservation 191, 10–19 (2015).
Lipa, J. J. & Triggiani, O. Apicystis gen nov and Apicystis bombi (Liu, Macfarlane & Pengelly) comb nov (Protozoa: Neogregarinida), a cosmopolitan parasite of Bombus and Apis (Hymenoptera: Apidae). Apidologie 27, 29–34 (1996).
Graystock, P., Yates, K., Darvill, B., Goulson, D. & Hughes, W. O. H. Emerging dangers: deadly effects of an emergent parasite in a new pollinator host. J. Invertebr. Pathol. 114, 114–119 (2013).
Evison, S. E. F. et al. Pervasiveness of parasites in pollinators. PLoS One 7, e30641 (2012).
Manley, R., Boots, M. & Wilfert, L. Emerging viral disease risk to pollinating insects: ecological, evolutionary and anthropogenic factors. J. Appl. Ecol. 52, 331–340 (2015).
McMahon, D. P. et al. A sting in the spit: widespread cross‐infection of multiple RNA viruses across wild and managed bees. J. Anim. Ecol. 84, 615–624 (2015).
Christensen, M. & Gilliam, M. Notes on the Ascosphaera species inciting chalkbrood in honey bees. Apidologie 14, 291–297 (1983).
Reynaldi, F. J., Lucia, M. & Garcia, M. L. G. Ascosphaera apis, the entomopathogenic fungus affecting larvae of native bees (Xylocopa augusti): First report in South America. Rev. Iberoam. Micol. 32, 261–264 (2015).
Anderson, D. L. & Gibson, N. L. New species and isolates of spore-cyst fungi (Plectomycetes: Ascosphaerales) from Australia. Aust. Syst. Bot. 11, 53–72 (1998).
Chorbinski, P. & Rypula, K. Studies on the morphology of strains Ascosphaera apis isolated from chalkbrood disease of the honey bees. Electron. J. Pol. Agric. Univ. 6 (2003).
Maxfield-Taylor, S. A., Mujic, A. B. & Rao, S. First detection of the larval chalkbrood disease pathogen Ascosphaera apis (Ascomycota: Eurotiomycetes: Ascosphaerales) in adult bumble bees. PLoS One 10, e0124868 (2015).
Graystock, P., Goulson, D. & Hughes, W. O. H. The relationship between managed bees and the prevalence of parasites in bumblebees. PeerJ 2, e522 (2014).
Maasen, A. Uber Bienenkrankheiten. Mitt. K. Biol. Anst. Land.-Fortsw. 16, 51–58 (1916).
Gilliam, M., Lorenz, B. J., Prest, D. B. & Camanzine, S. Ascosphaera apis from Apis cerana from South Korea. J. Invertebr. Pathol. 61, 111–112 (1993).
Gilliam, M., Lorenz, B. J. & Buchmann, S. L. Ascosphaera apis, the chalkbrood pathogen of the honey bee, Apis mellifera, from larvae of a carpenter bee, Xylocopa californica arizonensis. J. Invertebr. Pathol. 63, 307–309 (1994).
Pridal, P., Sedlácek, L. & Marvanová, L. Microbiology of Bombus terrestris L. larvae (Hymenoptera: Apoidea) from laboratory rearing. Acta Univ. Agric. Silvic. Mendel Brun. 8, 59–66 (1997).
Wynns, A. A., Jensen, A. B. & Eilenberg, J. Ascosphaera callicarpa, a new species of bee-loving fungus, with a key to the genus for Europe. PLoS One 8, e73419 (2013).
Genersch, E. American Foulbrood in honeybees and its causative agent, Paenibacillus larvae. J. Invertebr. Pathol. 103, S10–S19 (2010).
Wilson, G. G. & Burke, J. M. Nosema thomsoni n. sp., a microsporidian from Choristoneura conflictana (Lepidoptera: Tortricidae). Can. J. Zool. 49, 786–788 (1971).
Kyei‐Poku, G., Gauthier, D. & Van Frankenhuyzen, K. Molecular data and phylogeny of Nosema infecting lepidopteran forest defoliators in the genera Choristoneura and Malacosoma. J. Eukaryot. Microbiol. 55, 51–58 (2008).
Vilcinskas, A., Stoecker, K., Schmidtberg, H., Röhrich, C. R. & Vogel, H. Invasive harlequin ladybird carries biological weapons against native competitors. Science 340, 862–863 (2013).
Li, J. et al. Diversity of Nosema associated with bumblebees (Bombus spp.) from China. Int. J. Parasitol. 42, 49–61 (2012).
Ravoet, J. et al. Widespread occurrence of honey bee pathogens in solitary bees. J. Invertebr. Pathol. 122, 55–58 (2014).
Schoonvaere, K., Smagghe, G., Francis, F. & de Graaf, D. C. Study of the metatranscriptome of eight social and solitary wild bee species reveals novel viruses and bee parasites. Front. Microbiol. 9, 177 (2018).
Gisder, S. et al. Five-year cohort study of Nosema spp. in Germany: does climate shape virulence and assertiveness of Nosema ceranae? Appl. Environ. Microbiol. 76, 3032–3038 (2010).
Brown, M. J. F. Microsporidia: An emerging threat to bumblebees? Trends Parasitol. 33, 754–762 (2017).
Meeus, I., de Graaf, D. C., Jans, K. & Smagghe, G. Multiplex PCR detection of slowly‐evolving trypanosomatids and neogregarines in bumblebees using broad‐range primers. J. Appl. Microbiol. 109, 107–115 (2010).
Fernández, J. M. et al. Asymptomatic presence of Nosema spp. in Spanish commercial apiaries. J. Invertebr. Pathol. 111, 106–110 (2012).
Weiss, L. M. & Vossbrinck, C. R. In The microsporidia and microsporidiosis (Wittner, M., Weiss, L. M., editors) 129–171 (Washington, DC, USA: American Society of Microbiology, 1999).
James, R. R. & Skinner, J. S. PCR diagnostic methods for Ascosphaera infections in bees. J. Invertebr. Pathol. 90, 98–103 (2005).
Dobbelaere, W., de Graaf, D. C. & Peeters, J. E. Development of a fast and reliable diagnostic method for American foulbrood disease (Paenibacillus larvae subsp. larvae) using a 16S rRNA gene based PCR. Apidologie 32, 363–370 (2001).
Meeus, I., Smagghe, G., Siede, R., Jans, K. & de Graaf, D. C. Multiplex RT-PCR with broad-range primers and an exogenous internal amplification control for the detection of honeybee viruses in bumblebees. J. Invertebr. Pathol. 105, 200–203 (2010).
Sguazza, G. H., Reynaldi, F. J., Galosi, C. M. & Pecoraro, M. R. Simultaneous detection of bee viruses by multiplex PCR. J. Virol. Methods 194, 102–106 (2013).
Wang, H., Meeus, I., Piot, N. & Smagghe, G. Systemic Israeli acute paralysis virus (IAPV) infection in bumblebees (Bombus terrestris) through feeding and injection. J. Invertebr. Pathol. 151, 158–164 (2017).
Mommaerts, V., Sterk, G. & Smagghe, G. Hazards and uptake of chitin synthesis inhibitors in bumblebees Bombus terrestris. Pest Manag. Sci. 62, 752–758 (2016).
Acknowledgements
The authors are grateful to Biobest for providing pollen samples and bumble bee colonies. This research was supported by the National Council of Scientific and Technological Development (CNPq) through Science Without Borders Program – Brazil, for doctoral Grant (#201257/2014-9) to K.S.P. This research was funded by the Research Foundation-Flanders (FWO-Vlaanderen) and the Federal Agency for the Safety of the Food Chain – Belgium. We also thank support by COST (European Cooperation in Science and Technology) under grant agreement No. CA15223.
Author information
Authors and Affiliations
Contributions
K.S.P., I.V. and G.S. designed the research and analyzed the data. K.S.P. conducted the experiments; K.S.P. and I.V. drafted the paper. All authors were deeply engaged writing the main manuscript text.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Pereira, K.d.S., Meeus, I. & Smagghe, G. Honey bee-collected pollen is a potential source of Ascosphaera apis infection in managed bumble bees. Sci Rep 9, 4241 (2019). https://doi.org/10.1038/s41598-019-40804-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-40804-2
This article is cited by
-
Bacterial Strains Isolated from Stingless Bee Workers Inhibit the Growth of Apis mellifera Pathogens
Current Microbiology (2024)
-
Distribution of infectious and parasitic agents among three sentinel bee species across European agricultural landscapes
Scientific Reports (2024)
-
Bacterial and Fungal Symbionts in Pollen Provisions of a Native Solitary Bee in Urban and Rural Environments
Microbial Ecology (2023)
-
Longitudinal analysis on parasite diversity in honeybee colonies: new taxa, high frequency of mixed infections and seasonal patterns of variation
Scientific Reports (2020)
-
Evidence for and against deformed wing virus spillover from honey bees to bumble bees: a reverse genetic analysis
Scientific Reports (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
