
Abstract
Diseases of the gastrointestinal tract due to changes in the bacterial flora have been described with increasing incidence in the European brown hare. Despite extensive demographic and phylogeographic research, little is known about the composition of its gut microbiota and how it might vary based on potential environmental or host factors. We analysed the intestinal and faecal microbiota of 3 hare populations by Illumina MiSeq 16S rRNA gene amplicon sequencing. The phyla and OTU abundance composition differed significantly between intestinal and faecal samples (PERMANOVA: P = 0.002 and P = 0.031, respectively), but in both sample types Firmicutes and Bacteroidetes dominated the microbial community composition (45.51% and 19.30% relative abundance). Intestinal samples contained an enrichment of Proteobacteria compared with faecal samples (15.71-fold change, P < 0.001). At OTU level, a significant enrichment with best BLAST hits to the Escherichia-Shigella group, Eubacterium limosum, Sphingomonas kyeonggiensis, Flintibacter butyricus and Blautia faecis were detected in intestinal samples (P < 0.05). In our statistical model, geographic location and possibly associated environmental factors had a greater impact on the microbiota composition than host factors. Population had a significant effect on the composition of abundant intestinal and faecal OTUs, and on the abundance of potential pathogenic bacteria of the family Enterobacteriaceae, regularly associated with intestinal dysbiosis in hares, in faecal samples. Our study is the first to describe the microbiota in brown hares and provides a foundation to generate hypothesis aiming to test the role of gut health in population fluctuations of the species.
Similar content being viewed by others
Introduction
The European brown hare (EBH) (Lepus europaeus) is a widely distributed and important game species throughout Europe. Despite the classification of their population status as “least concern” by the International Union for Conservation of Nature (IUCN, red list 2017), significant population declines have been noted in the last 50–60 years leading to a “near threatened” or “threatened” status on national red lists for some regional populations, e.g.1. A range of factors including unfavourable climatic conditions2; predation3 and multiple disease epidemics such as the European brown hare syndrome (EBHS), Pasteurellosis, Pseudotuberculosis or Coccidiosis4,5,6 have contributed to regional threats. However, probably the most important threat for this species is the intensification of agriculture7,8,9,10. Habitat fragmentation, agricultural mechanization, monocultures, unpredictable food availability as well as pesticide and fertilizer use have been shown to have detrimental health effects on a variety of farmland wildlife species11,12,13 including substantial impact on hare population viability7,8. Species like the brown hare can be even more severely affected as the species’ diet is highly selective and adapted to maximise energy intake by positively selecting certain plants species14. Furthermore, lagomorphs are known to be very sensitive to any feed alterations and imbalances, causing disruption of the gut microbiota and resulting gastrointestinal disease15,16,17,18. The pathology is associated with an intestinal bacterial dysbiosis, which might lead to an increase of pathogenic bacteria such as e.g. Escherichia coli or Clostridium spp. and often results in reduced fitness, enteritis and other diseases or even death19,20. Results of recent health screenings of hare populations showed such changes in the gut microbiota21.
Recent research in human and veterinary science has highlighted the importance of the gut microbiota impact on host physiology22,23,24,25. A variety of host specific factors, such as age, sex, body condition, genetics and host phylogeny are known to shape the microbiota in an individual26,27,28,29,30. Additionally, the role of environmental factors, such as lifestyle, diet, climate and habitat conditions, as well as changes in land use are described to impact the microbial composition in the gastrointestinal tract (GIT)31,32,33. Nevertheless, this knowledge cannot be easily transferred to wildlife species. Wild animals face highly diverse and changing environments. They have established physiological plasticity and acclimatization mechanisms that differ greatly from domestic or laboratory animals34.
Generating physiological baseline data of populations living in their natural habitats is important in order to understand and predict what factors might have the most impact on a species. This requires studies of wildlife species within their natural habitats33,34. So far, studies investigating the gut microbiota of free-ranging wildlife are rare and baseline data of potentially influencing factors of microbial community composition, abundance of specific bacterial taxa associated with host health in wildlife species is lacking30,33,35,36. Studies comparing wild with captive wildlife populations have reported differing gut microbiota compositions associated with a highly co-evolutionary adapted microbiota in the investigated wildlife species and a loss of diversity in captive animals37. The importance of the gut microbiome has recently also been more and more considered in species conservation and management measures38,39.
In this study, we analysed the gut bacterial microbiota composition in European brown hares with 16S rRNA gene sequencing and assessed differences between intestinal and faecal samples. By the use of these molecular high-throughput techniques and community analysis we aimed to identify and describe the gut microbiota, a goal that could not be achieved by previous culture- and/or enrichment-dependent methods.
Furthermore we used a statistical model to investigate possible impacts of host factors, sampling population and potentially associated different land use on the gut microbiota composition of free-ranging European brown hare. Specifically, we tested the impact of host factors such as age, sex, body condition (heart fat) and gut health as well as sampling population on the abundance and diversity of the bacterial composition in intestinal and faecal samples of brown hares.
Material and Methods
Sample and data collection
A total of 25 brown hares (Lepus europaeus) of both sexes were included in this study. Metadata of all animals included is listed in Supplementary Table S1. The study including the experimental protocol was carried out and approved in accordance with all current laws of Austria and the guidelines and regulations of the institutional Ethics Committee and the institutional Good Scientific Practice Guidelines of the University of Veterinary Medicine, Vienna. Samples were all obtained during the hunting season in December 2015, in order to exclude any seasonal effects. In all hunting, respectively sampling areas, a systematized yearly drive-hunt using rifles according to the respective hunting law was performed. Sampling locations are spatially visualized in Fig. 1. The two Austrian populations are known to genetically diverse with geneflow occurring both locally and regionally40,41. The population on Pellworm has more restricted genetic diversity typical of an isolated island population (unpublished data). Sampled individuals were from well separated areas within each hunting ground to avoid the possibility of including individuals from the same family groups within each population sample.

Sampling locations of European brown hares (labelled with red triangles): Sampling area (a) Pellworm, Germany (Grassland type) is characterized by 70% grassland and 30% cropland of the total agricultural area; (b) Lower Austria, district Mistelbach, Austria (Cropland-type) by 79% of the agricultural area being used as cropland and 0.4% as grassland (c) Military Airport Langenlebarn; Tulln, Austria, (no agricultural use). For a detailed description see also Material and Methods.
As age could constitute an influential factor on the microbiota of hares, the age was determined by the weight of the dried eye lenses42. This method allows for the sensitive discrimination of subadult hares (i.e. young of the year or individuals not older than 9–10 months of age) that can otherwise not easily be distinguished from adult hares based on morphological characteristics. Hares that were younger than one year of age were classified as subadults. Digesta samples from the cranial flexure of the duodenum (intestinal samples) and faecal samples from the ampulla recti were collected in sterile sampling vials during a standardized necropsy by trained pathologists in the field within 20 minutes after death and immediately frozen at −20 °C, transported to the lab and frozen at −80 °C until use.
Heart fat (coronary fat deposit) as a parameter defining the nutritional status was assessed ranging from 1–6, such that 1 defined cachectic, 2 bad, 3 moderate, 4 good, 5 very good and 6 described obese. The parameter gut health was defined based on the following variables assessed by pathological examination of the gastrointestinal tract: 1: no macroscopic and/or no/very mild histopathological lesions (smooth mucosa, no signs of inflammation (0–+), no increase in inflammatory cells); 2: moderate dysbiosis signs of inflammation, ((+–++) inflammatory cells); 3: severe macroscopic and/or histopathological lesions (severe mucosal erythema, severe signs of inflammation, and (++–+++) inflammatory cells). The animals originated from three different geographical locations associated with different land use practices. As hares are highly selective feeders that prefer weeds/grasses and various crop types while avoiding cereals14, we chose the sampling sites according to the different land use types: (a) Grassland-type (Pellworm): Around 80% of the whole area of the island, Pellworm, northern Germany, are used as agricultural land; around 70% of the agricultural land consists of grassland (pastureland and green fodder for domestic species such as cattle and sheep); the remaining 30% are used as cropland; (main crop consists of winter wheat (Triticum aestivum), rapeseed (Brassica napus), as well as to a lower percentage, barley (Hordeum vulgare), oat (Avena sativa) and Flax (Linum usitatissimum)43,44. Around 8% are used for maize (Zea mays) plantation for a local biogas facility45. (b) Cropland-type (Lower Austria- District Mistelbach): Of the 89% agricultural land of the total area of the district, around 79% are used as cropland, where predominantly various species of grain and maize (Zea mays) are cultivated followed by oil seed, sugar beet (Beta vulgaris), wine and fruit plantation as well as fallow land; to a lower percentage, potatoes (Solanum tuberosum) and protein rich plants are cultivated. Only around 0.4% are defined as grassland46. (c) No agricultural use (Military Airport Langenlebarn, Tulln). As a control site a sampling site consisting of fallow land without any agricultural use was chosen (no land use; no fertilizer or pesticide use).
Genomic DNA extraction
Intestinal and faecal samples were thawed on ice and total genomic DNA was extracted from 250 mg using the Powersoil® DNA Isolation Kit (MoBio Laboratories Inc., Carlsbad, CA, USA) according to the manufacturer’s instructions with one modification: To ensure proper lysis of bacteria, a heating step at 70 °C for 10 min was introduced between mixing of samples with buffer C1 and bead-beating47. After extraction, all samples were quantified by spectrophotometry using the Qubit 2.0 Fluorometer (Thermo Fisher Scientific, Waltham, MA, USA) with the Qubit dsDNA HS Assay Kit (Thermo Fisher Scientific).
16S rRNA gene sequencing and sequence processing
For amplicon sequencing, we targeted the 16S rDNA hypervariable region V345, using an Illumina MiSeq sequencing platform with a 300-basepair paired-end read protocol. The universal primers 357F-HMP (5′-CCTACGGGAGGCAGCAG-3′) and 926R-HMP (5′-CCGTCAATTCMTTTRAGT-3′) were used to generate amplicons of ~570 base pairs48,49. Libraries were constructed by ligating sequencing adapters and indices onto purified PCR products using the Nextera XT Sample Preparation Kit (Illumina) and equimolar amounts of each of the libraries were pooled and sequenced. 16S rRNA gene PCRs, library preparation and sequencing, as well as demultiplexing and read stitching were performed by Microsynth (Microsynth AG, Balgach, Switzerland).
Process- and sequencing- controls
Prior to sampling, DNA extraction was attempted from blank tubes and a PCR with a long 16S rRNA gene primer panel was performed to verify that sampling tubes were not contaminated. A negative control was also included in the DNA extraction run. This negative control was also sequenced and should be a proxy for reagent contamination and cross-contamination between samples. Sequencing of the negative control revealed 5,000 sequences (average read count levels per sample was 77,000). Three non-template controls and one positive control (human stool sample) were included as internal controls in library preparation. These controls were identified as expected (3 negatives, one positive).The MiSeq Run was controlled by a PhiX control sample (https://emea.illumina.com/products/by-type/sequencing-kits/cluster-gen-sequencing-reagents/phix-control-v3.html?langsel=/it).
Microbial community analysis and predictive functional profiling
Analysis of the NGS data was conducted using the QIIME software package v1.9.150. A total of 3,856,491 demultiplexed sequences were produced, of which 2,700,987 (70.04%) passed the quality filter with a phred value of >9 and a minimum read length of 520 base pairs. A total of 7,947 chimeric sequences were filtered out before operational taxonomic units (OTUs) were clustered using 97.00% identity (=0.03 distance) against the USEARCH 6.1 database51 and de novo. Singletons were removed, and representative sequences were classified via the RDP classifier 2.252. Alpha diversity was calculated by subsampling the OTU table to the read depth of the sample with the lowest amount of sequences (n(seqlowest) = 8,671), resulting in 10 repetitions at 1,000 sequences up to 8,000 sequences/sample. Beta diversity was calculated by subsampling to an even read depth of 6,000 reads/sample, of which weighted UniFrac distance matrices53 and principal coordinate analysis were calculated54. Venn diagrams of OTUs were generated with the Venn diagram plotter (PNNL, Richland, WA) to display the number of OTUs shared by different populations. In addition, the core microbiota OTUs, defined as OTUs shared by over 90% of the samples, were identified. BLASTn analysis (NCBI GenBank database excluding uncultured/environmental sample sequences in the search set) of the top 50 most abundant OTUs and core microbiota OTUs were done using the highest identity matches of each unique OTUs representative sequence.
Quantification of bacteria in the gastrointestinal tract of hares
We conducted a quantitative PCR (qPCR) to assess the absolute abundance/copy numbers of bacterial cell equivalents (BCE) present in the intestinal and faecal flora. First, quantification of all bacteria was done by targeting a conserved 16S rRNA gene region (forward primer 5′-CCTACGGGAGGCAGCAG-3′, reverse primer 5′-ATTACCGCGGCTGCTGG-3′)55. Second, the primer pair Eco1457F (forward primer 5′-CATTGACGTTACCCGCAGAAGAAGC-3′) and Eco1652R (reverse primer 5′-CTCTACGAGACTCAAGCTTGC-3′) were used covering a multitude of Enterobacteriaceae, Erwiniaceae and Pectobaceriaceae- associated genera including Escherichia, Citrobacter, Cronobacter, Enterobacter, Shigella, Erwinia, Pantoea and Pectobacterium56. Standards for both primer sets were made by conducting a qPCR on three pooled intestinal and faecal samples that were quantified afterwards using the Qubit Fluorometer. Copy numbers of the standard curves with genomic DNA templates were calculated with the equation: DNA (molecules/μL) = [6.02 × 1023 (molecules/mol) × DNA amount (g/μL)]/[DNA length (bp) × 660 (g/mol/bp)], according to Li, Penner, Hernandez-Sanabria, Oba and Guan57. The 16S rRNA gene copy numbers (seven copies for Eco1457F -Eco1652R qPCR, four copies for the all-bacteria targeted qPCR58 were taken into account when extrapolating bacterial cell equivalents (BCE).
For qPCRs, DNA samples and negative controls were run in duplicate in a 20 µl reaction. The master mix contained 10 µl 2 × Brilliant III Ultra Fast SYBR Green qPCR Master Mix (Agilent, Vienna, Austria), 2 µl of 2.5 µM primers and 5 µl of water (nuclease-free). 1 µl DNA template including approximately 10 ng DNA (intestinal samples) and 1 ng DNA (faecal samples) was added. The following amplification protocol was used: Initial denaturation at 95 °C for 3 min, 40 cycles of 95 °C for 5 s and by 20 s at 61 °C and 63 °C for general bacteria and Eco1457F -Eco1652R qPCR, respectively. A melting curve (70 °C to 90 °C, with fluorescence measurements at 1 °C intervals), was done after each qPCR and specific amplicon peaks were received. qPCR results were analysed using the Stratagene MxPro software (QPCR Software, version 2.00).
Statistical analyses
Statistical analysis of the bacterial microbiota composition was implemented using the R statistical computing environment, version 4.3.359. Alpha diversity indices were assessed by calculating the Chao1 index and the observed OTU richness. We used lme models (linear mixed-effect models, R-package, nlme60) to analyse the effects of independent variables, i.e. population (geographical areas (Airport (A), Lower Austria (LA) and Pellworm (P)), sex (male vs. female), age class (sub adult vs. adult), and heart fat as a parameter defining the nutritional status of the animal (ranged 1–6, whereas 1 defined cachectic, 2 bad, 3 moderate, 4 good, 5 very good and 6 described obese) on diversity indices (dependent variable). Beside these fixed effects (geographical area, sex, age, heart fat and gut health), we also used the individual animal IDs (n = 25) as a random effect. All predictor variables in the full model were checked for multicollinearity by calculating the variance inflation factor. We used the Akaike Information criteria corrected for small sample size (AICc) to determine the best model with the most relevant fixed factors for each response variable. None of the models indicated primary collinearity issues (all values were below 2.661). The residuals of the models were assessed visually via histograms and Q-Q-plots as well as by calculation of the Shapiro-Wilk normality test. The dependent variable Chao1 calculated from the intestinal tract data was log transformed. The modelling procedure was carried out for both the faecal and intestinal datasets, although the effect of the faecal and intestinal samples on diversity indices itself was also calculated in an individual model run. We estimated the proportion of the variance of the fixed and random regressors using Pseudo-R-Squared (pseudo-r2-values62). In the next step, contrast coefficients of the explanatory variables were calculated among the response variables diversity indices by using a pairwise comparison of least-squares means, corrected for multiple comparison using the false discovery rate (FDR). The contrast calculation was implemented in R using the package lsmeans63. The level of significance was defined at P ≤ 0.050. The normality distribution of the abundance of phyla (n = 23), and most abundant OTUs (n = 50) were checked with the tests for multivariate data with the function manova (R Package stats) and the Cullen and Frey graph with the R package fitdistrplus and logspline64,65. Due to the non-normal distribution of the data, model residuals, and different identified optimal theoretical distributions of individual phyla and OTUs, the beta diversity of the microbiota of phyla and OTUs was assessed by applying a permutational multivariate analysis of variance (PERMANOVA) with the ADONIS function and 5000 permutations in R (R Package vegan66). To analyse whether independent variables (i.e. population, sex, age, heart fat and gut health) had also a significant effect on the composition of phyla and OTUs abundances, we calculated Bray-Curtis-dissimilarity matrix with the R function vegdist of the vegan package66. The distance matrix was applied as response variable. The modelling approach was carried out for faecal and intestinal samples, although the effect of the two on the composition of phyla and OTUs itself was also calculated in an individual ADONIS function. The multivariate homogeneity of group dispersions was performed with the betadisper function, a multivariate analogue of Levene’s test, followed by a permutation-based test of multivariate homogeneity of group dispersions with pairwise comparisons of group mean dispersions. The Kruskal-Wallis test was applied to identify potentially statistically significant differences between populations concerning each individual phylum, OTU, and independent variables, followed by a pairwise test for multiple comparison of mean rank sum (Dunn’s test), adjusted with Benjamini-Hochberg method (R Package PMCMR, FSA67,68).
Relative abundance plots of microbial communities were calculated with the transform_sample_counts function within the phyloseq package. The corresponding visualisation was conducted with the package ggplot2 in R69,70. The tool ‘Phylogenetic Investigation of Communities by Reconstruction of Unobserved States’ (PICRUSt) was applied to predict the metagenome functional content from amplicon sequencing71. Statistical analysis of metagenome predictions was carried out with STAMP72 by using an ANOVA with Benjamini-Hochberg FDR correction.
Additionally, the Kruskal Wallis test was applied to compare the different populations in terms of qPCR abundance of Eco1457F-Eco1652R and all bacteria in faecal samples and intestinal tract samples. A post-hoc test using Mann-Whitney tests with Bonferroni correction for multiple comparisons was used and the corresponding Z Values were computed to determine the effect size (r).
Results
The microbiota of intestinal and faecal hare samples
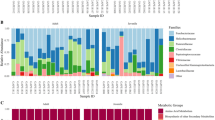
In total, 2,700,987 sequences (70.04%) passed our quality filter and were processed together for all downstream analysis. Reads generated for intestinal and faecal samples were distributed homogenously, with 53% reads belonging to intestinal and 47% belonging to faecal samples. At both microbiota sources analysed (intestinal and faecal samples) Firmicutes and Bacteroidetes dominated the microbial community composition (45.51% and 19.30% relative abundance over all samples respectively), followed by Spirochaetes and Proteobacteria (7.76% and 5.73% relative abundance over all samples) (Fig. 2A). In intestinal and faecal samples, 23 and 13 phyla were detected respectively. The 13 faecal-associated phyla corresponded to the highest abundance subset in intestinal samples. The phyla abundance composition differed significantly between intestinal and faecal samples (PERMANOVA: P = 0.002, pseudo r2 = 0.09; Permutest: P = 0.093). Intestinal samples contained an enrichment of Proteobacteria compared with faecal samples (15.71-fold change, P < 0.001) and more Actinobacteria (2.09-fold change, P = 0.002). Faecal samples contained significantly more Lentisphaerae (1.74-fold change, P = 0.050).

Taxonomic classification of 16S rRNA gene sequence reads parted by sampling type (i.e. intestinal vs. faecal) and population, whereas data represents average of OTU counts from replicate libraries for each category. (A) bacterial phyla (B) bacterial families in the gastrointestinal tract of hares. Phyla and families with less than 1% relative abundance were grouped together. Sequences that could not be assigned are depicted as “Unassigned”.
For intestinal and faecal samples 12,119 and 22,149 OTUs were detected over all hare populations, respectively. The overlapping pattern of abundant OTUs detected in both gut sites was high: From the 500 most abundant OTUs (89.50% of all sequences), only 1.40% (n = 7) were exclusively found in the faecal and 1.80% (n = 9) were exclusively found in intestinal samples. However, OTU abundance composition differed significantly between intestinal and faecal samples (PERMANOVA: P = 0.031, r2 = 0.04; Permutest: P = 0.855). In intestinal samples compared with faecal samples, a significant enrichment of OTUs with best BLAST hits to the Escherichia-Shigella group, Eubacterium limosum, Sphingomonas kyeonggiensis, Flintibacter butyricus and Blautia faecis were detected (Table 1). The core microbiota consisted of 66 shared OTUs, which all belonged to the 500 most abundant OTUs across all samples (Table 2).
Observed diversity richness OTU counts and diversity estimator Chao 1 counts are listed in Supplementary Table S2. Over all populations, the average observed diversity richness per sample was 711 OTUs in faecal and 510 OTUs in intestinal samples. Chao 1 estimated the diversity on average to contain 1,686 OTUs in faecal and 932 OTUs in intestinal samples. Additionally, a statistically significant effect between intestinal and faecal abundance for the diversity indices were observed (Chao 1, P < 0.001 and observed OTUs P < 0.001). The sampling site could explain approximately 22–32% of the variance of the diversity data, but most of the variance was explained at the individual level (OTU richness: conditional r2 = 0.57: Chao: conditional r2 = 0.56).
Factors influencing the bacterial microbiota of intestinal and faecal hare samples
The composition of the phyla abundance between different populations in intestinal samples differed by trend (PERMANOVA: P = 0.099, r2 = 0.14; Permutest: P = 0.148), whereas no significant effect in faecal samples could be observed (PERMANOVA: P = 0.610, r2 = 0.07; Permutest: P = 0.697). In particular, significant differences between different populations with respect to phyla abundance in intestinal samples were detected between Bacteroidetes, Lentisphaerae and WPS-2. Bacteroidetes were enriched as a trend in the Pellworm population compared with the Airport population (Airport – Pellworm, P = 0.054, 2.91-fold change), whereas Lentisphaerae and WPS-2 were significantly enriched in the Airport population (Lentisphaerae: Airport-LA, P = 0.044, 2.19-fold change; Airport-Pellworm, P = 0.046, 1.95-fold change; WPS-2: Airport-Pellworm, P = 0.049, 1.82-fold change; LA-Pellworm, P = 0.048, 1.70-fold change). The PERMANOVA analysis shows that age (Faecal: P = 0.523; Intestinal: P = 0.721), sex (Faecal: P = 0.708; Intestinal: P = 0.964), heart fat (Faecal: P = 0.482; Intestinal: P = 0.145) and gut health (Faecal: P = 0.821; Intestinal: P = 0.738) did not significantly affect phyla abundances. Population had further significant effects on the composition of intestinal (PERMANOVA: P = 0.002, r2 = 0.18; Permutest: P = 0.104) as well as faecal OTUs (PERMANOVA: P = 0.002, r2 = 0.19; Permutest: P = 0.258). Faecal OTUs were also significantly affected by age (PERMANOVA: P = 0.039, r2 = 0.08: Permutest: P = 0.441). In total, 12 and 13 OTUs from the most abundant 50 OTUs differed significantly between populations in intestinal and in faecal samples respectively: New.ReferenceOTU0 (NR0; best BLAST hit Sphingobacterium wenxiniae), NR11 (Ruminococcus albus) and NR43 (Oscillibacter valericigenes) were significantly enriched in the Airport population compared with the LA population (Table 3). OTU 296045 and OTU 1108377 (best BLAST hit Bacteroides sartorii and Muribaculum intestinale) had a significantly higher abundance in the Pellworm population compared with the Airport population (Table 3). NR1 (best BLAST hit Murimonas intestine) was higher in the Airport population compared with the Pellworm population (Table 3). Heart fat, sex and gut health parameter had no significant effect on intestinal or faecal-associated OTUs (P > 0.050). In this context, no significant effect of population, age, heart fat, sex and gut health on the diversity abundance in intestinal and faecal samples was observed. Our models of alpha diversity indices (Chao1 index and the observed OTU richness) could explain on average 89% of the variance of the data (conditional r2 = 0.88 and r² = 0.89 for intestinal and faecal samples, respectively). However, much of the variation in the data was due to the difference between individuals, since data variance explained by the fixed factors alone was considerably lower (mean marginal r2 = 0.08 and r² = 0.14 for intestinal and faecal samples, respectively). Abundant OTUs summed up at family level are shown in Fig. 2B.
The Venn diagram (Supplementary Fig. S1) displays an unequal amount of OTUs per population and a high number of unique OTUs in each population. In faecal samples, 7.69% of all OTUs, and in intestinal samples 8.97% of all OTUs were shared between the three populations examined. The phylogenetic distance measurement done with weighted UniFrac analysis did not reveal a clear clustering of populations, however the Airport population samples were more similar to each other compared with the other populations for both intestinal and faecal samples. The highest variability within a population was found for the LA population (Fig. 3). The PICRUSt analysis indicated one significant difference on KEGG (Kyoto Encyclopedia of Genes and Genomes) Level 3 in the population comparison: The linoleic acid metabolism was significantly higher in Airport and Pellworm populations compared with LA population (Effect size: 0.36, P < 0.005; Supplementary Table S3).

PCoA Plot based on weighted UniFrac distance matrices of faecal and intestinal samples. In Panel (A) faecal and in Panel (B) intestinal samples are depicted. UniFrac distances were calculated for all OTUs. Each point represents values from one individual with colors expressing population affiliation.
Bacterial cell equivalents in intestinal and faecal samples
In 1 g intestinal samples, between 3.2E + 07 (Airport population) and 4.1E + 07 BCE (Pellworm population) were detected with qPCR. In 1 g faecal samples, between 2.5E + 07 (LA population) and 7.3E + 08 (Airport population) BCE were found. The number of Enterobacteriaceae, Erwiniaceae and Pectobaceriaceae, covered with the primer panel Eco1457F- Eco1652R, varied in intestinal samples between 7.4E + 04 (Pellworm population) and 1.2E + 06 (LA population), and in the faecal samples between 5.1E + 03 (Pellworm population) and 3.2E + 04 (LA population) gene copies/g (Fig. 4). In faecal samples a significant effect was identified for the factor ‘population’ on the Eco1457F- Eco1652R qPCR (χ2 = 11.25, P = 0.003). Specifically, differences were detected between the Airport and LA (P = 0.005, effect size r = 0.70) and between Pellworm and LA (P = 0.030, effect size r = 0.58).

Bacterial cell equivalents (BCE) determined by qPCR. A primer panel covering all bacteria and a primer panel covering a multitude of Enterobacteriaceae, Erwiniaceae and Pectobaceriaceae- associated genera (Eco1457F-Eco1652R) were used for (A,B) faecal and (C,D) intestinal samples. (A,C) include absolute BCE counts, (B,D) show the relative abundance of Enterobacteriaceae, Erwiniaceae and Pectobaceriaceae- associated genera in proportion to all bacteria.
Discussion
This is to our knowledge the first study characterising the intestinal and faecal microbiota of the European brown hare based on 16S rRNA gene sequencing.
To date, studies describing the gastrointestinal microbiota are limited to one member of the family Leporoidae, the rabbit73,74. As sympatric relatives, often sharing the same habitat as well as feed preferences, the species do share similarities of their gastrointestinal physiology75. Leporidae are hindgut fermenters and practitioners of caecotrophy. Nevertheless, hares and rabbits show both morphological differences of their gastrointestinal tract (GIT) and different digestive strategies in terms of nutrient extraction14,75,76. Those differences in digestive function and strategies between the two species suggest that microbial communities in the GIT may also differ and potentially show divergent functionality or metabolic pathways. Indeed, we were able to identify that the core microbiota of the hare GIT diverges from that previously described in rabbits. Our study reveals that the gut microbiota of hares includes Clostridium, Bacteroides, and Ruminococcus species, but not Streptococcus and Enterobacter species as previously described to be abundant in rabbit caecum77,78,79,80,81,82. Despite these differences we also detected similarities between the gut microbiota of the two species, e.g. Eubacterium and Ruminococcus related OTUs, both found among the 50 most abundant and core microbiota OTUs in this study, have been described in the rabbit GIT as well83. Although the highly abundant phylotypes detected in this study were in accordance with phylotypes recently described in rex rabbits74, hare intestinal and faecal samples were highly enriched for Spirochaetes compared with rabbits. The majority of the phylum Spirochaetes consisted of the family Sphaerochaetaceae mainly comprising of OTUs affiliated to Sphaerochaeta pleomorpha or Sphaerochaeta globosa. These OTUs showed only 88% sequence similarity to reference 16S rRNA gene sequences but 93% to 96% sequence similarity to 16S rRNA gene sequences of uncultured Spirochete clones found in rumen of ruminants or the oral cavity of canines84,85. Interestingly, BLASTn database sequences include a large number of uncultured Sphaerochaeta species residing in a broad range of mammalian GIT or oral cavities. Unlike most other Spirochaetes, which usually have a helical morphology and flagella, Sphaerochaeta are spherical and not motile86. They are capable of heterofermentative growth on carbohydrates such as pentose and hexose monosaccharides, disaccharides and soluble starch86. In contrast to their closest relatives Spirochaeta, Treponema, and Borrelia, they are not thought to be pathogenic87.
Similar to the Sphaerochaeta associated OTUs, other high-abundance OTUs observed in this study had similarity scores to reference 16S rRNA genes below 90%, which is in accordance with a study on bacterial species in the rabbit caecum where the majority of sequences shared less than 97% identity to BLASTn database sequences83. Hence our data demonstrate that gastrointestinal tracts of hares harbour multiple bacterial species that that are yet to be described.
Studies in wildlife species are often restricted in sample availability due to limitations of access to free-ranging animals and restrictions in the application of invasive sampling methods. Non-invasive samples such as faeces are in many cases the only available sample rescource. In order to evalutae the representation of the gastrointestinal microbiota composition in faecal samples and therefore its potential use in future studies in hares, we tested the similarity between the faecal and intestinal microbial community composition.
Firmicutes and Bacteroidetes dominated the microbial community composition in both sample types, followed by Proteobacteria and Spirochaetes. Intestinal samples showed a higher number of phyla with a significantly different abundance pattern compared with faecal samples. Similarly, diversity indices and the abundance of OTUs differed significantly between sampling sites, indicating clear microbiota shifts along the hare’s gastrointestinal tract. Faecal samples were not representative for the description of the intestinal microbiota. This is in accordance with recent studies that identified a distinct microbial environment between luminal and mucosal sites and in respective compartments of the gastrointestinal tract of monogastric animals88,89. Therefore, future studies should consider using GIT samples representative for testing their specific hypothesis.
Land use practices associated with poorly diversified and restricted food availability is known to effect gut microbiota and individual fitness11. Highly specialized feeders, such as the brown hare, might be even more severely affected by a decrease in plant biodiversity14. Our three chosen sampling locations differ tremendously in land use and habitat type parameters. The agricultural land use in Pellworm consists predominantly of grassland and to a much lower percentage cropland; whereas the situation in LA is the opposite, with the majority of land used as cropland. The control site Airport consists of fallow land without any agricultural use. As highly selective feeders, hares prefer weeds and grasses, and specifically select for certain plant taxa14. Furthermore, they avoid crude fibre and select for high fat and energy content in their diet to provide essential polyunsaturated fatty acids (PUFA) such as linoleic acid and alpha-linolenic acid needed to reproduce and survive. A study by Popescu and colleagues found that European hares are able to selectively absorb PUFA in the gastrointestinal tract and excrete faeces that are highly depleted in PUFA and enriched in saturated fatty acids (FA)76. Interestingly, linoleic acid metabolism was significantly elevated in the Airport and Pellworm populations compared with the LA population (Supplementary Table S3), probably indicating the variation in food and nutrient variability between the populations in our study.
The overall phyla composition was relatively stable in hares (only intestinal samples varied between populations as a trend), but hare location had a significant impact on the OTU composition of faecal and intestinal samples between the different populations. In intestinal samples a number of specific taxa differed in abundance between the different populations. For example, there was a trend for a difference in the abundance of Bacteroidetes between LA and Airport and a significant difference in the abundance of OTU 296045 (best BLAST hit: Bacteroides sartorii), which was enriched in intestinal samples of the Pellworm population relative to the Airport population. Bacteroides species have been detected at high abundances in rabbit caecum in the past and are associated with healthy growth and high-weight gain74,90. Overall, the Pellworm population had the lowest OTU diversity. Population genetic data also suggest that Pellworm has lower diversity than the Austrian populations and its restricted geneflow is likely also associated with the low OTU diversity. Further research on associations between OTU diversity on gut health, respectively disease susceptibility and on physiological parameters is warranted. Hares of the Airport population showed lower abundances of the yet uncultured bacterial family S24-7 compared with the Pellworm and LA population. This family is being recognized as a predominant member of the intestinal tract of homeothermic animals, but data on their physiology, metabolic capacity, and interactions with the host are limited91. Plant glycan (hemicellulose and pectin) degradation has been described based on genomic characterization of the S24-7 family91. Our results suggest that both grassland and cropland agricultural areas are associated with increased abundances of S24-7 affiliated bacteria in the intestinal tracts of hares.
Population had a significant effect on both the composition of intestinal and faecal OTUs. Population was also the explanatory factor for the abundance of potential pathogenic bacteria of the family Enterobacteriaceae.
Many members of the family Enterobacteriaceae, which showed the highest abundance in the LA population and lowest abundance in hares of the Pellworm area, are connected to gastrointestinal disease92,93,94. The low abundance of Enterobacteriaceae and the high abundance of OTU 296045 (i.e, likely Bacteroides) in the GIT of Pellworm hares might indicate a healthier microbial composition in the intestinal tract. Host traits such as age, sex, body condition (heart fat) and gut health were not related to the abundance and diversity of the bacterial composition in intestinal and faecal samples of brown hares in this study.
Nevertheless, further investigations that look into associations of agricultural use, plant diversity and occurrence of dysbiosis are desirable. Considering the low number of sampled animals (n = 25) in the presented study, significant impact of independent variables should be interpreted with caution. A larger sample size in future studies would be desirable and would allow for more robust conclusions than those derived from the present study.
Conclusion
The current knowledge of gut microbiota composition and influencing factors in wildlife species under natural conditions is extremely limited. Our aim was to provide a broad foundation to generate specific hypothesis at the intersection of gut health, land use change and population viability for the EBH and other species impacted by rapid habitat modification. This combined information is lacking to date, despite being essential for understanding gut microbial variation within wildlife species to differentiate natural variability from that related to external stressors leading to functional dysbiosis affecting host health. Anthropogenic habitat modifications together with species-specific susceptibility to these modifications might also have an impact on wildlife gut microbiota.
This study generates fundamental baseline data on the diversity and composition of gut microbiota in the brown hare that will complement previous research and create a starting point for future research to understand the physiological and pathophysiological dynamics of gut microbiota as well as the causes of alterations to the intestinal microbial communities in this species.
Data Availability
Nucleotide sequence accession numbers: Sequencing data are available in the European Nucleotide Archive Database under the accession number PRJEB15166.
References
Reichlin, T., Klansek, E. & Hackländer, K. Diet selection by hares (Lepus europaeus) in arable land and its implications for habitat management. Eur. J Wildl. Res. 52, 109–118, https://doi.org/10.1007/s10344-005-0013-3 (2006).
van Wieren, S. E., Wiersma, M. & Prins, H. H. T. Climatic factors affecting a brown hare (Lepus europaeus) population. Lutra 49, 103–110 (2006).
Goszczyński, J. & Wasilewski, M. Predation of foxes on a hare population in central Poland. Acta Theriol. 37, 329–338 (1992).
Rattenborg, E. E. Diseases in the hare (Lepus europaeus) population in Denmark - epidemiological description and analyses of post-mortem data. (The Royal Veterinary & Agricultural University, 1994).
Haerer, G., Nicolet, J., Bacciarini, L., Gottstein, B. & Giacometti, M. Todesursachen, Zoonosen und Reproduktion bei Feldhasen in der Schweiz. Schweiz Arch. Tierheilkd. 143, 193–201, https://doi.org/10.5169/seals-591650 (2001).
Frölich, K. et al. Epizootiologic and ecologic investigations of European brown hares (Lepus europaeus) in selected populations from Schleswig-Holstein, Germany. J Wildl. Dis. 39, 751–761 (2003).
Homolka, M. & Zima, J. In The Atlas of European Mammals (eds Mitchell-Jones, A. J. et al.) (Academic Press, 1999).
Smith, R. K., Jennings, N. V. & Harris, S. A quantitative analysis of the abundance and demography of European hares Lepus europaeus in relation to habitat type, intensity of agriculture and climate. Mammal Rev. 35, 1–24, https://doi.org/10.1111/j.1365-2907.2005.00057.x (2005).
Schmidt, N. M., Asferg, T. & Forchhammer, M. C. Long-term patterns in European brown hare population dynamics in Denmark: effects of agriculture, predation and climate. BMC Ecol. 4, 15, https://doi.org/10.1186/1472-6785-4-15 (2004).
Smith, R. K., Jennings, N. V., Robinson, A. & Harris, S. Conservation of European hares Lepus europaeus in Britain: is increasing habitat heterogeneity in farmland the answer? J Appl. Ecol. 41, 1092–1102, https://doi.org/10.1111/j.0021-8901.2004.00976.x (2004).
Tissier, M. L., Handrich, Y., Dallongeville, O., Robin, J.-P. & Habold, C. Diets derived from maize monoculture cause maternal infanticides in the endangered European hamster due to a vitamin B3 deficiency. Proc. R. Soc. B 284, 20162168, https://doi.org/10.1098/rspb.2016.2168 (2017).
Medan, D., Torretta, J. P., Hodara, K., de la Fuente, E. B. & Montaldo, N. H. Effects of agriculture expansion and intensification on the vertebrate and invertebrate diversity in the Pampas of Argentina. Biodivers. Conserv. 20, 3077–3100, https://doi.org/10.1007/s10531-011-0118-9 (2011).
Butler, S. J., Vickery, J. A. & Norris, K. Farmland Biodiversity and the Footprint of Agriculture. Science 315, 381–384, https://doi.org/10.1126/science.1136607 (2007).
Schai-Braun, S. C. et al. The European Hare (Lepus europaeus): A Picky Herbivore Searching for Plant Parts Rich in Fat. PLoS ONE 10, e0134278, https://doi.org/10.1371/journal.pone.0134278 (2015).
Bagóné Vántus, V., Kovács, M. & Zsolnai, A. The rabbit caecal microbiota: development, composition and its role in the prevention of digestive diseases – a review on recent literature in the light of molecular genetic methods. Acta Agraria Kaposváriensis 18, 55–65 (2014).
Bennegadi, N., Gidenne, T. & Licois, D. Impact of fibre deficiency and sanitary status on non-specific enteropathy of the growing rabbit. Anim. Res. 50, 401–413, https://doi.org/10.1051/animres:2001135 (2001).
Michelland, R. J. et al. Rapid adaptation of the bacterial community in the growing rabbit caecum after a change in dietary fibre supply. Animal 5, 1761–1768, https://doi.org/10.1017/S1751731111001005 (2011).
Maslowski, K. M. & Mackay, C. R. Diet, gut microbiota and immune responses. Nat. Immunol. 12, 5–9, https://doi.org/10.1038/ni0111-5 (2011).
Lelkes, L. A review of rabbit enteric-diseases: A new perspective. J Appl. Rabbit Res. 10, 55–61 (1987).
Percy, D. H., Muckle, C. A., Hampson, R. J. & Brash, M. L. The enteritis complex in domestic rabbits: A field study. Can. Vet. J 34, 95–102 (1993).
Posautz, A. et al. Health screening of free-ranging European brown hares (Lepus europaeus) on the German North-Sea island Pellworm. Acta Vet. Scand. 57, 43, https://doi.org/10.1186/s13028-015-0132-0 (2015).
Clemente, J. C., Ursell, L. K., Parfrey, L. W. & Knight, R. The Impact of the Gut Microbiota on Human Health: An Integrative View. Cell 148, 1258–1270, https://doi.org/10.1016/j.cell.2012.01.035 (2012).
Hollister, E. B., Gao, C. & Versalovic, J. Compositional and Functional Features of the Gastrointestinal Microbiome and Their Effects on Human Health. Gastroenterology 146, 1449–1458, https://doi.org/10.1053/j.gastro.2014.01.052 (2014).
McKenna, P. et al. The Macaque Gut Microbiome in Health, Lentiviral Infection, and Chronic Enterocolitis. PLoS Pathog. 4, e20, https://doi.org/10.1371/journal.ppat.0040020 (2008).
Turnbaugh, P. J. et al. The Human Microbiome Project. Nature 449, 804–810, https://doi.org/10.1038/nature06244 (2007).
Kim, H. B. et al. Longitudinal investigation of the age-related bacterial diversity in the feces of commercial pigs. Vet. Microbiol. 153, 124–133, https://doi.org/10.1016/j.vetmic.2011.05.021 (2011).
Goodrich, J. K. et al. Human Genetics Shape the Gut Microbiome. Cell 159, 789–799, https://doi.org/10.1016/j.cell.2014.09.053 (2014).
Dominianni, C. et al. Sex, Body Mass Index, and Dietary Fiber Intake Influence the Human Gut Microbiome. PLoS ONE 10, e0124599, https://doi.org/10.1371/journal.pone.0124599 (2015).
Org, E. et al. Sex differences and hormonal effects on gut microbiota composition in mice. Gut Microbes 7, 313–322, https://doi.org/10.1080/19490976.2016.1203502 (2016).
Ley, R. E. et al. Evolution of Mammals and Their Gut Microbes. Science 320, 1647–1651, https://doi.org/10.1126/science.1155725 (2008).
Rothschild, D. et al. Environment dominates over host genetics in shaping human gut microbiota. Nature 555, 210–+, https://doi.org/10.1038/nature25973 (2018).
Tasnim, N., Abulizi, N., Pither, J., Hart, M. M. & Gibson, D. L. Linking the Gut Microbial Ecosystem with the Environment: Does Gut Health Depend on Where We Live? Front. Microbiol. 8, 1935, https://doi.org/10.3389/fmicb.2017.01935 (2017).
Menke, S. et al. Effects of host traits and land-use changes on the gut microbiota of the Namibian black-backed jackal (Canis mesomelas). FEMS Microbiol. Ecol. 93, fix123, https://doi.org/10.1093/femsec/fix123 (2017).
Fuller, A., Mitchell, D., Maloney, S. K. & Hetem, R. S. Towards a mechanistic understanding of the responses of large terrestrial mammals to heat and aridity associated with climate change. Clim. Change Resp. 3, 10, https://doi.org/10.1186/s40665-016-0024-1 (2016).
Phillips, C. D. et al. Microbiome analysis among bats describes influences of host phylogeny, life history, physiology and geography. Mol. Ecol. 21, 2617–2627, https://doi.org/10.1111/j.1365-294X.2012.05568.x (2012).
Salgado-Flores, A. et al. Rumen and Cecum Microbiomes in Reindeer (Rangifer tarandus tarandus) Are Changed in Response to a Lichen Diet and May Affect Enteric Methane Emissions. PLoS ONE 11, e0155213, https://doi.org/10.1371/journal.pone.0155213 (2016).
Clayton, J. B. et al. Captivity humanizes the primate microbiome. Proc. Natl. Acad. Sci. USA 113, 10376–10381, https://doi.org/10.1073/pnas.1521835113 (2016).
Redford, K. H., Segre, J. A., Salafsky, N., del Rio, C. M. & McAloose, D. Conservation and the Microbiome. Conserv. Biol. 26, 195–197, https://doi.org/10.1111/j.1523-1739.2012.01829.x (2012).
Bahrndorff, S., Alemu, T., Alemneh, T. & Nielsen, J. L. The Microbiome of Animals: Implications for Conservation Biology. Int. J Genomics, 5304028, https://doi.org/10.1155/2016/5304028 (2016).
Hartl, G. B., Suchentrunk, F., Nadlinger, K. & Willing, R. An integrative analysis of genetic differentiation in the brown hare Lepus europaeus based on morphology, allozymes, and mitochondrial DNA. Acta Theriol. 38, 33–57, https://doi.org/10.4098/AT.arch.93-40 (1993).
Sert, H., Suchentrunk, F. & Erdoğan, A. Genetic diversity within Anatolian brown hares (Lepus europaeus Pallas, 1778) and differentiation among Anatolian and European populations. Mamm. Biol. 70, 171–186, https://doi.org/10.1016/j.mambio.2004.10.003 (2005).
Suchentrunk, F., Willing, R. & Hartl, G. B. On eye lens weights and other age criteria of the Brown hare (Lepus europaeus Pallas, 1778). Z Säugetierk. 56, 365–374 (1991).
Hoffmann, D. Populationsdynamik und -enwicklung des Feldhasen in Schleswig-Holstein im Beziehungsgefüge von Klima, Prädation und Lebensraum. (Dr. rer.nat thesis, Universität Trier, 2003).
Nordsee-Insel Pellworm. Natur und Ökologie. Landwirtschaft auf Pellworm, http://www.pellworm.de/so-schoen-ist-es-hier/natur-und-oekologie/landwirtschaft.html (2018).
Verein Ökologisch Wirtschaften! e.V. Landwirtschaft auf Pellworm, http://oeko-verein-pellworm.de/cms/pellworm/landwirtschaft/64-landwirtschaft-auf-pellworm (2018).
Landwirtschaftskammer Niederösterreich. Bezirksspiegel. Bezirksbauernkammer Mistelbach. 11 (Bezirksbauernkammer Mistelbach, Mistelbach, 2017).
Metzler-Zebeli, B. U. et al. Changing Dietary Calcium-Phosphorus Level and Cereal Source Selectively Alters Abundance of Bacteria and Metabolites in the Upper Gastrointestinal Tracts of Weaned Pigs. Appl. Environ. Microbiol. 79, 7264–7272, https://doi.org/10.1128/aem.02691-13 (2013).
The NIH HMP Working Group. et al. The NIH Human Microbiome Project. Genom. Res. 19, 2317–2323, https://doi.org/10.1101/gr.096651.109 (2009).
Jumpstart Consortium Human Microbiome Project Data Generation Working Group. 16S 454 Sequencing Protocol. HMP Consortium (Vers. 4.2.2), https://www.hmpdacc.org/hmp/doc/16S_Sequencing_SOP_4.2.2.pdf (2010).
Caporaso, J. G. et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7, 335–336, https://doi.org/10.1038/nmeth.f.303 (2010).
Edgar, R. C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461, https://doi.org/10.1093/bioinformatics/btq461 (2010).
Wang, Q., Garrity, G. M., Tiedje, J. M. & Cole, J. R. Naïve Bayesian Classifier for Rapid Assignment of rRNA Sequences into the New Bacterial Taxonomy. Appl. Environ. Microbiol. 73, 5261–5267, https://doi.org/10.1128/Aem.00062-07 (2007).
Lozupone, C. & Knight, R. UniFrac: a New Phylogenetic Method for Comparing Microbial Communities. Appl. Environ. Microbiol. 71, 8228–8235, https://doi.org/10.1128/AEM.71.12.8228-8235.2005 (2005).
Vázquez-Baeza, Y., Pirrung, M., Gonzalez, A. & Knight, R. EMPeror: a tool for visualizing high-throughput microbial community data. GigaScience 2, 16, https://doi.org/10.1186/2047-217x-2-16 (2013).
Muyzer, G., de Waal, E. C. & Uitterlinden, A. G. Profiling of Complex Microbial Populations by Denaturing Gradient Gel Electrophoresis Analysis of Polymerase Chain Reaction-Amplified Genes Coding for 16S rRNA. Appl. Environ. Microbiol. 59, 695–700 (1993).
Bartosch, S., Fite, A., Macfarlane, G. T. & McMurdo, M. E. T. Characterization of Bacterial Communities in Feces from Healthy Elderly Volunteers and Hospitalized Elderly Patients by Using Real-Time PCR and Effects of Antibiotic Treatment on the Fecal Microbiota. Appl. Environ. Microbiol. 70, 3575–3581, https://doi.org/10.1128/AEM.70.6.3575-3581.2004 (2004).
Li, M., Penner, G. B., Hernandez-Sanabria, E., Oba, M. & Guan, L. L. Effects of sampling location and time, and host animal on assessment of bacterial diversity and fermentation parameters in the bovine rumen. J Appl. Microbiol. 107, 1924–1934, https://doi.org/10.1111/j.1365-2672.2009.04376.x (2009).
Větrovský, T. & Baldrian, P. The Variability of the 16S rRNA Gene in Bacterial Genomes and Its Consequences for Bacterial Community Analyses. PLoS ONE 8, e57923, https://doi.org/10.1371/journal.pone.0057923 (2013).
R Core Team. R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria, http://www.R-project.org/. (R Foundation for Statistical Computing, Vienna, Austria, 2017)
Pinheiro, J., Bates, D., DebRoy, S., Sarkar, D. & and the R Development Core team. nlme: Linear and Nonlinear Mixed Effects Models v. R Package Version 3.1-131, http://CRAN.R-project.org/package=nlme (2017).
O’Brien, R. M. A Caution Regarding Rules of Thumb for Variance Inflation Factors. Qual. Quant. 41, 673–690, https://doi.org/10.1007/s11135-006-9018-6 (2007).
Nakagawa, S. & Schielzeth, H. A general and simple method for obtaining R 2 from generalized linear mixed-effects models. Methods Ecol. Evol. 4, 133–142, https://doi.org/10.1111/j.2041-210x.2012.00261.x (2013).
Lenth, R. V. Least-Squares Means: The R Package lsmeans. J Stat. Softw. 69, 1–33, https://doi.org/10.18637/jss.v069.i01 (2016).
Delignette-Muller, M. L. & Dutang, C. fitdistrplus: An R Package for Fitting Distributions. J Stat. Softw. 64, 1–34, https://doi.org/10.18637/jss.v064.i04 (2015).
Kooperberg, C. logspline: Logspline Density Estimation Routines v. R Package Version 2.1.9, https://CRAN.R-project.org/package=logspline (2016).
Oksanen, J. et al. vegan: Community Ecology Package v. R package version 2.4-6, http://CRAN.R-project.org/package=vegan (2018).
Pohlert, T. PMCMR: Calculate Pairwise Multiple Comparisons of Mean Rank Sums v. R package Version 4.2, https://CRAN.R-project.org/package=PMCMR (2018).
Ogle, D. FSA: Simple Fisheries Stock Assessment Methods v. R package version 0.8.17, https://CRAN.R-project.org/package=FSA (2017).
McMurdie, P. J. & Holmes, S. phyloseq: An R Package for Reproducible Interactive Analysis and Graphics of Microbiome Census Data. PLoS ONE 8, e61217, https://doi.org/10.1371/journal.pone.0061217 (2013).
Wickham, H. ggplot2. Elegant Graphics for Data Analysis. (Springer, 2009).
Langille, M. G. I. et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotech. 31, 814, https://doi.org/10.1038/nbt.2676 (2013).
Parks, D. H., Tyson, G. W., Hugenholtz, P. & Beiko, R. G. STAMP: statistical analysis of taxonomic and functional profiles. Bioinformatics 30, 3123–3124, https://doi.org/10.1093/bioinformatics/btu494 (2014).
Crowley, E. J. et al. Comparison of the microbial population in rabbits and guinea pigs by next generation sequencing. PLoS ONE 12, e0165779, https://doi.org/10.1371/journal.pone.0165779 (2017).
Zeng, B. et al. The bacterial communities associated with fecal types and body weight of rex rabbits. Sci. Rep. 5, 9342, https://doi.org/10.1038/srep09342 (2015).
Stott, P. Comparisons of digestive function between the european hare (Lepus europaeus) and the european rabbit (Oryctolagus cuniculus): mastication, gut passage, and digestibility. Mamm. Biol. 73, 276–286, https://doi.org/10.1016/j.mambio.2007.07.002 (2008).
Popescu, F. D., Hackländer, K., Arnold, W. & Ruf, T. Effects of season and reproductive state on lipid intake and fatty acid composition of gastrointestinal tract contents in the European hare. J Comp. Physiol. B 181, 681–689, https://doi.org/10.1007/s00360-011-0559-1 (2011).
Bennegadi, N., Fonty, G., Millet, L., Gidenne, T. & Licois, D. Caecal microflora of growing-rabbits affected by a non specific enteropathy. Reprod. Nutr. Develop. 42, S15, https://doi.org/10.1051/rnd/200242001 (2002).
Sirotek, K., Santos, E., Benda, V. & Marounek, M. Isolation, Identification and Characterization of Rabbit Caecal Mucinolytic Bacteria. Acta Vet. Brno 72, 365–370, https://doi.org/10.2754/avb200372030365 (2003).
Belenguer, A., Balcells, J., Fondevila, M. & Torre, C. Caecotrophes intake in growing rabbits estimated either from urinary excretion of purine derivatives or from direct measurement using animals provided with a neck collar: effect of type and level of dietary carbohydrate. Anim. Sci. 74, 135–144, https://doi.org/10.1017/S1357729800052309 (2002).
Boulahrouf, A., Fonty, G. & Gouet, P. Establishment, Counts, and Identification of the Fibrolytic Microflora in the Digestive Tract of Rabbit. Influence of Feed Cellulose Content. Curr. Microbiol. 22, 21–25, https://doi.org/10.1007/bf02106208 (1991).
Padilha, M. T., Licois, D., Gidenne, T. & Carré, B. Caecal microflora and fermentation pattern in exclusively milk-fed young rabbits. Reprod. Nutr. Develop. 39, 223–230, https://doi.org/10.1051/rnd:19990207 (1999).
Jilge, B. & Meyer, H. Coprophagy-dependant changes of the anaerobic bacterial flora in stomach and small intestine of the rabbit. Z Versuchstierkd. 17, 308–314 (1975).
Abecia, L. et al. Molecular profiling of bacterial species in the rabbit caecum. FEMS Microbiol. Lett. 244, 111–115, https://doi.org/10.1016/j.femsle.2005.01.028 (2005).
Dewhirst, F. E. et al. The Canine Oral Microbiome. PLoS ONE 7, e36067, https://doi.org/10.1371/journal.pone.0036067 (2012).
Bekele, A. Z., Koike, S. & Kobayashi, Y. Phylogenetic diversity and dietary association of rumen Treponema revealed using group-specific 16S rRNA gene-based analysis. FEMS Microbiol. Lett. 316, 51–60, https://doi.org/10.1111/j.1574-6968.2010.02191.x (2011).
Ritalahti, K. M. et al. Sphaerochaeta globosa gen. nov., sp. nov. and Sphaerochaeta pleomorpha sp. nov., free-living, spherical spirochaetes. Int. J Syst. Evol. Microbiol. 62, 210–216, https://doi.org/10.1099/ijs.0.023986-0 (2012).
Abt, B. et al. Complete genome sequence of the termite hindgut bacterium Spirochaeta coccoides type strain (SPN1T), reclassification in the genus Sphaerochaeta as Sphaerochaeta coccoides comb. nov and emendations of the family Spirochaetaceae and the genus. Sphaerochaeta. Stand. Genomic Sci. 6, 194–209, https://doi.org/10.4056/sigs.2796069 (2012).
Holman, D. B., Brunelle, B. W., Trachsel, J. & Allen, H. K. Meta-analysis To Define a Core Microbiota in the Swine Gut. mSystems 2, e00004–00017, https://doi.org/10.1128/mSystems.00004-17 (2017).
Zhang, L., Wu, W., Lee, Y.-K., Xie, J. & Zhang, H. Spatial Heterogeneity and Co-occurrence of Mucosal and Luminal Microbiome across Swine Intestinal Tract. Front. Microbiol. 9, https://doi.org/10.3389/fmicb.2018.00048 (2018).
Combes, S., Fortun-Lamothe, L., Cauquil, L. & Gidenne, T. Engineering the rabbit digestive ecosystem to improve digestive health and efficacy. Animal 7, 1429–1439, https://doi.org/10.1017/S1751731113001079 (2013).
Ormerod, K. L. et al. Genomic characterization of the uncultured Bacteroidales family S24-7 inhabiting the guts of homeothermic animals. Microbiome 4, 36, https://doi.org/10.1186/s40168-016-0181-2 (2016).
Garrett, W. S. et al. Enterobacteriaceae Act in Concert with the Gut Microbiota to Induce Spontaneous and Maternally Transmitted Colitis. Cell Host Microbe 8, 292–300, https://doi.org/10.1016/j.chom.2010.08.004 (2010).
García, A., Fox, J. G. & Besser, T. E. Zoonotic Enterohemorrhagic Escherichia coli: A One Health Perspective. ILAR J 51, 221–232, https://doi.org/10.1093/ilar.51.3.221 (2010).
Swennes, A. G. et al. Enteropathogenic Escherichia coli prevalence in laboratory rabbits. Vet. Microbiol. 163, 395–398, https://doi.org/10.1016/j.vetmic.2013.01.006 (2013).
Acknowledgements
We would like to thank Renate Hengsberger for helping in preparing and revising the MS. This work was supported by research funds of the government of Lower Austria and the competence centre FFoQSI. FFoQSI is funded by the Austrian ministries BMVIT, BMDW and the Austrian provinces Lower Austria, Upper Austria and Vienna within the scope of COMET -Competence Centers for Excellent Technologies. The program COMET is handled by the Austrian Research Promotion Agency FFG. The funder had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
Conceptualization: G.S., E.M., A.P. and S.S.; Data sampling, curation: A.P., A.K.H., I.L. and D.H.; Laboratory work: D.J., I.L. and E.M.; Statistical analysis: B.P. and S.V.; Bioinformatic analysis: D.J., B.Z. and E.M.; Visualization: B.P., D.J., B.Z. and E.M.; Funding acquisition: G.S. and E.M.; Methodology: G.S., E.M., M.W., F.H., S.S. and I.L.; Project administration: G.S. and E.M.; Resources: G.S., E.M., A.K.H., M.W. and D.H.; Writing – original draft: G.S., E.M. and B.P.; Review & editing: all authors.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Stalder, G.L., Pinior, B., Zwirzitz, B. et al. Gut microbiota of the European Brown Hare (Lepus europaeus). Sci Rep 9, 2738 (2019). https://doi.org/10.1038/s41598-019-39638-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-39638-9
This article is cited by
-
Experimental validation of small mammal gut microbiota sampling from faeces and from the caecum after death
Heredity (2021)
-
Individual fate and gut microbiome composition in the European wild rabbit (Oryctolagus cuniculus)
Scientific Reports (2021)
-
Survival rates on pre-weaning European hares (Lepus europaeus) in an intensively used agricultural area
European Journal of Wildlife Research (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
