
Abstract
Culture-independent methods have contributed substantially to our understanding of global microbial diversity. Recently developed algorithms to construct whole genomes from environmental samples have further refined, corrected and revolutionized understanding of the tree of life. Here, we assembled draft metagenome-assembled genomes (MAGs) from environmental DNA extracted from two hot springs within an active volcanic ecosystem on the Kamchatka peninsula, Russia. This hydrothermal system has been intensively studied previously with regard to geochemistry, chemoautotrophy, microbial isolation, and microbial diversity. We assembled genomes of bacteria and archaea using DNA that had previously been characterized via 16S rRNA gene clone libraries. We recovered 36 MAGs, 29 of medium to high quality, and inferred their placement in a phylogenetic tree consisting of 3,240 publicly available microbial genomes. We highlight MAGs that were taxonomically assigned to groups previously underrepresented in available genome data. This includes several archaea (Korarchaeota, Bathyarchaeota and Aciduliprofundum) and one potentially new species within the bacterial genus Sulfurihydrogenibium. Putative functions in both pools were compared and are discussed in the context of their diverging geochemistry. This study adds comprehensive information about phylogenetic diversity and functional potential within two hot springs in the caldera of Kamchatka.
Similar content being viewed by others

Introduction
Terrestrial hydrothermal systems are of great interest to the general public and to scientists alike due to their unique and extreme conditions. Hot springs have been sought out by geochemists, astrobiologists and microbiologists around the globe who are interested in their chemical properties, which provide a strong selective pressure on local microorganisms. Drivers of microbial community composition in these springs include temperature, pH, in-situ chemistry, and biogeography1,2,3. The heated water streams contain substantial concentrations of carbon dioxide, nitrogen, hydrogen, and hydrogen sulphide. Moreover, high temperature subterranean erosion processes can result in elevated levels of soluble metals and metalloids. Microbes in these communities have evolved strategies to thrive in these conditions by converting hot spring chemicals into cellular energy4.
The Uzon Caldera is part of the Pacific Ring of Fire and is one of the largest active volcanic ecosystems in the world4. The geochemical properties of this system have been studied in detail5,6,7. The systematic study of Kamchatka thermophilic microbial communities was initiated by Georgy Zavarzin in the early 1980’s8. Briefly, the Uzon Caldera was created by a volcanic eruption. It is characterized by high water temperatures (20–95° Celsius), a wide range of pH (3.1–9.8), and many small lakes that are filled with sediment and pumice, dacite extrusions, and peatbog deposits9,10. Most of the hydrothermal springs lack dissolved oxygen, but harbour sulphides and rare trace elements (antimony, arsenic, boron, copper, lithium, and mercury)11. Many previously undiscovered bacteria have been isolated in this region using culture-dependent methods12,13,14, and 16S rRNA gene amplicon sequencing has revealed a diverse collection of new lineages of archaea15,16. Microorganisms from Uzon Caldera are well represented in culture collections17 and have also been previously studied using culture-independent genomic methods8,9,11,18,19,20,21,22,23.
In this study, we focus on two hydrothermal pools, Arkashin Schurf and Zavarzin Spring in the Uzon Caldera that were previously characterized using 16S ribosomal RNA gene sequencing and geochemical analysis by Burgess et al.9 (Fig. 1). Arkashin Schurf (ARK) is an artificial pool, approximately 1 m2 in size, in the central sector of the East Thermal Field (54°30′0″N, 160°0′20″E), which was dug during a prospecting expedition to Uzon by Arkadiy Loginov10. ARK has been generally stable in size and shape since its creation24. Flocs ranging in colour from pale yellow-orange to bright orange-red have been observed floating in ARK10. This pool is characterized by high concentrations of arsenic and sulphur, which result from the oxidation and cooling of magmatic waters as they reach the surface of the caldera9. Zavarzin Spring (ZAV) is a natural pool, approximately 10 m2 in size, in the Eastern Thermal Field (54°29′53″N, 160°0′52″E). Unlike ARK, the size and shape of ZAV is constantly in flux as vents collapse and emerge and as the amount of snowmelt changes24. Green microbial mats have been observed around the edge of ZAV and thicker brown and green mats have been found within the pool itself10.

Sampling locations in the Uzon Caldera, Kamchatka Russia. DNA had been extracted in 2009 by Burgess et al.9 from sediment samples of two active thermal pools Arkashin Schurf (b) and Zavarzin Spring (c). Photos were taken by Dr. Russell Neches (ORCID: 0000-0002-2055-8381) during an expedition in 2012 and he granted permission through a CC-BY license 4.0. Maps were plotted in R v. 3.4.0 with the package ‘ggmap’ v. 2.6.1101.
Burgess et al.9 found that the two pools differed geochemically with ARK containing higher amounts of total arsenic, rubidium, calcium, and caesium; and ZAV containing higher amounts of total vanadium, manganese, copper, zinc, strontium, barium, iron and sulphur10. Water temperatures in ARK ranged from 65 °C near the vent to 32 °C at the edge of the pool with temperatures as high as 99 °C at 10 cm depth into the vent sediments. ZAV showed relatively lower temperatures between 26 °C and 74 °C at different locations of the pool.
Using the same DNA sample that had been used in the Burgess et al. study9 as our starting material, we applied a metagenomic whole genome shotgun sequencing approach. We generated metagenomic assemblies from the shotgun sequence data for these environmental samples and then binned contigs into individual population-specific genomes and then identified and annotated taxonomic and functional genes for the microorganisms in the two pools. In contrast to the 16S rRNA gene amplicon approach, metagenomic sequencing avoids taxonomic primer bias25, provides more direct functional prediction information about the system26, and ultimately can result in a more precise taxonomy through multi-gene and whole-genome phylogenetic approaches27. However, at low sequencing depths, metagenomic sequencing and whole genome binning capture only the most abundant bacterial genes in the pools. Accordingly, we tested the following questions: (1) Can we recover metagenome-assembled genomes (MAGs) from the two pools? Are there any previously undiscovered or unobserved taxa that can be described using this approach? (2) How do any identified MAGs compare to Burgess et al.’s survey of the microbes in these pools? Do we find any archaea in the ARK pool from which Burgess et al. were unable to amplify any 16S rRNA gene sequences? (3) How do any MAGs found here fit into current views of the microbial tree of life? (4) Can we identify any differences in the functional genes or specific MAGs between the two pools that might be explained by their diverging geochemistry?
Results
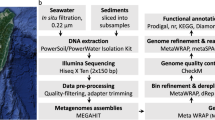
Quality filtering and assembly
DNA libraries were prepared and then sequenced using Solexa3 84 bp paired-end sequencing. For ARK, 52,908,626 Solexa reads (representing in total 4,444,324,584 bases) were processed while 77% of the reads were retained after adaptor removal and 76.32% passed trimming to Q10 (Supplementary Table S1). For ZAV, 58,334,696 Solexa reads were processed (representing in total 4,900,114,464 bases) while 59.91% were retained and 59.05% passed the cut-offs. Reads for each sample were then assembled using SPAdes28. The ARK assembly generated 103,026 contigs of sizes from 56 to 103,908 bp with an N50 of 3,059. The ZAV assembly generated 151,500 contigs of sizes from 56 to 791,131 base pairs with an N50 of 2637 (Supplementary Table S1). Sanger metagenomic reads were also generated from clone libraries for ARK and ZAV, but not used for metagenomic assemblies and binning.
Metagenome-assembled genome quality and taxonomic identification
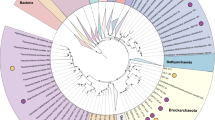
Using anvi’o29, we assembled 36 draft MAGs, 20 from ZAV (three high-quality, 12 medium-quality and five low-quality) and 16 from ARK (seven high-quality, seven medium-quality and two low-quality; Table 1, Supplementary Table S2). These MAGs include only 11.7% of the nucleotides present in the ZAV assembly and 19.2% of the nucleotides in the ARK assembly. MAGs from ZAV and ARK were taxonomically inferred to be bacteria (n = 22; Fig. 2, Supplementary Fig. S1) and archaea (n = 14). MAGs from ARK were taxonomically assigned to a diverse group of 12 phyla (Table 2). A similar range in taxonomic diversity, 13 phyla, is seen in MAGs binned from ZAV (Table 3).

Placement of the MAGs into their phylogenetic context. Taxonomy of the MAGs (metagenome-assembled genomes) was refined by placing them into a phylogenetic tree using PhyloSift v. 1.0.1 with its updated markers database for the alignment and RAxML v. 8.2.10 on the CIPRES web server for the tree inference. This tree includes the 36 MAGs (red dots), all taxa previously identified by Burgess et al. (2012) with complete genomes available on NCBI (n = 148)82, and 3,102 archaeal (yellow) and bacterial (grey) genomes previously used in Hug et al. (2016)79,80,81. The complete tree in Newick format and its alignment of 37 concatenated marker genes can be found on Figshare75,100. Branches with MAGs found in Arkashin Schurf (ARK) and Zavarzin Spring (ZAV) are enlarged (orange nodes). Blue: taxa from Burgess et al. (2012), black: taxa from Hug et al. (2016). GCA IDs from NCBI are shown for the closest neighbours of the MAGs. (a) Microbial tree of life, reconstructed with genomes representing taxa reported in Burgess et al. highlighting the placement of MAGs in this study; (b) Dictyoglomales, Thermoanaerobacteriales, Caldisericales, and Mesoaciditogales; (c) Nitrosphaera, Bathyarchaeota, Korarchaeota, and Crenarchaeota; (d) Chloroflexales; (e) Euryarchaeota; and (f) Deferribacteriales, Desulfobacteriales, and Aquificales. ARK-02, ARK-03, ARK-04, ZAV-04, ZAV-08, ZAV-09, and ZAV-15 can be found in Supplementary Figure S1.
Phylogenetic placement of MAGs
Phylogenetic analysis placed MAGs into the following bacterial clades: two in the Chloroflexales (ZAV-01, ZAV-02; Fig. 2), one in Deferribacteriales (ZAV-05), two in Desulfobacteriales (ARK-08, ZAV-10), four in Aquificales (ARK-05, ARK-13, ZAV-12, ZAV-16), one in Dictyoglomales (ZAV-14), one in Thermoanaerobacteriales (ARK-09), two in Caldisericales (ARK-10, ZAV-07), two in Mesoaciditogales (ARK-11, ZAV-03), three in Thermodesulfobacteria (ARK-04, ZAV-08, ZAV-15; Supplementary Fig. S1), two in Sphingobacteriales (ARK-03, ZAV-09), one in Acidobacteriales (ARK-02), and one in Thermodesulfovibrio and sister to Dadabacteria (ZAV-04). Within the archaea, phylogenetic analysis placed MAGs into one of the following groups or positions: one MAG in Candidatus Nitrosphaera (ARK-01; Fig. 2), three in Bathyarchaeota (ZAV-11, ZAV-13, ZAV-17), one in Korarchaeota (ZAV-18), one sister to Crenarchaeota (ARK-16), and seven in Crenarchaeota (ARK-12, ARK-14, and ZAV-06 most closely related to Fervidicoccus; and ARK-06, ARK-07, ZAV-19, and ZAV-20 most closely related to Caldisphaera). ARK-15 shared a common ancestor with an Aciduliprofundum species, nested within Thermoplasmatales and sister to Euryarchaeota. Compared to identification with CheckM there were two ambiguities: ARK-16 was assigned to Korarchaeota (Supplementary Table S3) vs. a sister to Crenarchaeota (Fig. 2); and ARK-02 was assigned to Candidatus Aminicenantes (Supplementary Table S3) vs. Acidobacteriales (Supplementary Fig. S1).
Taxonomic inference of 16S rRNA gene sequences from Burgess et al
Burgess et al.9 previously generated 16S rRNA gene sequences from clone libraries to investigate the archaeal and bacterial diversity of these pools. By downloading the Burgess et al.9 16S rRNA gene sequence data and analysing it using an updated database, we were able to infer taxonomy for some sequences that were previously unclassified (Tables S4, S5 and S6). We identified representatives of two new archaeal phyla (Aenigmarchaeota and Thaumarchaeota) in ZAV and saw a decrease in the proportion of unidentified archaeal sequences from the 13% reported by Burgess et al. to 7.7%. We also found no representatives of Euryarchaeota, which had previously been reported as 7% of the sequence library and suspect that these reads may have been reassigned to different taxonomic groups due to updates to the Ribosomal Database Project (RDP) database. Several previously unobserved phyla were identified as small proportions of the ZAV bacterial sequence library including Actinobacteria (0.3%), Atribacteria (4%), Elusimicrobia (1%), Ignavibacteriales (2.7%) and Microgenomates (0.3%). We saw only a moderate decrease in the unclassified bacteria from 24% to 17.3%. In comparison, we report a large decrease in the proportion of unclassified bacteria in the ARK sequence library from 19% to only 2.9%. This can be attributed to the identification of two previously unobserved phyla in the ARK bacterial library, Candidatus Aminicenantes (3.4%) and Thermotogae (13.1%).
Taxonomic comparison to 16S rRNA gene sequence libraries
Taxonomic assignments for seven of the nine bacterial MAGs found in ARK placed them in the same genera identified from the clone libraries prepared by Burgess et al.9 (Table 2). Since Burgess et al. were unable to amplify archaeal sequences from ARK, there were no 16S rRNA gene results on archaea to which to compare. Here, we were able to identify archaea from four different archaeal phyla including representatives of novel lineages. Eight of the thirteen bacterial and two of the seven archaeal MAGs found in ZAV match genera from the libraries constructed in Burgess et al.9 (Table 3).
Further comparing the 16S rRNA gene sequence libraries from ARK and ZAV to the inferred phyla present in the Sanger metagenomes prepared by TIGR and the quality-filtered Solexa reads, we found that the latter were able to detect additional phyla present in these hydrothermal systems (Tables S7 and S8). All of the assembled MAGs matched phyla observed using either all three methods (16S rRNA gene sequence libraries, Sanger metagenomes, Solexa reads) or using both the Sanger metagenomes and Solexa reads, but not the 16S rRNA gene sequence libraries (Fig. 3).

Shared phyla between MAGs and different sequencing methods. Venn diagrams depict the number of shared phyla observed between metagenome assembled genomes (MAGs) and different methods of sequencing and taxonomic assignment for (a) Arkashin Schurf (ARK) and (b) Zavarzin Spring (ZAV). The different methods include the Ribosomal Database Project v. 11.5 inferred taxonomy of the 16S rRNA gene Sanger clone libraries prepared by Burgess et al.9, the Kaiju v. 1.6.2 inferred taxonomy for the Sanger metagenomes prepared by TIGR and the Kaiju v. 1.6.2 inferred taxonomy for the Solexa reads which were later assembled to bin the MAGs. The different circles represent the 16S rRNA genes (blue), the MAGs (yellow), the Sanger metagenomes (orange) and the Solexa reads (magenta).
Comparison of genera found in both pools
Due to the diverging biogeochemistry between ARK and ZAV, we were interested in if shared genera between pools would be more similar to each other or to existing reference genomes. To investigate this question, we focused comparisons on two genera, Desulfurella and Sulfurihydrogenibium, for which draft MAGs were obtained in both pools with high completion (>90%).
For Desulfurella, the MAGs obtained from both pools at first visually appeared to be closely related to each other forming a clade with existing reference genomes for Desulfurella multipotens and D. acetivorans (Fig. 4). This was confirmed when we calculated pairwise average nucleotide identities (ANI) and found that ARK-08, ZAV-10, D. multipotens and D. acetivorans should all be considered the same species (ANI >95%; Supplementary Table S9). A threshold of greater than 95% ANI is generally considered appropriate for assigning genomes to the same species30.

Pangenomic comparison of shared genera between pools. Desulfurella genera (a) and Sulfurihydrogenibium genera (b) identified in both Arkashin Schurf (ARK) and Zavarzin Spring (ZAV) are visualized respectively in anvi’o against reference genomes downloaded from NCBI. ARK-08 and ZAV-10 were compared to three representative Desulfurella genomes including D. multipotens (GCA_900101285.1), D. acetivorans (GCA_000517565.1) and D. amilsii (GCA_002119425.1), while bins ARK-13 and ZAV-16 were compared to four representative Sulfurihydrogenibium genomes including S. subterraneum (GCA_000619805.1), S. yellowstonense (GCA_000173615.1), S. azorense (GCA_000021545.1) and S. sp. YO3AOP1 (GCA_000020325.1). Genomes are arranged based on a phylogenetic tree of shared single-copy core genes produced in anvi’o using FastTree v. 2.1. Gene clusters have been grouped into categories based on presence/absence including: ‘Single-copy core genes’ (gene clusters representing # genes from Campbell et al.)64, ‘Universally shared’ (gene clusters present in all genomes), ‘Often Shared’ (gene clusters present in two or more genomes) and ‘Unique’ (gene clusters present in only one genome). Gene calls were annotated in anvi’o using NCBI’s Clusters of Orthologous Groups (COG’s). Gene clusters with an assigned NCBI COG are indicated in black.
For Sulfurihydrogenibium, the two MAGs appear to be more closely related to each other than to existing reference genomes and appear to form their own clade (Fig. 4b). This inference is supported by high ANI values from which we infer that the two MAGs are actually the same species (ANI = 97.2%; Supplementary Table S10). The ANI values of these MAGs suggest that they comprise a distinct species for this genus when compared to the four existing reference genomes (ANI <76%).
Functional annotations; qualitative differences between ARK and ZAV
High concentrations of arsenic have previously been found in Arkashin Schurf, hence we searched specifically for homologs of genes involved in arsenic biotransformation and compared them between the two pools. Homologs of genes encoding proteins that are predicted to be involved in the arsenic biogeochemical cycle were present in both pools (n = 86 for ARK and n = 73 for ZAV; Supplementary Table S11). These included homologs of ArsA, ArsB, ArsC and ArsH, ACR3, Arsenite_ox_L, Arsenite_ox_S, and the ArsR regulator (Supplementary Table S12). Homologs of ArsH were restricted to Arkashin Schurf. ACR3 could be assigned to MAG ARK-10; ArsA to ARK-07, ARK-11, ARK-16, and ZAV-03; Arsenite_ox_L to ARK-07 and ARK-16; and Arsenite_ox_S to ARK-01 and ARK-07.
We searched for complete chemical pathways in both pools and linked them to the MAGs. In total, 222 complete KEGG gene pathways were predicted to be present in ARK and ZAV combined. Completeness of a pathway is defined as including all necessary components (gene blocks) to complete a metabolic cycle. Nine complete pathways were predicted to be present exclusively in ARK (Supplementary Table S13) and 14 pathways in ZAV. We grouped the 119 shared gene pathways that could be found in both pools based on KEGG orthologies into carbohydrate and lipid metabolism (n = 31; Supplementary Table S14), energy metabolism (n = 16; Table 4), and environmental information processing (n = 31; Supplementary Table S15). An exhaustive list of all predicted KEGG pathways in both pools and KEGG pathway maps can be found in Supplementary Table S16. Detailed diagrams of ARK-15 (Aciduliprofundum) and ZAV-18 (Korarchaeota) can be found in Supplementary Fig. S2. Metabolic pathway summary pie charts were made for these bins because of their high completion estimates and relative novelty.
Discussion
We recovered 36 MAGs (20 in ZAV and 16 in ARK) comprising a broad phylogenetic range of archaeal and bacterial phyla. Moreover, the MAG’s we constructed from two volcanic hot springs fill in some phylogenetic gaps in the collection of available genomes and thus can be useful for inferring details about microbial phylogenetic relationships. These include draft MAGs for several candidate phyla including archaeal Korarchaeota, Bathyarchaeota and Aciduliprofundum; and bacterial Aminicenantes. Korarchaeota and Crenarchaeota have been found in thermal ecosystems previously using 16S rRNA gene amplicon sequencing15,31,32 but with very little genomic information so far19,33. Korarchaeota have been described exclusively in hydrothermal environments34. They belong to one of the three major supergroups in the Euryarchaeota, together with the Thaumarchaeota, Aigarchaeota and the Crenarchaeota (TACK, proposed name Eocyta)35. TACK make up a deeply branching lineage that does not seem to belong to the main archaeal groups. Bathyarchaeota are key players in the global carbon cycle in terrestrial anoxic sediments36,37. They appear to be methanogens and can conserve energy via methylotrophic methanogenesis (see below). Aciduliprofundum spp. have only been found in hydrothermal vents and have one cultivated representative; Aciduliprofundum boonei38. This taxon is an obligate thermoacidophilic sulphur and iron reducing heterotroph. Aminicenantes (candidate phylum OP8) is a poorly characterized bacterial lineage that can be found in various environments, such as hydrocarbon-contaminated soils, hydrothermal vents, coral-associated, terrestrial hot springs, and groundwater samples39. A high-level of intraphylum diversity with at least eight orders has been proposed for this group40.
The phylogenetic tree constructed here using 37 single-copy marker genes recapitulates and confirms the structure seen in other recent studies using different sets of single-copy genes27,41. The placement of the MAGs into this phylogeny was in agreement with their taxonomic assignments based on CheckM’s marker set with two exceptions42.
The first exception is ARK-16 clustering with Crenarchaeota in the tree but getting assigned to Korarchaeota with CheckM. Only one genomic representative is currently available for the phylum Candidatus Korarchaeota (other than ZAV-18). It is possible that ARK-16 is still a member of this phylum but is also distantly related to the available genome leading to the branching pattern observed here (Fig. 2). Other possibilities include that ARK-16 represents a novel phylum of archaea that is sister to Crenarchaeota or that ARK-16 is a new group within the Crenarchaeota.
The second exception is ARK-02 clustering next to Acidobacterium spp. (Acidobacteriales) in the tree but getting assigned to the Aminicenantes group with CheckM. Previous phylogenomic studies have placed Candidatus Aminicenantes as sister to the Acidobacteriales43. Thus, the placement of ARK-02 as sister to Acidobacteriales here is likely not an ambiguity and instead further supports Candidatus Aminicenantes as its proper taxonomic placement (Supplementary Fig. S1). Originally, we had included three Candidatus Aminicenantes species when building Fig. 2, but they were removed by trimAl because they were missing a substantial number of the 37 single-copy marker genes.
Generally, many of the same taxa as the MAGs assembled here were also observed in the analysis of the 16S rRNA gene sequence library prepared by Burgess et al.9. Using an updated version of the RDP database decreased the proportion of unclassified sequences in the ARK bacterial library and the ZAV archaeal library, identifying several new phyla in both sequence libraries. The proportion of unclassified sequences in the ZAV bacterial library decreased slightly but remained relatively high (17.3%; Supplementary Table S6), indicating that there is still a large amount of bacterial novelty in ZAV. It is possible that this proportion can be partially explained by the five bacterial ZAV MAGs that do not represent genera identified in the clone library, but which were observed in the Sanger metagenomic reads. These include members of the phyla Aquificae, Bacteriodetes, Thermodesulfobacteria and Thermotogales (Table 3; Supplementary Table S8).
A recent 16S rRNA gene sequencing study by Merkel et al.8 found the most abundant members of ARK to be the archaea Thermoplasmataceae group A10 (phylum Euryarchaeota) and Caldisphaera at 34% and 30% relative abundance respectively. Here, we generated two medium quality draft MAGs that were assigned to Caldisphaera (ARK-06; ARK-07). We did not generate any draft MAGs for members of the Thermoplasmataceae group A10, but we did bin a high-quality draft MAG from a candidate group in the same phylum, Candidatus Aciduliprofundum (ARK-15). In Fig. 2, the closest relative to ARK-15 is Aciduliprofundum sp. MAR08-339 and together they form a sister group to Picrophilus oshimae, a member of the Thermoplasmataceae. Candidatus Aciduliprofundum has been previously placed next to Thermoplasmataceae based on a maximum likelihood phylogenetic tree using 16S rRNA genes38 and a Bayesian phylogeny constructed from the concatenation of 57 ribosomal proteins44. However, the relationship of Aciduliprofundum to other archaea is still unresolved45.
Even though the number of archaeal taxa represented in genome data has increased since Burgess et al.9, the current understanding of archaeal phylogenetic diversity is still limited. Primer bias is known to historically plague archaeal amplicon sequencing studies46 and additionally may explain why Burgess et al. were unable to amplify archaeal sequences from ARK. The novel archaeal MAGs assembled here, combined with the additional archaeal and bacterial phyla identified in the Sanger metagenomes, provides a good argument for re-examining previously characterized environments using new methods to further expand the view of the tree of life (Fig. 3).
After investigating the pangenomes of the draft MAGs that were assigned to the Desulfurella genus (ARK-08 and ZAV-10) relative to reference genomes, we propose that these MAGs, D. multipotens and D. acetivorans should all be considered the same species. The collapse of D. multipotens and D. acetivorans into one species has been previously suggested by Florentino et al. based on both ANI and DDH (DNA-DNA hybridization) values47. Two species of Desulfurella were previously isolated from Kamchatka, D. kamchatkensis and D. propionica48. Neither strain has a publicly available genome sequence, although Miroshnichenko et al. performed DDH between these strains and D. acetivorans finding values of 40% and 55% respectively indicating that these are unique strains. However, the authors also found that there was high sequence similarity (>99%) between full length 16S rRNA genes for D. multipotens, D. acetivorans, D. kamchatkensis and D. propionica. Given this, and that the MAGs, D. multipotens and D. acetivorans appear to be one species, we wonder what the genomes of D. kamchatkensis and D. propionica might reveal about the relationships within this genus. This situation highlights the need for an overhaul in microbial taxonomy based on whole genome sequences, a concept which has been proposed and discussed by many for years (e.g., Hugenholtz et al.)49, but which has been particularly highlighted recently in Parks et al.41.
Meanwhile, the MAGs assigned to the Sulfurihydrogenibium genus (ARK-13 and ZAV-16) appear to be from a previously unsequenced species for this genus when compared to existing reference genomes. It is possible that these MAGs represent draft genomes of S. rodmanii, a novel species of Sulfurihydrogenibium that was previously cultured from hot springs in the Uzon Caldera, but for which a reference genome does not yet exist13. Sulfurihydrogenibium rodmanii is a strict chemolithoautotroph, it is microaerophilic and utilizes sulphur or thiosulfate as its only electron donors and oxygen as its only electron acceptor. ARK-13 has a GC content of 34.23% and ZAV-16 has a GC content of 34.32% (Table 1). These closely match the GC content estimate reported for S. rodmanii of 35%13. Additionally, S. rodmanii is the best match for several of the Sulfurihydrogenibium sequences in the Burgess et al. clone library, providing further support for the hypothesis that the MAGs identified in this study may represent members of this species.
Out of the total 222 predicted complete KEGG pathways, nine were unique to ARK and 15 to ZAV. Interestingly, homologs of the complete denitrification pathway M00319 and anoxygenic photosystem II M00597 were only found in ZAV. Denitrification is a respiration process in which nitrate or nitrite is reduced as a terminal electron acceptor under low oxygen or anoxic conditions and in which organic carbon is required as an energy source8. Denitrification has been predicted to be carried out mostly by Thiobacillus spp., Micrococcus spp., Pseudomonas spp., Achromobacter spp., and Calditerrivibrio spp. in the Uzon Caldera16. The last of these, Calditerrivibrio, is represented here by ZAV-05 which could have contributed homologs to this complete, predicted KEGG pathway. This is in agreement with Burgess et al.’s stable isotope analysis of 15N.
Anoxic photosynthesis is performed by obligate and facultative anaerobes such as Chloroflexus and Roseiflexus and requires energy in the form of sunlight. Both genera were present in ZAV. Moreover, we found three versions of the complete anoxygenic photosystem II pathway M00597 in the ZAV pool overall, and one version of the chlorophyll metabolism pathway M00680 could be assigned to bin ZAV-02 (Chloroflexus).
We reconstructed 31 different predicted carbohydrate and lipid metabolism KEGG pathways using homologous genes in ARK and ZAV including cell wall component biosynthesis; e.g., isoprenoids and other lipopolysaccharides. Both pools are predicted to contain genes that encode proteins for all major aerobic energy cycles, including the complete citrate cycle, Entner-Doudoroff, Leloir, and Embden-Meyerhof pathway. Carbon fixation through autotrophic CO2 fixation was represented by four major pathways in both pools: crassulacean acid metabolism (CAM), Wood-Ljungdahl pathway, Arnon-Buchanan cycle, and reductive pentose phosphate cycle. There are many variants of the Wood-Ljungdahl pathway, one of which is preferred by sulphate-reducing microbial organisms that grow by means of anaerobic respiration50. Coupled with methanogenesis; i.e., the Acetyl-CoA and the F420 pathway reducing CO2,51. Recently it has been shown that Bathyarchaeota possess the archaeal Wood-Ljungdahl pathway36,37. Similarly, the Arnon-Buchanan cycle is commonly found in anaerobic or microaerobic microbes present at high temperatures, such as Aquificae and Nitrospirae52.
We found homologs of three predicted, complete major nitrogen pathways and three complete major sulphur pathways in both pools. These included assimilatory nitrate reduction to ammonia, dissimilatory nitrate reduction to ammonia and nitrogen fixation from nitrogen to ammonia. With regard to sulphur, the metabolisms included thiosulfate oxidation to sulphate, assimilatory sulphate reduction to H2S, and dissimilatory sulphate reduction to H2S. These two pathways; i.e., aerobic sulphur oxidation and anaerobic hydrogen oxidation coupled with sulphur compound reduction can be performed by aerobic Sulfurhydrogenibium and anaerobic Caldimicrobium8. We assembled MAGs assigned to both of these taxa in ZAV (ZAV-16 and ZAV-15) and of the former taxon in ARK (ARK-13). Sulphate-reducing bacteria can also change the concentration of arsenic in a pool by generating hydrogen sulphide, which leads to reprecipitation of arsenic53. Hence, the presence of sulphur oxidizers and sulphur reducers in a pool can significantly impact the fate of environmental arsenic.
Homologs of predicted protein families that play a role in the biotransformation of arsenic were found in both pools. This includes homologs of the predicted genes arsB and ACR3, which code for arsenite (As(III)) pumps that remove reduced arsenic from the cell54. Early microorganisms originated in anoxic environments with high concentrations of reduced As(III)55. Most microbes have evolved efflux systems to get rid of As(III) from their cells54. Hence, nearly every extant microbe is armed with As(III) permeases, such as ArsB or ACR353. Some organisms evolved genes encoding anaerobic respiratory pathways utilizing As(III) as an electron donor to produce energy while oxidizing As(III) to As(V)56. This type of arsenic cycling has been predicted to be carried out by members of Hydrogenobaculum spp., Sulfurihydrogenibium spp., Hydrogenobacter spp., and other Aquificales57. We found MAGs assigned to Hydrogenobaculum and Sulfurihydrogenibium in both pools, with two high quality drafts in ARK. In addition to the Aquificales, we also found several copies of ACR3 in ARK-10, Caldisericum exile.
With increasing atmospheric oxygen concentrations, As(III) is oxidized to As(V), a toxic compound which can enter the cells of most organisms via the phosphate uptake systems54. Consequently, organisms needed to find ways to survive with these environmental toxins inside their cells. This was the advent of several independently evolved As(V) reductases, such as the arsC system53. The only protein homolog which was found exclusively in ARK is ArsH, which is presumably involved in arsenic methylation. In addition to oxidation and reduction of inorganic arsenic species, arsenic methylation is another strategy to detoxify As(V)53. Common methylation pathways include ArsM and ArsH and are regulated by the As(III)-responsive transcriptional repressor ArsR, which was common in both pools. Coupled with ATP hydrolysis, some microbes also developed an energy-dependent process where As(III) is actively pumped out of the cell53,58. Driven by the membrane potential, ArsA can bind to ArsB and pump out As(III). Homologs of predicted ArsB proteins were found in both pools with no assignment to any of the MAGs. However, homologs of arsA could be found in ARK-07, ARK-11, ARK-16, and ZAV-03 which correspond to a Korarchaeota representative, a Caldispaera sp. and two Mesoaciditoga species, one in each pool.
The breadth of undiscovered microbial diversity on this planet is extreme, particularly when it comes to uncultured archaea whose abundance has likely been underestimated because of primer bias for years46. By incorporating metagenomic computational methods into the wealth of pre-existing knowledge about these ecosystems, we can begin to putatively characterize the ecological roles of microorganisms in these hydrothermal systems. Future work should aim to isolate and characterize the novel microorganisms in these pools so that we can fully understand their biology, confirm the ecological roles they play, and complement and expand the current models regarding the tree of life.
Methods
Sample collection and DNA extraction
The DNA used here is the same DNA that was used in Burgess et al.9. In short, Burgess et al. extracted DNA from sediment from ARK (Fig. 1), collected in the field in 2004 (sample A04) using the Ultra-Clean® Soil DNA Kit (MoBio Laboratories, Inc., Carlsbad, CA, USA) following the manufacturer’s instructions. They then extracted DNA from sediment from ZAV (Fig. 1) collected in the field in 2005 (sample Z05) using the PowerMax® Soil DNA Isolation Kit (MoBio Laboratories, Inc.) following the manufacturer’s instructions. DNA was sequenced with two approaches: Sanger sequencing of clone libraries at TIGR (The Institute for Genomic Research) and paired-end Solexa3 sequencing of 84 bp at the UC Davis Genome Center. Details on sequencing and clone library construction can be found in the Supplementary Material.
Sequence processing and metagenomic assembly
Quality filtering was performed on Solexa reads using bbMap v. 36.9959 with the following parameters: qtrim = rl, trimq = 10, minlength = 70; i.e., trimming was applied to both sides of the reads, trimming the reads back to a Q10 quality score and only keeping reads with a minimum length of 70 bp. Adaptors were removed from the Solexa reads and samples were assembled into two metagenomes (one for ARK and one for ZAV) using SPAdes v. 3.9.060 with default parameters for the metagenome tool (metaspades). Sanger metagenomic reads were processed using phred61 to make base calls and assign quality scores, and Lucy62 to trim vector and low quality sequence regions.
Metagenomic binning and gene calling
Metagenomic data was binned using anvi’o v. 2.4.029, following a modified version of the workflow described by Eren et al.29. First, a contigs database was generated for each sample from the assembled metagenomic data using ‘anvi-gen-contigs-database’ which calls open reading frames using Prodigal v. 2.6.263. Single-copy bacterial64 and archaeal65 genes were identified using HMMER v. 3.1b266. Taxonomy was assigned to contigs using Kaiju v. 1.5.067 with the NCBI BLAST non-redundant protein database nr including fungi and microbial eukaryotes v. 2017-05-16. In order to visualize the metagenomic data with the anvi’o interactive interface, a blank-profile for each sample was constructed with contigs >1 kbp using ‘anvi-profile’, which hierarchically clusters contigs based on their tetra-nucleotide frequency profiles. Contigs were manually clustered into bins using a combination of hierarchical clustering, taxonomic identity, and GC content using ‘anvi-interactive’ to run the anvi’o interactive interface. Clusters were then manually refined using ‘anvi-refine’ and bins were continuously assessed for completeness and contamination using ‘anvi-summarize’ and the CheckM v. 1.0.742 lineage-specific workflow. Although multiple strains likely contribute to each bin68, we did not investigate strain variability in this study. For a detailed walk-through of the analyses used to bin the metagenomic data, please refer to the associated Jupyter notebooks for ZAV69 and for ARK70.
Taxonomic and phylogenetic inference of MAGs
Using the standards suggested by Bowers et al.71, bins were defined as high-quality draft (>90% complete, <5% contamination), medium-quality draft (>50% complete, <10% contamination) or low-quality draft (<50% complete, <10% contamination) MAGs.
Taxonomy was tentatively assigned to MAGs using a combination of inferences by Kaiju67 and CheckM’s lineage-specific workflows42. Taxonomy was refined and confirmed by placing MAGs in a phylogenetic context using PhyloSift72 v. 1.0.1 with the updated PhyloSift markers database (version 4, 2018-02-1273). For this purpose, MAGs, all taxa previously identified by Burgess et al.9 with complete genomes available on NCBI (downloaded 2017-09-06), and all archaeal and bacterial genomes previously used in Hug et al. (2016) were placed in a phylogenetic tree27. Details on how this tree was constructed can be found in the Supplementary Material. Briefly, PhyloSift builds an alignment of the concatenated sequences for a set of core marker genes for each taxon. We used 37 of these single-copy marker genes (Supplementary Material) to build an amino acid alignment, which was trimmed using trimAl v.1.274. Columns with gaps in more than 5% of the sequences were removed, as well as taxa with less than 75% of the concatenated sequences. The final alignment75 comprised 3,240 taxa (Supplementary Table S3) and 5,459 amino acid positions. This alignment was then used to build a new phylogenetic tree in RAxML v. 8.2.10 on the CIPRES Science Gateway web server76 with the LG plus CAT (after Le and Gascuel)77 AA substitution model. One hundred fifty bootstrap replicates were conducted. The full tree inference required 2,236 computational hours on the CIPRES supercomputer. The Interactive Tree Of Life website iTOL was used to finalize and polish the tree for publication78.
All genomes used in this tree and a mapping file can be found on Figshare (genomes in Hug et al.’s version of the tree of life (2016)79,80,81 and genomes representing taxa identified in Burgess et al. (2012))82.
New analysis of 16S rRNA gene sequences
The 16S rRNA gene sequences generated by Burgess et al.9 were downloaded from NCBI. Using the same parameters and method as described in Burgess et al., but with an updated database, we inferred the taxonomy of these sequences. Briefly, sequences were uploaded to the RDP (Ribosomal Database Project) website and aligned to the RDP database (v. 11.5)83. Then, the SeqMatch tool was used to identify the closest match using all good quality sequences ≥1200 bp in length.
Taxonomic inference of metagenomic reads
Taxonomy was assigned to the quality-filtered Solexa reads for each sample using Kaiju v. 1.6.267 with the NCBI BLAST non-redundant protein database nr including fungi and microbial eukaryotes v. 2017-05-16. Kaiju was run using greedy mode with five substitutions allowed with an e-value cut-off for taxonomic assignment of 0.05. Taxonomy for each sample was summarised by collapsing taxonomic assignments to the phylum level. This process was repeated to infer taxonomy for the metagenomic reads from the Sanger clone libraries. Inferred taxonomy for the Solexa reads, Sanger metagenomes, MAGs and the RDP results for the 16S rRNA genes sequences from Burgess et al. were then imported into R v. 3.4.3 and compared using the ‘VennDiagram’ package v.1.6.2084.
Pangenomic comparison of pools and investigation of arsenic metabolizing genes
In order to characterize gene functions in ARK and ZAV, we identified gene clusters within the two thermal pools and visualized them in anvi’o using their pangenomic workflow85. We also used this workflow to investigate whether shared genera between pools would be more similar to each other or to reference genomes. We focused our comparisons on the genera Desulfurella and Sulfurihydrogenibium as we were able to obtain draft MAGs for these genera in both pools with high completion (>90%). Bins ARK-08 and ZAV-10 were compared to all three representative Desulfurella genomes available on NCBI (GCA_900101285.1, GCA_000517565.1, and GCA_002119425.1), while bins ARK-13 and ZAV-16 were compared to all four representative Sulfurihydrogenibium genomes (GCA_000619805.1, GCA_000173615.1, GCA_000021545.1, and GCA_000020325.1).
To quantify pairwise similarities, we used DIAMOND v. 0.9.9.11086, which calculates similarities between proteins. Then, we applied the MCL algorithm v. 14–13787 to construct gene clusters with an inflation value of 2.0 when comparing all MAGs from both pools and 10.0 when investigating close relatives, and muscle v. 3.8.1551 to align protein sequences88. Gene calls were annotated during this workflow with NCBI’s Clusters of Orthologous Groups (COGs)89 and Kyoto Encyclopedia of Genes and Genomes (KEGG) orthologies downloaded from GhostKOALA90 following the workflow for anvi’o by Elaina Graham (as described in http://merenlab.org/2018/01/17/importing-ghostkoala-annotations/). Functional pathways for ARK-15 and ZAV-18 were visualised using the online Rapid Annotation using Subsystem Technology (RAST)91 server which annotates them with SEED92.
Given unusually high concentrations of arsenic in Arkashin Schurf, we decided to look specifically for homologs of genes involved in arsenic biotransformations and compare them between the two pools. We manually downloaded HMMs (Hidden Markov Models) for protein families with a functional connection to arsenic from the TIGRFAM repository. Our selection of proteins was based on Zhu et al. (2017)53 (i.e., ArsA, ArsB, ArsC, ArsH, ArsR, and ACR3). We searched all open reading frames (ORFs) in both pools against them using blastx v. 2.6.0. Hits with at least 85% coverage on the length of the match, an e-value of 1e-10 and 85% identity were kept. These hits were searched using blastx v. 2.6.0 with an e-value threshold of 1e-4 against the MAGs to find out which organisms possess genes involved in arsenic biotransformations93.
R v. 3.4.0 with the package ‘plyr’ v. 1.8.494 was used to summarise homologs of shared and unique genes and predicted metabolic pathways qualitatively between the two pools. When investigating close relatives, phylogenetic trees were built in anvi’o using FastTree95 on the single-copy core genes identified to order taxa during visualization. Average nucleotide identity (ANI) values were calculated between close relatives and representative genomes using autoANI (https://github.com/osuchanglab/autoANI)30,96,97,98. Adobe Photoshop CS6 was used to finalize figures.
Data Availability
Sanger reads were deposited on NCBI’s GenBank under SRA IDs SRS3441489 (SRX4275258) and SRS3441490 (SRX4275259). Raw Solexa reads were deposited on NCBI’s GenBank under BioProject ID PRJNA419931 and BioSample IDs SAMN08105301 and SAMN08105287; i.e., SRA IDs SRS2733204 (SRX3442520) and SRS2733205 (SRX3442521). Draft MAGs were deposited in GenBank under accession numbers SAMN08107294 - SAMN08107329 (BioProject ID PRJNA419931). NCBI performed their Foreign Contamination Screen and removed residual sequencing adaptors prior to publication. The complete contamination screen is described in the supplementary material. Draft MAGs and associated anvi’o files can be found on DASH99. The alignment and raw tree file in Newick format used for Fig. 2 can be found on Figshare75,100.
Change history
21 February 2020
An amendment to this paper has been published and can be accessed via a link at the top of the paper.
References
Skirnisdottir, S. et al. Influence of Sulfide and Temperature on Species Composition and Community Structure of Hot Spring Microbial Mats. Appl. Environ. Microbiol. 66, 2835–2841 (2000).
Mathur, J. et al. Effects of abiotic factors on the phylogenetic diversity of bacterial communities in acidic thermal springs. Appl. Environ. Microbiol. 73, 2612–2623 (2007).
Pearson, A. et al. Factors controlling the distribution of archaeal tetraethers in terrestrial hot springs. Appl. Environ. Microbiol. 74, 3523–3532 (2008).
Cowan, D., Tuffi, M., Mulako, I. & Cass, J. Terrestrial Hydrothermal Environments. In Life at Extremes: Environments, Organisms and Strategies for Survival (ed. Bell, E.) 1, 220–241 (CABI Press, London, UK, 2012).
Beskrovnyy, N. S. et al. Presence of oil in hydrothermal systems associated with volcanism. Int. Geol. Rev. 15, 384–393 (1973).
Migdisov, A. A. & Bychkov, A. Y. The behaviour of metals and sulphur during the formation of hydrothermal mercury–antimony–arsenic mineralization, Uzon caldera, Kamchatka, Russia. J. Volcanol. Geotherm. Res. 84, 153–171 (1998).
Karpov, G. A. & Naboko, S. I. Metal contents of recent thermal waters, mineral precipitates and hydrothermal alteration in active geothermal fields, Kamchatka. J. Geochem. Explor. 36, 57–71 (1990).
Merkel, A. Y. et al. Microbial diversity and autotrophic activity in Kamchatka hot springs. Extremophiles 21, 307–317 (2017).
Burgess, E. A., Unrine, J. M., Mills, G. L., Romanek, C. S. & Wiegel, J. Comparative geochemical and microbiological characterization of two thermal pools in the Uzon Caldera, Kamchatka, Russia. Microb. Ecol. 63, 471–489 (2012).
Burgess, E. A. Geomicrobiological description of two contemporary hydrothermal pools in Uzon, Caldera, Kamchatka, Russia as models for sulfur biogeochemistry. (University of Georgia, USA, 2009).
Rozanov, A. S. et al. Molecular analysis of the benthos microbial community in Zavarzin thermal spring (Uzon Caldera, Kamchatka, Russia. BMC Genomics 15(Suppl 12), S12 (2014).
Miroshnichenko, M. L. et al. Ammonifex thiophilus sp. nov., a hyperthermophilic anaerobic bacterium from a Kamchatka hot spring. Int. J. Syst. Evol. Microbiol. 58, 2935–2938 (2008).
O’Neill, A. H., Liu, Y., Ferrera, I., Beveridge, T. J. & Reysenbach, A.-L. Sulfurihydrogenibium rodmanii sp. nov., a sulfur-oxidizing chemolithoautotroph from the Uzon Caldera, Kamchatka Peninsula, Russia, and emended description of the genus Sulfurihydrogenibium. Int. J. Syst. Evol. Microbiol. 58, 1147–1152 (2008).
Slobodkin, A. I. et al. Dissulfurimicrobium hydrothermale gen. nov., sp. nov., a thermophilic, autotrophic, sulfur-disproportionating deltaproteobacterium isolated from a hydrothermal pond. Int. J. Syst. Evol. Microbiol. 66, 1022–1026 (2016).
Auchtung, T. A., Shyndriayeva, G. & Cavanaugh, C. M. 16S rRNA phylogenetic analysis and quantification of Korarchaeota indigenous to the hot springs of Kamchatka, Russia. Extremophiles 15, 105–116 (2011).
Mardanov, A. V. et al. Uncultured archaea dominate in the thermal groundwater of Uzon Caldera, Kamchatka. Extremophiles 15, 365–372 (2011).
Bonch-Osmolovskaya, E. A. Studies of Thermophilic Microorganisms at the Institute of Microbiology, Russian Academy of Sciences. Microbiology 73, 551–564 (2004).
Perevalova, A. A. et al. Distribution of Crenarchaeota representatives in terrestrial hot springs of Russia and Iceland. Appl. Environ. Microbiol. 74, 7620–7628 (2008).
Reigstad, L. J., Jorgensen, S. L. & Schleper, C. Diversity and abundance of Korarchaeota in terrestrial hot springs of Iceland and Kamchatka. ISME J. 4, 346–356 (2010).
Gumerov, V. M., Mardanov, A. V., Beletsky, A. V., Bonch-Osmolovskaya, E. A. & Ravin, N. V. Molecular analysis of microbial diversity in the Zavarzin Spring, Uzon Caldera, Kamchatka. Microbiology 80, 244–251 (2011).
Chernyh, N. A. et al. Microbial life in Bourlyashchy, the hottest thermal pool of Uzon Caldera, Kamchatka. Extremophiles 19, 1157–1171 (2015).
Zarafeta, D. et al. Metagenomic mining for thermostable esterolytic enzymes uncovers a new family of bacterial esterases. Sci. Rep. 6, 38886 (2016).
Wemheuer, B., Taube, R., Akyol, P., Wemheuer, F. & Daniel, R. Microbial diversity and biochemical potential encoded by thermal spring metagenomes derived from the Kamchatka Peninsula. Archaea 2013, 136714 (2013).
Karpov, G. A. Uzon, A Protected Land. (Petropavlovsk-Kamchatskiy, Logata, Kamchatprombank, 1998).
Jovel, J. et al. Characterization of the Gut Microbiome Using 16S or Shotgun Metagenomics. Front. Microbiol. 7, 459 (2016).
Ojeda Alayon, D. I. et al. Genetic and genomic evidence of niche partitioning and adaptive radiation in mountain pine beetle fungal symbionts. Mol. Ecol. 26, 2077–2091 (2017).
Hug, L. A. et al. A new view of the tree of life. Nat Microbiol 1, 16048 (2016).
Bankevich, A. et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477 (2012).
Eren, A. M. et al. Anvi’o: an advanced analysis and visualization platform for ‘omics data. PeerJ 3, e1319 (2015).
Goris, J. et al. DNA-DNA hybridization values and their relationship to whole-genome sequence similarities. Int. J. Syst. Evol. Microbiol. 57, 81–91 (2007).
Kvist, T., Ahring, B. K. & Westermann, P. Archaeal diversity in Icelandic hot springs. FEMS Microbiol. Ecol. 59, 71–80 (2007).
Meyer-Dombard, D. R., Shock, E. L. & Amend, J. P. Archaeal and bacterial communities in geochemically diverse hot springs of Yellowstone National Park, USA. Geobiology 3, 211–227 (2005).
Elkins, J. G. et al. A korarchaeal genome reveals insights into the evolution of the Archaea. Proc. Natl. Acad. Sci. USA 105, 8102–8107 (2008).
Castelle, C. J. & Banfield, J. F. Major New Microbial Groups Expand Diversity and Alter our Understanding of the Tree of Life. Cell 172, 1181–1197 (2018).
Lake, J. A., Henderson, E., Oakes, M. & Clark, M. W. Eocytes: a new ribosome structure indicates a kingdom with a close relationship to eukaryotes. Proc. Natl. Acad. Sci. USA 81, 3786–3790 (1984).
He, Y. et al. Genomic and enzymatic evidence for acetogenesis among multiple lineages of the archaeal phylum Bathyarchaeota widespread in marine sediments. Nat Microbiol 1, 16035 (2016).
Lazar, C. S. et al. Genomic evidence for distinct carbon substrate preferences and ecological niches of Bathyarchaeota in estuarine sediments. Environ. Microbiol. 18, 1200–1211 (2016).
Reysenbach, A.-L. et al. A ubiquitous thermoacidophilic archaeon from deep-sea hydrothermal vents. Nature 442, 444–447 (2006).
Farag, I. F., Davis, J. P., Youssef, N. H. & Elshahed, M. S. Global patterns of abundance, diversity and community structure of the Aminicenantes (candidate phylum OP8). PLoS One 9, e92139 (2014).
Hugenholtz, P., Pitulle, C., Hershberger, K. L. & Pace, N. R. Novel Division Level Bacterial Diversity in a Yellowstone Hot Spring. J. Bacteriol. 180, 366–376 (1998).
Parks, D. H. et al. A proposal for a standardized bacterial taxonomy based on genome phylogeny. bioRxiv, https://doi.org/10.1101/256800 (2018).
Parks, D. H., Imelfort, M., Skennerton, C. T., Hugenholtz, P. & Tyson, G. W. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 25, 1043–1055 (2015).
Eichorst, S. A. et al. Genomic insights into the Acidobacteria reveal strategies for their success in terrestrial environments. Environ. Microbiol. 20, 1041–1063 (2018).
Brochier-Armanet, C., Forterre, P. & Gribaldo, S. Phylogeny and evolution of the Archaea: one hundred genomes later. Curr. Opin. Microbiol. 14, 274–281 (2011).
Zuo, G., Xu, Z. & Hao, B. Phylogeny and Taxonomy of Archaea: A Comparison of the Whole-Genome-Based CVTree Approach with 16S rRNA Sequence Analysis. Life 5, 949–968 (2015).
Eloe-Fadrosh, E. A., Ivanova, N. N., Woyke, T. & Kyrpides, N. C. Metagenomics uncovers gaps in amplicon-based detection of microbial diversity. Nat Microbiol 1, 15032 (2016).
Florentino, A. P., Stams, A. J. M. & Sánchez-Andrea, I. Genome Sequence of Desulfurella amilsii Strain TR1 and Comparative Genomics of Desulfurellaceae Family. Front. Microbiol. 8 (2017).
Miroshnichenko, M. L. et al. Desulfurella kamchatkensis sp. nov. and Desulfurella propionica sp. nov., new sulfur-respiring thermophilic bacteria from Kamchatka thermal environments. Int. J. Syst. Bacteriol. 48, 475–479 (1998).
Hugenholtz, P., Skarshewski, A. & Parks, D. H. Genome-Based Microbial Taxonomy Coming ofAge. Cold Spring Harb. Perspect. Biol. 8 (2016).
Berg, I. A. Ecological aspects of the distribution of different autotrophic CO2 fixation pathways. Appl. Environ. Microbiol. 77, 1925–1936 (2011).
Borrel, G., Adam, P. S. & Gribaldo, S. Methanogenesis and the Wood–Ljungdahl Pathway: An Ancient, Versatile, and Fragile Association. Genome Biol. Evol. 8, 1706–1711 (2016).
Levicán, G., Ugalde, J. A., Ehrenfeld, N., Maass, A. & Parada, P. Comparative genomic analysis of carbon and nitrogen assimilation mechanisms in three indigenous bioleaching bacteria: predictions and validations. BMC Genomics 9, 581 (2008).
Zhu, Y.-G., Xue, X.-M., Kappler, A., Rosen, B. P. & Meharg, A. A. Linking Genes to Microbial Biogeochemical Cycling: Lessons from Arsenic. Environ. Sci. Technol. 51, 7326–7339 (2017).
Yan, Y., Ding, K., Yu, X.-W., Ye, J. & Xue, X.-M. Ability of Periplasmic Phosphate Binding Proteins from Synechocystis sp. PCC 6803 to Discriminate Phosphate Against Arsenate. Water Air Soil Pollut. Focus 228 (2017).
Oremland, R. S., Saltikov, C. W., Wolfe-Simon, F. & Stolz, J. F. Arsenic in the Evolution of Earth and Extraterrestrial Ecosystems. Geomicrobiol. J. 26, 522–536 (2009).
Zhu, Y.-G., Yoshinaga, M., Zhao, F.-J. & Rosen, B. P. Earth Abides Arsenic Biotransformations. Annu. Rev. Earth Planet. Sci. 42, 443–467 (2014).
Hamamura, N. et al. Linking microbial oxidation of arsenic with detection and phylogenetic analysis of arsenite oxidase genes in diverse geothermal environments. Environ. Microbiol. 11, 421–431 (2009).
Lin, Y.-F., Walmsley, A. R. & Rosen, B. P. An arsenic metallochaperone for an arsenic detoxification pump. Proc. Natl. Acad. Sci. USA 103, 15617–15622 (2006).
Bushnell, B. BBMap short read aligner. Available at: http://sourceforge.net/projects/bbmap (2014).
Bankevich, A. et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477 (2012).
Ewing, B., Hillier, L., Wendl, M. C. & Green, P. Base-calling of automated sequencer traces using phred. I. Accuracy assessment. Genome Res. 8, 175–185 (1998).
Chou, H. H. & Holmes, M. H. DNA sequence quality trimming and vector removal. Bioinformatics 17, 1093–1104 (2001).
Hyatt, D. et al. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 11, 119 (2010).
Campbell, J. H. et al. UGA is an additional glycine codon in uncultured SR1 bacteria from the human microbiota. Proc. Natl. Acad. Sci. USA 110, 5540–5545 (2013).
Rinke, C. et al. Insights into the phylogeny and coding potential of microbial dark matter. Nature 499, 431–437 (2013).
Eddy, S. R. Accelerated Profile HMM Searches. PLoS Comput. Biol. 7, e1002195 (2011).
Menzel, P., Ng, K. L. & Krogh, A. Fast and sensitive taxonomic classification for metagenomics with Kaiju. Nat. Commun. 7, 11257 (2016).
Kiefl, E., Delmont, T. O. & Eren, A. M. Analyzing sequence variants with anvi’o., http://merenlab.org/2015/07/20/analyzing-variability/ (2015).
Ettinger, C. L. Kamchatka Zavarzin Spring Metagenome Analysis Notebook. Figshare. Code., https://doi.org/10.6084/m9.figshare.6873743.v1
Wilkins, L. G. E. Kamchatka Arkashin Schurf Metagenome Analysis Notebook. Figshare. Code., https://doi.org/10.6084/m9.figshare.6874925.v1 (2018).
Bowers, R. M. et al. Minimum information about a single amplified genome (MISAG) and a metagenome-assembled genome (MIMAG) of bacteria and archaea. Nat. Biotechnol. 35, 725–731 (2017).
Darling, A. E. et al. PhyloSift: phylogenetic analysis of genomes and metagenomes. PeerJ 2, e243 (2014).
Jospin, G. Markers database for PhyloSift. Figshare (2018).
Capella-Gutiérrez, S., Silla-Martínez, J. M. & Gabaldón, T. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25, 1972–1973 (2009).
Wilkins, L. G. E., Ettinger, C. L., Jospin, G. & Eisen, J. A. Sequence alignment for phylogenetic tree construction. Figshare. Dataset., https://doi.org/10.6084/m9.figshare.6916298.v1
Miller, M. A., Pfeiffer, W. & Schwartz, T. Creating the CIPRES Science Gateway for inference of large phylogenetic trees. In2010 Gateway Computing Environments Workshop (GCE), https://doi.org/10.1109/gce.2010.5676129 (2010).
Le, S. Q. & Gascuel, O. An improved general amino acid replacement matrix. Mol. Biol. Evol. 25, 1307–1320 (2008).
Letunic, I. & Bork, P. Interactive tree of life (iTOL)v3: an online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res. 44, W242–5 (2016).
Wilkins, L. G. E., Ettinger, C. L., Jospin, G. & Eisen, J. A. Genomes used to infer a tree of life in Hug et al. - part II. Figshare, https://doi.org/10.6084/m9.figshare.6863744.v2 (2016)
Wilkins, L. G. E., Ettinger, C. L., Jospin, G. & Eisen, J. A. Genomes used to infer a tree of life in Hug et al. - manually downloaded. Figshare, https://doi.org/10.6084/m9.figshare.6863813.v1 (2016)
Wilkins, L. G. E., Ettinger, C. L., Jospin, G. & Eisen, J. A. Genomes used to infer a tree of life in Hug et al. - part I. Figshare, https://doi.org/10.6084/m9.figshare.6863594.v1 (2016)
Wilkins, L. G. E., Ettinger, C. L., Jospin, G. & Eisen, J. A. Genomes of taxa analyzed in Burgess et al. from two hot springs in Kamchatka, Russia. Figshare, https://doi.org/10.6084/m9.figshare.6863798.v1 (2012)
Cole, J. R. et al. Ribosomal Database Project: data and tools for high throughput rRNA analysis. Nucleic Acids Res. 42, D633–42 (2014).
Chen, H. & Boutros, P. C. VennDiagram: a package for the generation of highly-customizable Venn and Euler diagrams in R. BMC Bioinformatics 12, 35 (2011).
Delmont, T. O. & Murat Eren, A. Linking pangenomes and metagenomes: the Prochlorococcus metapangenome. PeerJ 6, e4320 (2018).
Buchfink, B., Xie, C. & Huson, D. H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 12, 59–60 (2015).
van Dongen, S. & Abreu-Goodger, C. Using MCL to extract clusters from networks. Methods Mol. Biol. 804, 281–295 (2012).
Edgar, R. C. MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics 5, 113 (2004).
Galperin, M. Y., Makarova, K. S., Wolf, Y. I. & Koonin, E. V. Expanded microbial genome coverage and improved protein family annotation in the COG database. Nucleic Acids Res. 43, D261–9 (2015).
Kanehisa, M., Sato, Y. & Morishima, K. BlastKOALA and GhostKOALA: KEGG Tools for Functional Characterization of Genome and Metagenome Sequences. J. Mol. Biol. 428, 726–731 (2016).
Aziz, R. K. et al. The RAST Server: rapid annotations using subsystems technology. BMC Genomics 9, 75 (2008).
Overbeek, R. et al. The SEED and the Rapid Annotation of microbial genomes using Subsystems Technology (RAST). Nucleic Acids Res. 42, D206–14 (2014).
Altschul, S. F. et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402 (1997).
Wickham, H. The Split-Apply-Combine Strategy for Data Analysis. J. Stat. Softw. 40 (2011).
Price, M. N., Dehal, P. S. & Arkin, A. P. FastTree 2 – Approximately Maximum-Likelihood Trees for Large Alignments. PLoS One 5, e9490 (2010).
Davis, E. W. et al. Gall-ID: tools for genotyping gall-causing phytopathogenic bacteria. PeerJ 4, e2222 (2016).
Stajich, J. E. et al. The Bioperl toolkit: Perl modules for the life sciences. Genome Res. 12, 1611–1618 (2002).
Camacho, C. et al. BLAST+: architecture and applications. BMC Bioinformatics 10, 421 (2009).
Ettinger, C. L., Wilkins, L. G. E., Jospin, G. & Eisen, J. A. Metagenome-assembled genomes (MAG’s) from two thermal pools in Uzon Caldera, Kamchatka, Russia. DASH repository, https://doi.org/10.25338/B8N01R (2018).
Wilkins, L. G. E., Ettinger, C. L., Jospin, G. & Eisen, J. A. Tree in Fig. 2 - Archaeal and bacterial genomes used by Hug et al. in 2016 to construct a microbial tree of life; MAGs isolated from two hot springs in the Uzon Caldera, Kamchatka, Russia; and all genomes of taxa analyzed in Burgess et al. (2012) with one representative genome on NCBI (Newick file). Figshare, https://doi.org/10.6084/m9.figshare.6874928.v3 (2018).
Kahle, D. & Wickham, H. ggmap: Spatial Visualization withggplot2. R J. 5, 144–161 (2013).
Acknowledgements
We would like to thank Russell Neches (ORCID: 0000-0002-2055-8381) for the use of photographs taken on a trip to Kamchatka, Russia in 2012. We also would like to thank Elizabeth A. Burgess and Juergen Wiegel for providing JAE with the DNA used here. We are also grateful to Christopher Brown (ORCID: 0000-0002-7758-6447) and Laura Hug (ORCID: 0000-0001-5974-9428) for their help getting access to the genomes used in Figure 2. A special thank you goes to Alexandros Stamatakis (ORCID: 0000-0003-0353-0691), Wayne Pfeiffer and Mark Miller for offering their help with getting the CIPRES Science Gateway server to run RAxML-HPC2 v.8 on XSEDE. We also thank two anonymous reviewers for comments on earlier versions of this manuscript. A. Murat Eren (ORCID: 0000-0001-9013-4827) provided constructive feedback on the publicly available preprint version of this article. LGEW was supported by a fellowship of the Swiss National Science Foundation P2LAP3_164915. Funding for the sequencing at TIGR was provided by a a subcontract to JAE for a grant from the National Science Foundation (MCB-MO 0238407). Funding for some of the work on this project was also provided by grant from the Gordon and Betty Moore Foundation (GBMF5603) to JAE.
Author information
Authors and Affiliations
Contributions
L.G.E.W. and C.L.E. analysed the data, prepared figures and/or tables, wrote, edited and reviewed drafts of the paper. G.J. advised on data analysis and reviewed drafts of the paper. J.A.E. contributed reagents/materials/analysis tools, advised on data analysis, reviewed and edited drafts of the paper.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wilkins, L.G.E., Ettinger, C.L., Jospin, G. et al. Metagenome-assembled genomes provide new insight into the microbial diversity of two thermal pools in Kamchatka, Russia. Sci Rep 9, 3059 (2019). https://doi.org/10.1038/s41598-019-39576-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-39576-6
This article is cited by
-
Environmental activity-based protein profiling for function-driven enzyme discovery from natural communities
Environmental Microbiome (2024)
-
Exploration of genes encoding KEGG pathway enzymes in rhizospheric microbiome of the wild plant Abutilon fruticosum
AMB Express (2024)
-
Disentangling direct vs indirect effects of microbiome manipulations in a habitat-forming marine holobiont
npj Biofilms and Microbiomes (2024)
-
Radiation impacts gene redundancy and biofilm regulation of cryoconite microbiomes in Northern Hemisphere glaciers
Microbiome (2023)
-
Baseline metagenome-assembled genome (MAG) data of Sikkim hot springs from Indian Himalayan geothermal belt (IHGB) showcasing its potential CAZymes, and sulfur-nitrogen metabolic activity
World Journal of Microbiology and Biotechnology (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
