
Abstract
Insulin resistance (IR) is a hallmark of type 2 diabetes, metabolic syndrome and cardiometabolic risk. An epigenetic phenomena such as DNA methylation might be involved in the onset and development of systemic IR. The aim of this study was to explore the genetic DNA methylation levels in peripheral white blood cells with the objective of identifying epigenetic signatures associated with IR measured by the Homeostatic Model Assessment of IR (HOMA-IR) following an epigenome-wide association study approach. DNA methylation levels were assessed using Infinium Methylation Assay (Illumina), and were associated with HOMA-IR values of participants from the Methyl Epigenome Network Association (MENA) project, finding statistical associations for at least 798 CpGs. A stringent statistical analysis revealed that 478 of them showed a differential methylation pattern between individuals with HOMA-IR ≤ 3 and > 3. ROC curves of top four CpGs out of 478 allowed differentiating individuals between both groups (AUC≈0.88). This study demonstrated the association between DNA methylation in some specific CpGs and HOMA-IR values that will help to the understanding and in the development of new strategies for personalized approaches to predict and prevent IR-associated diseases.
Similar content being viewed by others


Introduction
Type 2 diabetes (T2D) is a worldwide major health concern and the most predominant type of diabetes1. According to the World Health Organisation, the global prevalence of diabetes among adults over 18 years old has risen from 4.7% in 1980 to 8.5% in 2014. Furthermore, in 2015 about 1.6 million deaths were directly attributed to diabetes1.
T2D is a multifactorial disease defined by the interaction of genetics and environmental factors2. The heritability for T2D is estimated to be between 15 and 85%. However, the genetic loci identified to date only explain 5–10% of this heritability3. In this context, available evidences suggest that epigenetics may be contributing to variations in gene expression and the risk for this metabolic disease4. In fact, recent investigations have associated the onset and progression of diabetes with specific changes in the epigenome3,5.
Insulin resistance (IR) is a pathological condition in which cells fail to respond properly to insulin6. IR is one of the most important precursors of T2D and other adversely associated cardiometabolic conditions, such as obesity, hypertension, cardiovascular disease (CVD)7, and metabolic syndrome8. IR is specifically associated with a low-grade inflammation, as well as with chronic enhancement of oxidative stress, triggering endothelial dysfunction and promoting atherogenesis4. Furthermore, both genetic and epigenetic factors are involved in the development of systemic IR9. The validated method Homeostatic Model Assessment of IR (HOMA-IR) is usually employed for measuring IR and β-cell function10.
Epigenetic marks are heritable changes that cannot be explained through variations in DNA nucleotide sequence11. These modifications are potentially reversible and can be altered by environmental factors2, resulting in alterations of gene expression and providing an interactive connection among genetics, specific diseases and the environment12.
Among the different epigenetic modifications, DNA methylation has been widely searched13. Some epigenome-wide association studies (EWASs) have revealed significant associations between DNA methylation and glucose homeostasis5,14,15,16,17,18,19,20, but only four of them studied some relationships with IR in different populations and approaches5,14,15,18. Therefore, the aim of the current work was to explore DNA methylation levels in peripheral white blood cells (PWBCs) by using an EWAS strategy with the objective of identifying epigenetic signatures associated with HOMA-IR and specifically identifying potential biomarkers that allow the discrimination of potentially hazardous HOMA-IR levels.
The assessment of epigenetic phenomena may help to understand the basis of metabolic pathway regulation, as well as the relationships between genomics and the environment influence, to promote new strategies to better understand human health and to develop novel biomarker panels related to T2D, obesity and accompanying comorbidities20,21.
Results
Participant characteristics
Anthropometric and biochemical characteristics of the participants are reported (Table 1).
DNA methylation was significantly associated with HOMA-IR
Methylation values of CpGs were analysed for Linear Models for Microarray Data (LIMMA) regression with HOMA-IR in 332 subjects. Significant CpGs were selected by a False Discovery Rate (FDR) cut-off of 0.05 and a slope ≥ |0.1|, obtaining 798 CpGs (Supplementary Material Table S1). The top 10 CpGs were further analysed for robustness. Spearman correlations were performed, and six CpGs were selected by having higher rho coefficient. Then, multiple linear regressions were performed adjusting by sex, age, study and body mass index (BMI), remaining three of the six CpGs significant (Table 2). These CpGs were cg13133503 (corresponding gene according to Illumina CG database CLCA4), cg07638362 (NA), and cg16462528 (LECT1), which are highlighted in a Manhattan plot (Fig. 1). Linear regression graphs between methylation values and HOMA-IR for these three CpGs are also represented (Fig. 2).

Manhattan plot of HOMA-IR-associated CpGs selected by a slope ≥|0.1|. Points above the dot line showed a False Discovery Rate (FDR) < 0.05. The three CpGs selected by slope ≥|0.1|, FDR < 0.05, Spearman’s rho, and by multiple linear regressions adjusting by sex, age, study and body mass index are marked.

Linear regression graphs between HOMA-IR and methylation β values of the significant three CpGs selected by slope ≥|0.1|, False Discovery Rate (FDR) <0.05, and Spearman’s rho. Adjusted by study, sex, age and body mass index. Dot lines on both sides of the solid line (linear regression for correlation) represent 95% confidence band.
Individuals with HOMA-IR > 3 showed a differential methylation pattern
Participants were classified according to the HOMA-IR cut-off of 3 in order to analyse whether methylation was differential between both groups. There were 78 individuals with HOMA-IR > 3 and 254 with HOMA-IR ≤ 3 (Table 1). Methylation values of the 798 CpGs were compared between both HOMA-IR groups. After applying the Bonferroni correction for multiple comparisons, a total of 478 CpGs showed statistically significant differences (Supplementary Material Table S2).
The resulting 478 CpGs were clustered in a heat map according to methylation patterns (Fig. 3). Two main clusters of 61 and 271 individuals were generated. The first cluster contained 62.3% of individuals with HOMA-IR > 3. However, the second cluster only included 14.8% of HOMA-IR > 3. The difference in HOMA-IR proportions of the clusters was statistically significant (p < 0.001).

Heat map of 478 CpGs selected by Student’s t-test between HOMA-IR ≤3 and >3 (p < 6.26·10−5 after Bonferroni correction).
Differentially methylated CpGs between HOMA-IR groups were related to glucose and insulin pathways
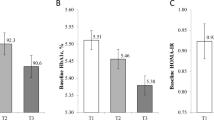
Canonical pathways were obtained from Ingenuity Pathway Analysis (IPA) for these 478 CpGs (Fig. 4). Some of the statistically significant pathways were related to insulin and glucose, such as Protein Kinase A Signalling, Sirtuin Signalling Pathway, G-Protein Coupled Receptor Signalling, Rac Signalling, Mature Onset Diabetes of Young (MODY) Signalling, RhoA Signalling, and Leptin Signalling in Obesity. The top four CpGs differentially methylated between HOMA-IR ≤3 and >3 were cg23475244 (NA), cg06115835 (SH3RF3), cg16278828 (MAN2C1) and cg16639311 (NA) as illustrated (Fig. 5).

Canonical pathways from Ingenuity Pathway Analysis of 478 CpGs selected by Student’s t-test between HOMA-IR ≤3 and >3 (p < 6.26·10−5 after Bonferroni correction).

Box plots of top four CpGs selected by Student’s t-test between HOMA-IR ≤3 and >3 (p < 6.26·10−5 after Bonferroni correction). Whiskers represent minimum and maximum values.
The top four CpGs allow to differentiate between HOMA-IR ≤3 and >3
In order to further analyse whether methylation could differentiate between both HOMA-IR groups, the Receiver Operating Characteristic (ROC) curves adjusted by study, sex, age and BMI for the top four CpGs (cg23475244, cg06115835, cg16278828, and cg16639311) were calculated. The areas under the curve (AUC) of these CpGs were around 0.90 (AUC cg23475244 = 0.8965, AUC cg06115835 = 0.9026, AUC cg16278828 = 0.8989, and AUC cg16639311 = 0.8952), and after an internal validation (Fig. 6), the values were around 0.88 (AUC cg23475244 = 0.8865, AUC cg06115835 = 0.8919, AUC cg16278828 = 0.8893, and AUC cg16639311 = 0.8826).

ROC curves of the top four CpGs (cg23475244, cg06115835, cg16278828, and cg16639311). Optimism corrected value was calculated using the Tibshirani’s enhanced bootstrap method described by Harrell64.
Discussion
This study involving the Methyl Epigenome Network Association (MENA) project demonstrated the association between DNA methylation in specific CpGs and HOMA-IR values. Our results also provided evidence of a differential methylation pattern between individuals with a HOMA-IR ≤3 and >3. Additionally, these data have led to the identification of four CpGs that allow us to differentiate individuals between HOMA-IR ≤ 3 and >3 with an approximate AUC of 0.88. This assay adds further insights and knowledge about the relationship between T2D-related traits and epigenetic DNA modifications.
As aforementioned, IR is a hallmark of several diseases and unhealthy cardiometabolic conditions such as T2D, CVD, hypertension, obesity7 and metabolic syndrome8. Epigenetic mechanisms have been involved in the onset and development of IR9. Indeed, several studies have related methylation of specific genes with HOMA-IR3,7,8,22,23,24,25,26,27,28,29,30,31. Nevertheless, few EWAS have been performed to date5,14,15,18. In line with these studies, this EWAS of the MENA project showed an association of 798 CpGs with HOMA-IR (slope ≥ |0.1| and FDR < 0.05). In our study, from the top 10 CpGs, selected ones with better association and significant after linear regressions adjusting by study, age, sex, and BMI were cg07638362 (according to Illumina CG database this CpG was not associated to any gene), cg13133503 (CLCA4) and cg16462528 (LECT1). These CpGs, to our knowledge, have not been previously described in other EWAS. However, some of the mentioned genes have been found in the list of one study. Specifically, differentially methylated regions of LECT1 and CLCA4 have been significantly different between diabetics and non-diabetics32. Both CLCA4 and LECT1 have been related to methylation regulation33,34,35. CLCA4 has been involved in the activation of cAMP-dependent protein kinase A [www.genecards.org]. This pathway is intimately connected to glucose homeostasis36. On the other hand, LECT1 plays a role as antiangiogenic factor in cardiac valves, preventing valvular heart diseases37. Methylation of this gene may be associating IR with CVD. Thus, the association of several CpGs between DNA methylation and IR detected in our study adds further support for a potential role of abnormal DNA methylation in IR7.
Since IR is a key feature of T2D, obesity and metabolic syndrome7,8, it is interesting to analyse other EWAS and methylation studies related to these adverse metabolic conditions. These investigations have been performed in several tissues such as pancreatic islets, liver, adipose tissue, skeletal muscle and blood cells38. There are five genes in our list that were previously related to insulin resistance (CXCR1, HDAC4, IGFR1, LEPR, and ABCG1)4,5,18. On the other hand, T2D and glycaemic traits have been associated with the following genes found in our selection NR4A332, KCNQ139, IRS139,40, SREBF114,16,17,20, SOCS314,16,17,20, ZNF518B8, SAMD1215,19, LY6G6E16, PHGDH20, and ABCG15,14,15,16,18,41. Additionally, IRS140, SREBF118,20,42, ABCG117,20,43,44,45, SOCS317,44,46, LY6G6E43 and PHGDH45,47 have also been found in EWAS analysing BMI or obesity traits. Other genes from our list that are related to obesity or BMI were AOC348, c7orf5043, NOD220,42, and SLC1A542. Regarding genes associated with age, ZNF42349 and THRB50 were found in our list. In the case of smoking-associated genes, ECE1, ATP8B2, c7orf50, IGF1R, RPL23A, SFRS151, RPTOR, RARA52, c6orf4853, and IER354 appeared in the selection. Interestingly, the specific CpGs described for ABCG1 (cg06500161)5,14,15,16,17,18,20,42,43,44,45, SREBF1 (cg11024682)14,16,17,18,20,42,43, SOCS3 (cg18181703)14,16,17,20,46, and PHGDH (cg14476101)20,42,45,47 were also found in our list. These four mentioned CpGs probably represent the widest described ones in relationship with T2D, obesity and other metabolic impairments in several studies with different tissues such as skeletal muscle, liver, pancreas and blood cells. Our investigation adds some new CpGs and genes to the previously described list, contributing to the knowledge and the management of IR-associated diseases.
As a novelty, our results have shown that individuals with HOMA-IR ≤ 3 or >3 exhibited a differential methylation pattern for at least 478 CpGs. Furthermore, the clustering showed that 62.3% of individuals in the first cluster had a HOMA-IR > 3. Thus, more than half of the people with similar methylation patterns presented a HOMA-IR > 3. However, the distribution of some cohorts was not heterogeneous. This situation is due to the specific recruitment requirements for each cohort. Indeed, cohorts such as RESMENA, where all the patients had metabolic syndrome, is completely found in the first cluster.
Furthermore, these 478 CpGs corresponded to some genes involved in glucose and insulin-related pathways according to IPA. For example, Protein Kinase A Signalling, where protein kinase A activation triggers insulin secretion in β-cells55; Sirtuin Signalling Pathway, where sirtuins influence many steps of glucose metabolism in liver, pancreas, muscle and adipose tissue56; and G-Protein Coupled Receptor Signalling, where insulin and glucagon secretion is affected by factors binding to G-protein coupled receptors on the surface of β- and α-cells57. Other pathways were Rac Signalling, which is involved in the regulation of insulin-stimulated glucose uptake58; RhoA Signalling, pathway that has been implicated in the pathogenesis of diabetes59; and Leptin Signalling in Obesity, since leptin is a regulator of glycaemic control60. Furthermore, Maturity Onset Diabetes of Young (MODY) Signalling represents the pathway of another type of diabetes that accounts for less than 2% of all diabetic cases. MODY is a monogenic form of diabetes characterized by an early onset, autosomal dominant mode of inheritance and a primary defect in pancreatic β-cell function61.
Only two of the top four CpGs with statistically significant differences between HOMA-IR ≤3 and >3 individuals presented associated genes according to Illumina CG database. Those genes were SH3RF3 and MAN2C1. The function of SH3RF3 is not well known, whereas MAN2C1 is related to glycosaminoglycan (GAG) metabolism. The GAGs are heteropolysaccharides formed by a chain of repeating disaccharide units62. Changes in GAGs structure and function have been reported in the kidney, liver, arteries and retinal vessels of diabetics63.
Since methylation patterns of the 478 CpGs were able to cluster HOMA-IR individuals, we analysed the ability of the top four CpGs to differentiate between HOMA-IR ≤3 and >3 individuals. These top four CpGs distinguished HOMA-IR groups with a valuable AUC around 0.88 after an internal validation based on the optimistic correction model described by Harrell64, suggesting these CpGs as potential valuable biomarkers of IR.
This study was not devoid of limitations. Firstly, methylation is tissue-specific and the ideal tissue for this study would have been the pancreatic β-cells or cells from recognized insulin sensitive tissues such as skeletal muscle or white adipose tissue65. However, peripheral blood is the best non-invasive alternative tissue that reflects multiple metabolic and inflammatory pathways66, and relevant studies have demonstrated that epigenetic reprogramming may serve as a surrogate marker for metabolic disorders41. Interestingly, gene methylation parallelisms between peripheral blood cells and pancreatic islets have been recently reported, suggesting that blood may be used as a marker for islet DNA methylation67. Secondly, type I and type II error cannot be discarded, although multiple comparison tests and statistical adjustments for potential confounding factors such as sex, age, cohorts, DNA methylation chips, and cell composition heterogeneity have been performed. Thirdly, a validation sample would have been useful to corroborate the results in the selected genes. Unfortunately, this sample was not available. However, in order to resolve this issue and correct the overestimation of AUC, an internal validation using a bootstrap method64 was performed, obtaining similar results. Further studies are needed to verify the relationship between the selected CpGs and HOMA-IR. Finally, due to the cross-sectional feature of the study, methylation cannot be defined as a cause or consequence of cardiometabolic conditions. Remarkably, although there is an epigenetic programming during the first stages of human development68, Wahl et al. have described methylation alterations as a cause of higher BMI and adiposity20.
Epigenetic gene regulation, and specifically, DNA methylation, is playing a role in the pathogenesis of many complex disorders, including T2D, obesity or metabolic syndrome22. There is great interest to perform methylation profiling in peripheral blood to find potential methylation disease-related associations and use specific DNA methylated regions as biomarkers69. In summary, this study found associations between DNA methylation and IR, a hallmark of T2D, with a differential methylation pattern between individuals with HOMA-IR ≤ 3 and > 3 in genes that are mainly involved in glucose and insulin-related pathways, and suggested four CpGs as biomarkers of IR. These results will hopefully contribute to the understanding of some epigenetic mechanisms that may regulate glycaemic traits, such as HOMA-IR, and the risk of T2D, as well as provide the basis for creating personalized strategies to predict, prevent and treat IR-associated diseases.
Subjects and Methods
Participants
The MENA project was conducted in 523 adult participants from available cohorts at the University of Navarra (UNAV): DiOGenes-UNAV with n = 5870, OBEPALIP with n = 2971, Food4Me-UNAV with n = 4272, GEDYMET with n = 5773, ICTUS with n = 774, NUGENOB-UNAV with n = 4275, PREDIMED-UNAV with n = 12976,77, RESMENA with n = 4778, OBEKIT with n = 10079 and NormoP with n = 12. However, only 474 final samples were available after the data processing explained in detail below.
Study designs, characteristics, inclusion and exclusion criteria were described for each study cohort, except for NormoP, whose design has not yet been described. All of them were approved by the Research Ethics Committee of the University of Navarra (CEI-UN, Pamplona, Spain), except for GEDYMET, which was approved by the Ethics committee of the School of Medicine, Pontificia Universidad Católica de Chile (Santiago, Chile), in compliance with the Helsinki Declaration of ethical principles for medical research involving human subjects. All participants provided written informed consent.
The NormoP cohort participants recruitment started in 2016 in the University of Navarra (Pamplona, Spain). Eligible participants were self-declared healthy individuals, >18 years old, and had a BMI of between 18.5 and 24.9 kg/m2. Exclusion criteria included pregnancy, type I diabetes, severe renal and digestive diseases, hydroelectrolitic disorders, acute CVD, cardiac arrhythmias, ictus, neoplasia, anaemia, eating disorders, pharmacological treatment, and dietary supplements that may affect the results.
Study variables
Anthropometric measurements and the metabolic profile were obtained from databases of the aforementioned cohorts, which followed validated protocols. Data of some characteristics were not available for all the 474 participants. IR was estimated using the validated HOMA-IR index method10.
DNA extraction and DNA methylation analysis
Venous blood samples were drawn on EDTA tubes. Genomic DNA was extracted from PWBCs using the MasterPureTM DNA Purification kit (Epicenter, Madison, WI), whose quality was assessed with the Pico Green dsDNA Quantitation Reagent (Invitrogen, Carlsbad, CA). High-quality DNA samples (500 ng) were treated with bisulfite using the EZ-96 DNA Methylation Kit (Zymo Research Corporation, Irvine, CA) according to the manufacturer’s instructions, converting cytosine into uracil. DNA methylation levels were measured by microarray with the Infinium Human Methylation 450 K bead chip technology (Illumina, San Diego, CA, USA) in all the cohorts, except OBEKIT, which was performed with Infinium MethylationEPIC beadchip (Illumina). This analysis was conducted in the Unidad de Genotipado y Diagnóstico Genético from Fundación Investigación Clínico de Valencia, as detailed elsewhere80.
Treatment of methylation raw data
Beta-values have been used as metrics to measure methylation levels. Beta-value in methylation experiments is the estimate of the methylation level using the ratio of the methylation probe intensity and the overall intensity, corresponding to the percentage of methylation on a specific site81. After obtaining intensity data using ChAMP package for R v.1.11.082 as described elsewhere83, the filtering process was performed in probes with a detection p-value above 0.01 in one or more samples, probes with a beadcount <3 in at least 5% of samples, non-CpG probes, probes with SNPs84, probes that align to multiple locations84 and probes located on the X or Y chromosomes.
From the 523 initial participants, samples with a failed CpG fraction above 0.01 were eliminated (n = 20), leaving 503 individuals. After filtering probes, intra-cell type normalization was done using Subset-quantile Within Array Normalization (SWAN) method to avoid the bias introduced by the Infinium type 2 probe design85. In order to assess the similarity of normalized methylation samples in both batches and the pooled data, multidimensional scaling plots based on top of 1000 most variable probes were performed. A total of 29 samples failed to fulfil this requisite, which left 474 participants for the subsequent analyses.
After SWAN normalization, magnitude of batch effects were assessed and corrected using the ComBat normalization method, which is an empirical Bayes based method to correct for technical variation related to the slide86,87. Furthermore, differences in methylation resulting from differences in cellular heterogeneity were corrected using the Houseman procedure88.
The data discussed in this publication have been deposited in NCBI’s Gene Expression Omnibus89 and are accessible through GEO Series accession number GSE115278 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE115278).
Statistical analysis
After pre-processing, LIMMA package from the R statistical software82 was used to compute a linear regression between DNA methylation values and HOMA-IR. A total of 332 subjects from the MENA project showed data for both variables (Table 1). This analysis was adjusted by the effect of confounding factors such as sex, age, study and bead chip. Raw p-values were corrected using the Benjamini-Hochberg procedure for multiple comparisons, and a FDR cut-off of 0.05 and a slope ≥ |0.1| were used as statistically significant thresholds. The top 10 CpGs were analysed for robustness with Spearman correlations and then, linear regressions between HOMA-IR and methylation adjusted for study, sex, age, and BMI were also performed for the six selected CpGs.
The cut-off for HOMA-IR differs for different races, ages, genders, diseases, complications, etc.90 and no reference value has been established91. Since there is no consensus for the HOMA-IR cut point and in order to facilitate the analysis of this metabolically heterogeneous group, a cut-off of HOMA-IR = 3 was chosen, corresponding to a value between the 75th and 80th percentiles, which are established as cut points by International Diabetes Federation (IDF) and Adult Treatment Panel III (ATPIII) for metabolic syndrome92. No influences in terms of races were considered, since more than 92% of the individuals were Caucasian in the MENA project and additionally, the study has been considered as a covariate in the analyses. Moreover, some studies have previously used this cut-off for HOMA-IR93,94. Differentially methylated CpGs between individuals with HOMA-IR > 3 and HOMA-IR ≤ 3 were explored using two-tailed Student’s t-test with Bonferroni correction. A p-value < 6.26·10−5 was considered significant. Adjusted (for study, sex, age, and BMI) ROC curves were performed to determine the AUC of the top selected CpGs distinguishing individuals between HOMA-IR ≤ 3 or > 3. Furthermore, an internal validation using a correction for optimistic prediction was performed according to Tibshirani’s enhanced bootstrap method described by Harrell64 in order to evaluate the overestimation of the model.
Statistical calculations were performed with STATA version 12.0 (Stata Corp, College Station, TX, USA), unless otherwise indicated. Manhattan plots, correlation graphs and box plots were produced using GraphPad Prism 6 (Graph-Pad Software, CA, USA). The heat map was created with the R software82 using library gplots and the heatmap.2 function.
Ingenuity Pathway Analysis
Differentially methylated CpGs between individuals with HOMA-IR > 3 and HOMA-IR ≤ 3 were analysed by IPA software (Qiagen Redwood City, CA, USA, www.ingenuity.com) as defined in the package. Predefined pathways and functional categories of the Ingenuity Knowledge Base were used in order to detect associated pathways and relevant gene regulatory networks95. Pathway analyses were performed with IPA’s Core Analysis module. Canonical pathways with a p < 0.05 after Fisher’s test were defined as a statistically significant overrepresentation of input genes in a given process.
Data Availability
The data have been deposited in NCBI’s Gene Expression Omnibus89 and are accessible through GEO Series accession number GSE115278 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE115278).
References
(WHO), W. H. O. Diabetes, http://www.who.int/diabetes/en/ (2018).
Barres, R. & Zierath, J. R. The role of diet and exercise in the transgenerational epigenetic landscape of T2DM. Nat Rev Endocrinol 12, 441–451 (2016).
Canivell, S. et al. Differential methylation of TCF7L2 promoter in peripheral blood DNA in newly diagnosed, drug-naive patients with type 2 diabetes. PLoS One 9, e99310 (2014).
De Rosa, S. et al. Type 2 Diabetes Mellitus and Cardiovascular Disease: Genetic and Epigenetic Links. Front Endocrinol (Lausanne) 9, 2 (2018).
Hidalgo, B. et al. Epigenome-wide association study of fasting measures of glucose, insulin, and HOMA-IR in the Genetics of Lipid Lowering Drugs and Diet Network study. Diabetes 63, 801–807 (2014).
Schinner, S., Scherbaum, W. A., Bornstein, S. R. & Barthel, A. Molecular mechanisms of insulin resistance. Diabet Med 22, 674–682 (2005).
Zhao, J., Goldberg, J., Bremner, J. D. & Vaccarino, V. Global DNA methylation is associated with insulin resistance: a monozygotic twin study. Diabetes 61, 542–546 (2012).
Sookoian, S. et al. Epigenetic regulation of insulin resistance in nonalcoholic fatty liver disease: impact of liver methylation of the peroxisome proliferator-activated receptor gamma coactivator 1alpha promoter. Hepatology 52, 1992–2000 (2010).
Arner, P. et al. The epigenetic signature of systemic insulin resistance in obese women. Diabetologia 59, 2393–2405 (2016).
Matthews, D. R. et al. Homeostasis model assessment: insulin resistance and beta-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia 28, 412–419 (1985).
Milagro, F. I. et al. High fat diet-induced obesity modifies the methylation pattern of leptin promoter in rats. J Physiol Biochem 65, 1–9 (2009).
Corella, D. & Ordovás, J. M. Biomarkers: Background, classification and guidelines for applications in nutritional epidemiology. Nutrición Hospitalaria 31, 177–188 (2015).
Tammen, S. A., Friso, S. & Choi, S. W. Epigenetics: the link between nature and nurture. Mol Aspects Med 34, 753–764 (2013).
Chambers, J. C. et al. Epigenome-wide association of DNA methylation markers in peripheral blood from Indian Asians and Europeans with incident type 2 diabetes: a nested case-control study. Lancet Diabetes Endocrinol 3, 526–534 (2015).
Kulkarni, H. et al. Novel epigenetic determinants of type 2 diabetes in Mexican-American families. Hum Mol Genet 24, 5330–5344 (2015).
Al Muftah, W. A. et al. Epigenetic associations of type 2 diabetes and BMI in an Arab population. Clin Epigenetics 8, 13 (2016).
Dayeh, T. et al. DNA methylation of loci within ABCG1 and PHOSPHO1 in blood DNA is associated with future type 2 diabetes risk. Epigenetics 11, 482–488 (2016).
Kriebel, J. et al. Association between DNA Methylation in Whole Blood and Measures of Glucose Metabolism: KORA F4 Study. PLoS One 11, e0152314 (2016).
Soriano-Tarraga, C. et al. Epigenome-wide association study identifies TXNIP gene associated with type 2 diabetes mellitus and sustained hyperglycemia. Hum Mol Genet 25, 609–619 (2016).
Wahl, S. et al. Epigenome-wide association study of body mass index, and the adverse outcomes of adiposity. Nature 541, 81–86 (2017).
Hernandez-Aguilera, A. et al. Epigenetics and nutrition-related epidemics of metabolic diseases: Current perspectives and challenges. Food Chem Toxicol 96, 191–204 (2016).
Gemma, C. et al. Methylation of TFAM gene promoter in peripheral white blood cells is associated with insulin resistance in adolescents. Mol Genet Metab 100, 83–87 (2010).
Canivell, S. et al. Gastric inhibitory polypeptide receptor methylation in newly diagnosed, drug-naive patients with type 2 diabetes: a case-control study. PLoS One 8, e75474 (2013).
Desgagne, V. et al. Epigenetic dysregulation of the IGF system in placenta of newborns exposed to maternal impaired glucose tolerance. Epigenomics 6, 193–207 (2014).
Garcia-Cardona, M. C. et al. DNA methylation of leptin and adiponectin promoters in children is reduced by the combined presence of obesity and insulin resistance. Int J Obes (Lond) 38, 1457–1465 (2014).
Gillberg, L., Jacobsen, S. C., Ronn, T., Brons, C. & Vaag, A. PPARGC1A DNA methylation in subcutaneous adipose tissue in low birth weight subjects–impact of 5 days of high-fat overfeeding. Metabolism 63, 263–271 (2014).
Kammel, A. et al. Early hypermethylation of hepatic Igfbp2 results in its reduced expression preceding fatty liver in mice. Hum Mol Genet 25, 2588–2599 (2016).
Jeon, J. P. et al. Differential DNA methylation of MSI2 and its correlation with diabetic traits. PLoS One 12, e0177406 (2017).
Ramos-Lopez, O., Riezu-Boj, J. I., Milagro, F. I., Martinez, J. A. & Project, M. DNA methylation signatures at endoplasmic reticulum stress genes are associated with adiposity and insulin resistance. Mol Genet Metab 123, 50–58 (2018).
Ramos-Lopez, O. et al. Association of low dietary folate intake with lower CAMKK2 gene methylation, adiposity, and insulin resistance in obese subjects. Nutr Res 50, 53–62 (2018).
van Dijk, S. J. et al. DNA methylation in blood from neonatal screening cards and the association with BMI and insulin sensitivity in early childhood. Int J Obes (Lond) 42, 28–35 (2018).
Volkov, P. et al. Whole-Genome Bisulfite Sequencing of Human Pancreatic Islets Reveals Novel Differentially Methylated Regions in Type 2 Diabetes Pathogenesis. Diabetes 66, 1074–1085 (2017).
Aoyama, T. et al. Cell-specific epigenetic regulation of ChM-I gene expression: crosstalk between DNA methylation and histone acetylation. Biochem Biophys Res Commun 365, 124–130 (2008).
Hwang, J. H. et al. DNA methylation patterns and gene expression associated with litter size in Berkshire pig placenta. PLoS One 12, e0184539 (2017).
Long, H. Y. et al. Blood DNA methylation pattern is altered in mesial temporal lobe epilepsy. Sci Rep 7, 43810 (2017).
Yang, H. & Yang, L. Targeting cAMP/PKA pathway for glycemic control and type 2 diabetes therapy. J Mol Endocrinol 57, R93–R108 (2016).
Yoshioka, M. et al. Chondromodulin-I maintains cardiac valvular function by preventing angiogenesis. Nat Med 12, 1151–1159 (2006).
Davegardh, C., Garcia-Calzon, S., Bacos, K. & Ling, C. DNA methylation in the pathogenesis of type 2 diabetes in humans. Mol Metab, https://doi.org/10.1016/j.molmet.2018.01.022 (2018).
Nilsson, E. et al. Altered DNA methylation and differential expression of genes influencing metabolism and inflammation in adipose tissue from subjects with type 2 diabetes. Diabetes 63, 2962–2976 (2014).
Ronn, T. et al. Impact of age, BMI and HbA1c levels on the genome-wide DNA methylation and mRNA expression patterns in human adipose tissue and identification of epigenetic biomarkers in blood. Hum Mol Genet 24, 3792–3813 (2015).
Cheng, Z., Zheng, L. & Almeida, F. A. Epigenetic reprogramming in metabolic disorders: nutritional factors and beyond. J Nutr Biochem 54, 1–10 (2018).
Mendelson, M. M. et al. Association of Body Mass Index with DNA Methylation and Gene Expression in Blood Cells and Relations to Cardiometabolic Disease: A Mendelian Randomization Approach. PLoS Med 14, e1002215 (2017).
Demerath, E. W. et al. Epigenome-wide association study (EWAS) of BMI, BMI change and waist circumference in African American adults identifies multiple replicated loci. Hum Mol Genet 24, 4464–4479 (2015).
Wilson, L. E., Harlid, S., Xu, Z., Sandler, D. P. & Taylor, J. A. An epigenome-wide study of body mass index and DNA methylation in blood using participants from the Sister Study cohort. Int J Obes (Lond) 41, 194–199 (2017).
Geurts, Y. M. et al. Novel associations between blood DNA methylation and body mass index in middle-aged and older adults. Int J Obes (Lond) 42, 887–896 (2018).
Ali, O. et al. Methylation of SOCS3 is inversely associated with metabolic syndrome in an epigenome-wide association study of obesity. Epigenetics 11, 699–707 (2016).
Aslibekyan, S. et al. Epigenome-wide study identifies novel methylation loci associated with body mass index and waist circumference. Obesity (Silver Spring) 23, 1493–1501 (2015).
Agha, G. et al. Adiposity is associated with DNA methylation profile in adipose tissue. Int J Epidemiol 44, 1277–1287 (2015).
Park, J. L. et al. Identification and evaluation of age-correlated DNA methylation markers for forensic use. Forensic Sci Int Genet 23, 64–70 (2016).
Nilsson, E. & Ling, C. DNA methylation links genetics, fetal environment, and an unhealthy lifestyle to the development of type 2 diabetes. Clin Epigenetics 9, 105 (2017).
Zhu, X. et al. Genome-Wide Analysis of DNA Methylation and Cigarette Smoking in a Chinese Population. Environ Health Perspect 124, 966–973 (2016).
Wilson, R. et al. The dynamics of smoking-related disturbed methylation: a two time-point study of methylation change in smokers, non-smokers and former smokers. BMC Genomics 18, 805 (2017).
Tsai, P. C. et al. Smoking induces coordinated DNA methylation and gene expression changes in adipose tissue with consequences for metabolic health. Clin Epigenetics 10, 126 (2018).
Kondratyev, N., Golov, A., Alfimova, M., Lezheiko, T. & Golimbet, V. Prediction of smoking by multiplex bisulfite PCR with long amplicons considering allele-specific effects on DNA methylation. Clin Epigenetics 10, 130 (2018).
Gao, Z. et al. Protein kinase A translocation and insulin secretion in pancreatic beta-cells: studies with adenylate cyclase toxin from Bordetella pertussis. Biochem J 368, 397–404 (2002).
Turkmen, K., Karagoz, A. & Kucuk, A. Sirtuins as novel players in the pathogenesis of diabetes mellitus. World J Diabetes 5, 894–900 (2014).
Layden, B. T., Durai, V. & Lowe, J. W.L. G-Protein-Coupled Receptors, Pancreatic Islets, and Diabetes. Nature Education 3, 13 (2010).
Satoh, T. Rho GTPases in insulin-stimulated glucose uptake. Small GTPases 5, e28102 (2014).
Begum, N., Sandu, O. A. & Duddy, N. Negative regulation of rho signaling by insulin and its impact on actin cytoskeleton organization in vascular smooth muscle cells: role of nitric oxide and cyclic guanosine monophosphate signaling pathways. Diabetes 51, 2256–2263 (2002).
Meek, T. H. & Morton, G. J. The role of leptin in diabetes: metabolic effects. Diabetologia 59, 928–932 (2016).
Kim, S. H. Maturity-Onset Diabetes of the Young: What Do Clinicians Need to Know? Diabetes Metab J 39, 468–477 (2015).
Nelson, D. L. & Cox, M. M. Lenhninger. Principios de bioquímica. 5 edition, (Omega, 2009).
Gowd, V., Gurukar, A. & Chilkunda, N. D. Glycosaminoglycan remodeling during diabetes and the role of dietary factors in their modulation. World J Diabetes 7, 67–73 (2016).
Harrell, F. E. Jr. Regression Modeling Strategies. With Applications to Linear Models, Logistic and Ordinal Regression, and Survival Analysis. Second edition, https://doi.org/10.1007/978-3-319-19425-7 (Springer, Springer Series in Statistics, 2015).
Bansal, A. & Simmons, R. A. Epigenetics and developmental origins of diabetes: correlation or causation? Am J Physiol Endocrinol Metab 315, E15–E28 (2018).
Muka, T. et al. The role of global and regional DNA methylation and histone modifications in glycemic traits and type 2 diabetes: A systematic review. Nutr Metab Cardiovasc Dis 26, 553–566 (2016).
Bacos, K. et al. Blood-based biomarkers of age-associated epigenetic changes in human islets associate with insulin secretion and diabetes. Nat Commun 7, 11089 (2016).
Panzeri, I. & Pospisilik, J. A. Epigenetic control of variation and stochasticity in metabolic disease. Mol Metab, https://doi.org/10.1016/j.molmet.2018.05.010 (2018).
Heyn, H. & Esteller, M. DNA methylation profiling in the clinic: applications and challenges. Nat Rev Genet 13, 679–692 (2012).
Larsen, T. M. et al. The Diet, Obesity and Genes (Diogenes) Dietary Study in eight European countries - a comprehensive design for long-term intervention. Obes Rev 11, 76–91 (2010).
Huerta, A. E., Navas-Carretero, S., Prieto-Hontoria, P. L., Martinez, J. A. & Moreno-Aliaga, M. J. Effects of alpha-lipoic acid and eicosapentaenoic acid in overweight and obese women during weight loss. Obesity (Silver Spring) 23, 313–321 (2015).
Celis-Morales, C. et al. Design and baseline characteristics of the Food4Me study: a web-based randomised controlled trial of personalised nutrition in seven European countries. Genes Nutr 10, 450 (2015).
Santos, J. L. et al. Development and assessment of the disposition index based on the oral glucose tolerance test in subjects with different glycaemic status. J Physiol Biochem 72, 121–131 (2016).
Abete, I. et al. Epigenetic Changes in the Methylation Patterns of KCNQ1 and WT1 after a Weight Loss Intervention Program in Obese Stroke Patients. Curr Neurovasc Res 12, 321–333 (2015).
Petersen, M. et al. Randomized, multi-center trial of two hypo-energetic diets in obese subjects: high- versus low-fat content. Int J Obes (Lond) 30, 552–560 (2006).
Estruch, R. et al. Primary prevention of cardiovascular disease with a Mediterranean diet. N Engl J Med 368, 1279–1290 (2013).
Martinez-Gonzalez, M. A. et al. Cohort profile: design and methods of the PREDIMED study. Int J Epidemiol 41, 377–385 (2012).
Zulet, M. A. et al. The reduction of the metabolyc syndrome in Navarra-Spain (RESMENA-S) study: a multidisciplinary strategy based on chrononutrition and nutritional education, together with dietetic and psychological control. Nutr Hosp 26, 16–26 (2011).
Ramos-Lopez, O. et al. Differential lipid metabolism outcomes associated with ADRB2 gene polymorphisms in response to two dietary interventions in overweight/obese subjects. Nutr Metab Cardiovasc Dis 28, 165–172 (2018).
Arpón, A. et al. Adherence to Mediterranean diet is associated with methylation changes in inflammation-related genes in peripheral blood cells. J Physiol Biochem 73, 445–455 (2017).
Du, P., Kibbe, W. A. & Lin, S. M. lumi: a pipeline for processing Illumina microarray. Bioinformatics 24, 1547–1548 (2008).
Team, R. RStudio: Integrated Development for R, http://www.rstudio.com/ (2015).
Morris, T. J. et al. ChAMP: 450k Chip Analysis Methylation Pipeline. Bioinformatics 30, 428–430 (2014).
Nordlund, J. et al. Genome-wide signatures of differential DNA methylation in pediatric acute lymphoblastic leukemia. Genome Biol 14, r105 (2013).
Maksimovic, J., Gordon, L. & Oshlack, A. SWAN: Subset-quantile within array normalization for illumina infinium HumanMethylation450 BeadChips. Genome Biol 13, R44 (2012).
Teschendorff, A. E., Zhuang, J. & Widschwendter, M. Independent surrogate variable analysis to deconvolve confounding factors in large-scale microarray profiling studies. Bioinformatics 27, 1496–1505 (2011).
Johnson, W. E., Li, C. & Rabinovic, A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics 8, 118–127 (2007).
Houseman, E. A. et al. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinformatics 13, 86 (2012).
Edgar, R., Domrachev, M. & Lash, A. E. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res 30, 207–210 (2002).
Tang, Q., Li, X., Song, P. & Xu, L. Optimal cut-off values for the homeostasis model assessment of insulin resistance (HOMA-IR) and pre-diabetes screening: Developments in research and prospects for the future. Drug Discov Ther 9, 380–385 (2015).
Ziaee, A., Esmailzadehha, N., Oveisi, S., Ghorbani, A. & Ghanei, L. The threshold value of homeostasis model assessment for insulin resistance in Qazvin Metabolic Diseases Study (QMDS): assessment of metabolic syndrome. J Res Health Sci 15, 94–100 (2015).
Gayoso-Diz, P. et al. Insulin resistance (HOMA-IR) cut-off values and the metabolic syndrome in a general adult population: effect of gender and age: EPIRCE cross-sectional study. BMC Endocr Disord 13, 47 (2013).
Khalaf, N. et al. Coffee and Caffeine Are Associated With Decreased Risk of Advanced Hepatic Fibrosis Among Patients With Hepatitis C. Clin Gastroenterol Hepatol 13, 1521–1531 e1523 (2015).
Kumar, K., Manrai, M., Sood, A. K. & Sharma, R. A clinical study of insulin resistance in patients with chronic pancreatitis. Diabetes Metab Syndr 11(Suppl 1), S283–S286 (2017).
Volkmar, M. et al. DNA methylation profiling identifies epigenetic dysregulation in pancreatic islets from type 2 diabetic patients. EMBO J 31, 1405–1426 (2012).
Acknowledgements
The authors wish to thank other members of MENA project for their contribution: Abete I, Crujeiras AB, Cuervo M, Goni L, Marti A, Martinez-Gonzalez MA, Navas-Carretero S, San-Cristobal R, and Zulet MA. We thank Nora Goodwin (from Dublin Institute of Technology, Dublin, Ireland) for reviewing the manuscript and Marta García-Granero for her statistical assistance. We also would like to thank all the volunteers who took part in the study, the personnel of the primary care centres, and other investigators of the different research groups for their participation in the recruitment process and the acquisition of the data. This work was supported by grants from the Government of Navarra (PT024), CIBERobn (CB12/03/30002) and Ministerio de Economía y Competitividad (AGL2013-45554-R). A.A was supported by a “Formación de Profesorado Universitario” predoctoral fellowship from Ministerio de Educación, Cultura y Deporte (FPU15/02790). O.R.L. was supported by a postdoctoral grant from The Nacional Council of Science and Technology from Mexico (CONACyT, Num. CVU. 444175) in collaboration with the Doctoral Program in Sciences in Molecular Biology in Medicine (CONACyT, PNPC 000091).
Author information
Authors and Affiliations
Contributions
A.A. performed most of the data analysis and wrote the first version of the paper. O.R.L. and M.L.M. helped in the analysis. J.L.S. critical discussed all aspects of the manuscript and contributed with a cohort. J.I.R.B., F.I.M. and J.A.M. supervised data analysis and helped with interpretation and with manuscript elaboration, as well as provide the conceptual design and financial support. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Arpón, A., Milagro, F.I., Ramos-Lopez, O. et al. Epigenome-wide association study in peripheral white blood cells involving insulin resistance. Sci Rep 9, 2445 (2019). https://doi.org/10.1038/s41598-019-38980-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-38980-2
This article is cited by
-
A comprehensive map of the aging blood methylome in humans
Genome Biology (2024)
-
Development of an epigenetic clock resistant to changes in immune cell composition
Communications Biology (2024)
-
Culture expansion of CAR T cells results in aberrant DNA methylation that is associated with adverse clinical outcome
Leukemia (2023)
-
Genetic associations vary across the spectrum of fasting serum insulin: results from the European IDEFICS/I.Family children’s cohort
Diabetologia (2023)
-
Multi-tissue epigenetic analysis identifies distinct associations underlying insulin resistance and Alzheimer’s disease at CPT1A locus
Clinical Epigenetics (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
