
Abstract
Cancer remains one of the biggest threats to human society. There are massive demands for compounds to selectively kill cancerous cells. Earlier studies have shown that bovine α -lactalbumin made lethal to tumor cells (BAMLET) becomes cytotoxic against cancer cells in complex with oleic acid {Hoque, M. et. al., PLoSOne 8, e68390 (2013)}. In our study, we obtained bovine α-lactalbumin complexed with lanthanum ion (La3+-B-α-LA) and determined its high resolution crystal structure. The natural calcium binding site of bovine α-lactalbumin is replaced by lanthanum. The La3+ complex formation by B-α-apo-LA was also supported by various biophysical methods. Interestingly, our complex, La3+-B-α-LA exhibits much greater anticancer activity against breast cancer cells as compared to the reported BAMLET-oleic acid complex. This study shows that La3+-B-α-LA complex is preferentially more toxic to MCF-7 cells as compared to KB (oral cancer) and HeLa (cervical) cells, while almost non-toxic to the healthy cells that we studied. Our data indicates that the cytotoxicity of La3+-B-α-LA against cancer cells is through apoptotic path way. The higher anticancer activity of La3+-B-α-LA is attributable to the requisite structural changes induced in the protein by La3+ binding as supported by the crystal structure of the complex.
Similar content being viewed by others

Introduction
Development of new methods for cancer treatment and prevention are always in high demand1. Human α-lactalbumin (H-α-LA) made lethal to tumor cells (HAMLET) and bovine α-lactalbumin (B-α-LA) made lethal to tumor cells (BAMLET), both cause death to the cancer cells through apoptosis but sparing the healthy cells2,3,4,5,6,7. Conversion of native α-lactalbumin into the apoptosis inducing form, BAMLET/HAMLET, involves partial unfolding of the protein followed by complexing with oleic acid followed by stabilising the partially folded conformation8,9,10,11,12,13,14,15,16,17. The detailed mechanism by which HAMLET/BAMLET acts as anti-cancer agent is under explored18. Some recent studies report the binding of B-α-LA to nanoclusters19,20 and nanoparticles21 of gold. Such nano-species show alternative pathways for stabilizing the unfolded state of B-α-LA so that the resulting protein-metal nano-composites express their required properties22. Though the BAMLET like structure could be synthesized from B-α-LA/ H-α-LA by different methods11,12,19,23, to our knowledge no such method uses any metal ion, further no crystal structure of BAMLET/HAMLET is known. The formation of protein-inorganic hybrids is greatly influenced by the exposed residues, binding core, coordination characteristics of the inorganic ion and the medium. The protein-inorganic hybrids show wide range of applications in catalysis, as drug carriers and in drug delivery, and in killing cancer cells24,25,26.
In the present study, we focused on the metallation of apo-B-α-LA by La3+ in order to complex as well to induce structural changes into the protein. The metallation was studied by spectroscopy and calorimetry. The coordination, protein conformation and structural aspects of La3+ bound B-α-LA were established by analyzing the crystal structure. The selective anti-cancer activity of this complex was studied using different types of cancer cell lines. The details of apoptosis were probed in one case, and the activity was compared accordingly with that reported in the literature.
Results and Discussion
Binding of La3+ to apo-B-α-LA
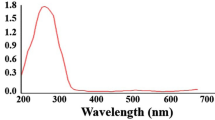
The interaction followed by binding of La3+ with apo-B-α-LA was studied by analytical methods, thermodynamics and spectroscopy. The ICP-AES data (see Supplementary Data, S01) of the isolated complex carried out as per the details given in the section on Methods fits well with one La3+ ion per protein. The spectra exhibited an increase in the absorbance of both the 280 and 215 nm bands as the concentration of the added La3+ increases (Fig. 1a). While the 280 nm band suggests the influence of binding on aromatic amino acid residues, the 215 nm band reflects the interaction of La3+ ion with the peptide bond as well as the free carboxylate groups. The fluorescence emission spectra were recorded upon addition of La3+ to apo-B-α-LA at λex = 295 nm and only a maximum of ~20% quenching was observed (Fig. 1b), supporting that the La3+ binding to protein does not substantially alter the structure where the aromatic side chains are present. The CD spectra exhibited marginal ellipticity changes in 222 and 208 nm bands (Fig. 1c) and thus supports minimum conformational changes in the secondary structure of the protein. If an ion binds in the pocket meant for Ca2+ binding, such binding is expected to bring only minimal spectral changes.

Spectral data of apo-bovine-α-LA titrated with different La3+ as per the details given in the section on “Methods”: (a) Absorption spectra obtained during the titration. The inset is the absorbance vs. concentration plots at 215 and 280 nm. (b) Fluorescence emission spectra (λex = 295 nm). The inset is the I/I0 vs. concentration plot. (c) Circular dichroism spectra. The inset is the ellipticity vs. concentration plots at 222 and 208 nm. (d) Calorimetric titration isotherm. The concentration of La3+ used in each of these experiment can be seen from the x-axis of the inset of the corresponding figure.
The ITC titration exhibited clear cut isotherm supporting the binding of La3+ to the protein (Fig. 1d) and the data fits well to one La3+ ion per protein molecule with a binding constant (Ka) of (7.3 ± 1.8) × 104 M−1. The enthalphy and entropy components of the La3+ binding are (1.25 ± 0.1) × 104 cal/mol and 64.4 cal/mol/deg respectively as derived from the ITC data. The La3+ binding pocket was identified based on the crystal structure established as given in this paper.
Structural proof for the binding of La3+ in the Ca2+ binding site of apo-B-α-LA
The La3+ complex of apo-B-α-LA was crystallized as per the details given in the experimental section and the structure of the complex was established by single crystal X-ray diffraction at a resolution of 1.85 Å with one molecule in the asymmetric unit in P3121 space group (Table 1). The binding of La3+ in the Ca2+ pocket was confirmed. Further, we have also addressed the conformational changes noticed in the protein backbone upon forming the complex.
The numbering of the residues in La3+-B-α-LA structure is same as that given in the crystal structure of Ca2+-B-α-LA (PDB ID = 1F6S)27. The overall structural fold of the La3+-B-α-LA complex is almost identical to the structure of apo-B-α-LA27. The structural superposition of La3+-B-α-LA with that of Ca2+-B-α-LA produced a root mean square deviation (r.m.s.d.) value of 0.54 Å. The structure of the complex La3+-B-α-LA is composed of four α-helices and three antiparallel β-strands (Fig. 2a). A flexible loop (residues 105–110) below the cleft region adopts a helical conformation in La3+-B-α-LA complex (Fig. 2a). The structure can be divided into α and β sub-domains and the presence of a cleft region clearly demarcates the existence of these two sub-domains. The α−sub-domain (residues 1–34 and 86–122) is bigger than the β-sub-domain (residues 35–85). The smaller β-sub-domain comprises of antiparallel β-sheets, irregular loops and a 310 helix (Fig. 2a). In addition, the two loop regions of the β-sub-domain composed of amino acid residues, i.e., Gln43-Ser47 (loop 1) and Lys62-Gln65 (loop 2), are exposed to the solvent in the Ca2+-B-α-LA structure. On the contrary, those two loop regions of the β-sub-domain are stabilized by three symmetry related molecules in the lattice of La3+-B-α-LA structure (Fig. 2b). The average thermal B-factors of the residues from the loop 1 and loop 2 of the La3+-B-α-LA structure are 19.1 and 15.1 Å2 respectively. Conversely, the average B-factor of the residues from loop 1 and loop 2 of Ca2+-B-α-LA complex are 73.6 and 66.7 Å2 respectively. The La3+-B-α-LA forms an interfacial contact with the symmetry related molecules at the edge of the two domains by forming hydrogen bonds (Fig. 2b). There are 62 water molecules in the crystal structure of La3+-B-α-LA with an average B-factor of ~34.9 Å2, but ranges from 8.1 to 48.6 Å2.

(a) The overall structure of La3+-B-α-LA represented as cartoon and the bound La3+ion is shown as a cyan sphere. The secondary structural elements are marked as H, α-helix; h, 310 helix; S, β-strand and the disulphide bonds are shown in yellow. The residues involved in La3+ ion binding and disulphide bridge formation are shown as ball and stick. The polar interactions with bound La3+ ion are shown as dotted lines. (b) The contacts between the symmetry related molecules of La3+-B-α-LA complex in the lattice are shown. The secondary structural elements of the four molecules are shown in green, magenta, yellow and wheat colour. The residues are shown as sticks. The polar interactions are presented with distances in Å.
Coordination characteristics of La3+ bound to protein in the crystal structure
The clear electron density defines the correct position of the La3+ ion in the Ca2+-binding cleft of holo-B-α-LA structure. The La3+ ion is located in the same binding core as described previously for other metal ion bound structures of B-α-LA28,29,30. In the present structure, the La3+ is bound to the protein through the side chain carboxylate groups of Asp82, Asp87 and Asp88, and the main chain amide carbonyl groups of Lys79 and Asp84 (Fig. 3a,c) in addition to three water molecules (W1, W2 and W3). The corresponding La3+-O bond distances are 2.48, 2.63, 2.41, 2.1, 2.42, 2.7, 2.5, and 2.56 Å, and the bond angles range from 56.9 to 158.6° (see Supplementary Data, S02).

La3+ primary coordination as obtained from the crystal structure of La3+-B-α-LA complex: (a) Electron density map (Fo-Fc map contoured at 4σ level) for La3+ in the calcium binding cleft. (b) Dodecahedral geometry of La3+ ion with eight coordination as observed in the La3+-B-α-LA crystal structure. (c) Zoomed in stereo view showing the interactions of La3+ ion with residues and the water molecules in case of La3+-B-α-LA structure. The protein residues are shown as ball and stick model. The water molecules are shown as small red spheres. The bound La3+ ion is shown as sky blue sphere.
In the structure of La3+-B-α-LA, the La3+ shows eight coordination with dodecahedral geometry (Fig. 3b) while it is pentagonal bipyramidal w.r.t. Ca2+ in case of the Ca2+-B-α-LA structure. In the La3+-B-α-LA structure, the five coordinations, viz., Asp82, Asp87, Asp88 and two water molecules (W2 and W3) form a pentagon while the Lys79, Asp84 and W1 forms a triangle. In this triangle, the Asp 84 is on one side of the pentagon and the Lys79 and W1 are on the opposite side to this, while La3+ sits at the centre. The O···O bond distances in this pentagon range from 2.48 to 3.48 Å and O···O···O angles range from 95.6 to 113.5° supporting that the pentagon is highly puckered.
Comparison between the Ca2+ and La3+ coordination cores
The coordination cores of La3+ and Ca2+ bound to protein (B-α-LA) were compared. There are only two water molecules in the Ca2+-B-α-LA structure. The average La3+···O distance observed in the present structure (~2.5 Å) is longer than that observed for the average Ca2+···O distance (2.37 Å) by ~0.18 Å, supporting that the metal ion binding core expands in case of La3+ bound structure. The analysis revealed that La3+ is coordinated in the metal binding cleft of B-α-LA with an additional water molecule as compared to that in the Ca2+ bound structure. Additionally, some unique conformational changes are observed for the amino acid side chains involved upon binding to La3+ (Fig. 4c). The Asp82 and Asp87 side chains are in a similar plane for both the apo-B-α-LA and Ca2+-B-α-LA structures, while in La3+-B-α-LA, the side chain of Asp82 is rotated and hence the carboxylate group is almost perpendicular to what was observed in case of apo- (Fig. 4a) and Ca2+ structures (Fig. 4b).

Comparison of the La3+-B-α-LA structure with that of the apo-B-α-LA and Ca2+-B-α-LA
The structural comparison of La3+-B-α-LA with its apo- and holo-(Ca2+)-B-α-LA was performed and the analysis revealed some changes in the metal binding residues of the La3+-B-α-LA structure. Apart from these differences, there are some conformational changes in the protein backbone of La3+-B-α-LA as given in Fig. 5. The disulphide bond between Cys6-Cys120 in La3+-B-α-LA is flipped into an opposite orientation as compared to apo-B-α-LA and Ca2+-B-α-LA. The flexible loop region consisting of the residues Val42 to Ser47 exhibited conformational change as compared to two other B-α-LA structures reported27. The overall difference in r.m.s.d. values calculated after superposition of La3+-B-α-LA with apo-B-α-LA and Ca2+-B-α-LA resulted in values of 0.89 and 0.54 Å, respectively. The average B-factors of the main chain atoms are 29.0, 38.3 and 48.4 Å2 respectively for La3+-B-α-LA, apo-B-α-LA and Ca2+-B-α-LA. The average B factors suggest that the structure of the polypeptide in the La3+-B-α-LA complex is more rigid as compared to the other two structures. This implicates that the binding of La3+ ion and polar interfacial contacts among the symmetry related molecules stabilize the overall structure of B-α-LA when compared to the apo-form and or the Ca2+ bound form. When these structures were overlaid, several conformational changes were revealed in both the main chain as well in the side chains in case of La3+-B-α-LA structure when compared to its apo-form, as can be noticed from the encircled regions in Fig. 5.

Superposition of the backbone structures of La3+-B-α-LA (green) with apo-B-α-LA (grey) and Ca2+-B-α-LA (magenta) showing major structural changes at disulphide linkage represented as dashed rectangle (zoomed view shown in inset), and the dotted circle represents a major change in loop region.
Biocompatibility of the La3+-B-α-LA complex
The La3+-B-α-LA complex reported in this paper was examined for its biocompatibility with normal mouse fibroblast cells (L929) and N1H3 cells by performing the SRB assay. While the La3+ salt alone showed cell viability of 82–85%, the La3+-B-α-LA complex showed >95%. In order to check the effect of concentration, four different concentrations were used for the cell viability studies. In the concentration range of 4.5 to 35 µM studied, the La3+-B-α-LA complex showed cell viability of >90–95% (Fig. 6) suggesting the non-toxic nature of this complex and hence the studies were extended to evaluate the anticancer property of La3+-B-α-LA complex.

Bar diagram for the cell viability vs. different concentrations of samples treated with the healthy cells, viz., (a) L929 cells and (b) N1H3 cell lines.
Cytotoxicity of the La3+-B-α-LA complex
The native form of the calcium bound protein (Ca2+-B-α-LA) does not exhibit any anti-cancer activity, however, exhibits enhanced effect when it undergoes conformational change to that of BAMLET that is stabilised by oleic acid as reported in the literature11. Therefore, herein, we explore the anticancer activity of La3+-B-α-LA on HeLa (cervical cancer cells), KB cells (oral cancer cells) and MCF-7 cells (breast cancer cells), since the binding of La3+ induces some conformational changes and stabilizes a structure that is different from that of apo- and or Ca2+ structure. It was observed that La3+-B-α-LA shows anti proliferation effect on KB, HeLa and MCF-7 cells (Fig. 7). In the concentration range 4–35 µM, the La3+-B-α-LA brings cell death of ~15–20% in case of KB cells and ~30–35% in case of HeLa cell lines. The anti-cancer activity of La3+-B-α-LA complex on MCF-7 cell lines showed ~70–93% of anticancer effect, whereas the simple La3+ salt and the apo-protein showed only 35–40% and 1–2% respectively, suggesting that the anti-cancer effect results primarily from the conformational/structural changes induced in the protein by La3+ binding, since the contribution from the bare La3+ is less than half of that exhibited by the La3+-B-α-LA complex. Thus, the cell culture studies reveal that La3+-B-α-LA complex at a concentration of 0.35 µM specifically kills the breast cancer cells sparing the healthy ones and its anticancer property follows a trend, i.e., MCF-7 (~93%)≫ > HeLa (~40%) > KB (~20%).

Bar diagram for the cell viability vs. the cells (KB cells, HeLa cell and MCF-7 cell lines) that get treated by the samples. The colour coding: apo-B-α-LA (blue), Ca2+-B-α-LA (magenta), La3+ (olive green) and La3+-B-α-LA (maroon).
Fluorescence microscopy studies of the La3+-B-α-LA complex in MCF-7 cells
The apoptotic cell death was demonstrated in MCF-7 cells by fluorescence microscopy in the presence of La3+-B-α-LA complex, by carrying out the experiments using caspase 3/7 dye31. The microscopy studies carried out in this regard include, (i) untreated cells, and cells treated with (ii) 15 µM apo-B-α-LA, (iii) 15 µM Ca2+-B-α-LA, (iv) 200 µM of La3+ salt and (v) 15 µM La3+-B-α-LA complex. In case of (i), (ii) and (iii), no green fluorescence was observed suggesting that there is no role in activating caspase 3/7 driven apoptosis. However, La3+ treated cells, i.e., (iv), exhibited weak green fluorescence indicating that La3+ treatment alone can activate caspase 3/7 mediated apoptosis to some extent. Though the cells adopted round shape due to the treatment, these have not undergone apoptosis. In case of the cells treated with La3+-B-α-LA complex, i.e., (v), almost all the cells exhibited typical green fluorescence as well as distorted cell morphology (Fig. 8) indicating the activation of caspase 3/7 pathway which is characteristic of apoptotic cell death32. The integrated green fluorescence intensity is >3 times higher when the MCF-7 cells were treated with La3+-B-α-LA complex as compared to the same when treated with simple La3+ salt (Fig. 8p).

Fluorescence microscopy images of MCF-7 cells: (a–c) Are untreated. (d–f) Are for apo-B-α-LA. (g–i) Are for Ca2+-B-α-LA. (j–l) Are for just La3+ salt. (m–o) Are for La3+-B-α-LA. (p) Relative green fluorescence intensity for the studies carried out with MCF-7 cells when treated with that mentioned in the figure.
Concluding Remarks
Both the absorption and emission spectra clearly showed the interaction of La3+ with apo-B-α-LA, and ICP-AES and the ITC showed 1:1 protein: La3+ complex. The crystal structure of La3+-B-α-LA complex was determined at a resolution of 1.85 Å. To our knowledge, this is the first La3+ complex of B-α-LA. The structure revealed the presence of La3+ occupying the Ca2+ binding site of native B-α-LA but with some primary coordination sphere changes as compared to that of the Ca2+- and or apo- protein. Thus, the La3+ exhibit eight coordination with dodecahedral geometry, while the Ca2+ was in distorted pentagonal bipyramidal geometry with seven coordination. As the La3+ ion occupies the Ca2+ cleft in the protein, this is not expected to alter the spectral features to any great extent. The CD spectra indeed showed that the loss of α-helical structure is minimum, which can also be gauzed from the crystal structure. The average B-factors of the main chain atoms are 29.0, 38.3 and 48.4 Å2 respectively for La3+-B-α-LA, apo-B-α-LA and Ca2+-B-α-LA structures supporting that La3+ complex of protein is more rigid than the apo- as well as holo (Ca2+)- proteins. The La3+-B-α-LA complex at a concentration of 0.5 mg/ml specifically kills the breast cancer cells (MCF-7 cells) to an extent of >90% sparing the healthy ones, and hence this protein – La3+ complex is specific to the cancer type. The differential cell proliferation results obtained among these three cancer cells seems to be dependent on the nature of the receptor sites present on the corresponding cells, though the details of the same deserves a specialized study. Recently, we found greater anticancer activity in case of MCF-7 cells over the other cancer cells even in case of α-LA coated on AuNPs where the conformation of the protein would be different from the apo- or holo- protein, supporting that the structural change in the protein has role in the anticancer activity20.
The cell death has been confirmed to arise from apoptosis based on the studies carried out in presence of caspase 3/7 dye by fluorescence microscopy. The anticancer activity exhibited by the La3+ complex is much greater as compared to the literature reported BAMLET, which is only ~70%. While the oleic acid stabilizes the partial unfolded structure of the protein to result in BAMLET, in the present case the La3+ induces structural changes which are more prone for anticancer activity. Our extended fluorescence microscopy studies reveal that the green fluorescence intensity observed for La3+-B-α-LA complex is greater by 3 fold as compared to the same when these were treated with simple La3+ salt. In fact, the average B-factor and r.m.s.d. supports this when the corresponding parameters of the La3+-B-α-LA complex are compared with apo-/holo(Ca2+)-protein and or the lanthanide ion alone. Comparison of the CD spectra of La3+-B-α-LA complex with that of the literature reported BAMLET33 supported that the La3+ induces a different structure and this supports the greater anticancer activity exhibited by the La3+-B-α-LA complex as compared to the BAMLET in MCF-7 cells.
Methods
Apo-B-α-LA was procured from Sigma Aldrich Chem. Co., and used in all the experiments without further purification. The lanthanum perchlorate salt was prepared starting from lanthanum oxides followed by treating the reaction mixture with perchloric acid and recrystallizing the product. A 10 mM Tris-HCl buffer at pH 7.4 was used for all the experiments unless otherwise mentioned.
Both the healthy and the cancer cells used in the present study were obtained from National Centre for Cell Science, Pune, India. Dulbeco’s Modified Eagle Medium (DMEM) and DMEM without phenol red, Dulbecco’s Phosphate Buffered Saline (DPBS), Fetal Bovine Serum (FBS) and coverslips for fluorescence microscopy were purchased from Sigma-Aldrich, USA. Caspase 3/7 green detection reagent was procured from Thermo Fischer scientific.
Spectroscopy
UV-Visible absorption studies were performed on Varian instrument. One mL of 0.015 mM (25 µl of 1 mg/mL) apo-B-α-LA was taken into a cuvette and titrated against 5 mM La3+ such that the metal ion to protein mole ratio varied from 0 to 60 folds (by adding incremental addition of 0.5 µl metal ion solution at each time) and subtracted from the background at a scan rate 200 nm/min. Fluorescence spectral studies were performed on a Varian instrument at the same concentrations and mole ratios. The experiments were performed in quartz cuvette of path length 1 cm and scan speed of 200 nm/min. Fluorescence spectra were measured by exciting the solutions at 280 nm and 295 nm. Other details are same as that used for the absorption spectra. Far–UV CD spectra were recorded on JASCO-810 using Quartz cuvette of 0.1 cm path length. The CD spectra were accumulated at room temperature at a scan speed of 100 nm/min between 190–270 nm. Each time, 0.5 mL of 35 µM apo-B-α-LA was taken for CD measurements. For inductively coupled plasma atomic emission spectroscopy (ICPAES), a 1 mg/mL of apo-B-α-LA and La3+ (at 1:10 ratio) were taken and incubated overnight and then the samples were diluted to 10 mL and were dialysed in 10 mM Tris-HCl buffer at pH 7.4 for 6 hours. The samples were analysed with ICPAES before and after the dialysis, and the metal concentration was evaluated.
Isothermal titration calorimetry (ITC)
The calorimetric titrations were performed at 25 °C with a microcal isothermal titration calorimeter from GE Healthcare (Northampton, MA, USA). The concentration of the protein used was 70 µM and this was titrated against 20 µl of 5 mM of La3+salt solution. The buffer was similarly titrated with same metal ion concentration and this was subtracted to give the final thermogram. All the solutions were degassed for 30 minutes prior to the start of the experiment. A good fit to these data was done using the Origin software 8.0 version.
Crystallization
The protein, apo-B-α-LA was dissolved in metal ion free water. The final concentration of the protein used for crystallization was 25 mg/mL. Apo-B-α-LA with La3+ ion was crystallized using hanging drop vapour diffusion method at 295 K. The crystallization drops were set up by mixing 2 µl of protein solution with 2 µl of mother liquor and 0.5 µl of 2 mM lanthanum perchlorate. The crystallization drops were equilibrated against 300 µl of mother liquor. Optimization of protein to La3+ ion concentration ratio was performed prior to crystallization set up to avoid precipitation in the crystallization drop. The best crystals were obtained using precipitant containing 0.1 M Tris-HCl at pH 6.0 and 2 M ammonium sulphate. The crystals appeared within one week of crystallization set up and these grew to their maximum size of 0.3 × 0.25 × 0.15 mm in three weeks.
Diffraction data collection and data processing
The crystals were cryo-protected using the reservoir solution also containing 30% (v/v) of glycerol. A single crystal was picked up from the crystallization drop using cryo-loop and quickly transferred to the cryo-protectant solution. Immediately after that, the crystal was flash cooled by transferring it to the liquid nitrogen stream at 100 K. The diffraction data set was collected by rotation method with 0.5° oscillation per image. The data set was collected at the Protein Crystallography Facility of Indian Institute of Technology Bombay using Cu Kα X-ray source generated by a Rigaku Micro Max-007 HF diffractometer fitted with R-Axis IV++ image plate detector. The indexing, integration and scaling of the data set were performed by XDS software package34. The intensities were converted to structure factors with program modules F2MTZ and CAD of CCP435. The data collection statistics is presented in Table 1.
Structure determination and refinement
The structure of La3+bound apo-B-α-LA was determined by molecular replacement method using the molecular replacement module of the PHASER program36. The A-chain of Ca2+-B-α-LA crystal structure (PDB ID = 1F6S)27 was used as a search model for the initial phase determination. The calculation of Matthews’ coefficient37 showed the presence of one molecule in the asymmetric unit with VM value of 2.06 A3 Da−1 which corresponds to 40% solvent content. After finding correct orientation of the protein molecule by PHASER, initial few cycles of refinement of the model was done by REFMAC38. The analysis of the initial sigma weighted Fo-Fc electron density map showed the presence of lanthanum ion in the metal binding cleft of B-α-LA. After placing the La3+ ion inside the protein molecule, repeated cycles of refinement were done using REFMAC and manual model building was performed using COOT39. Water and glycerol molecules, and sulphate ions were progressively added at peaks of the sigma-A weighted Fo-Fc electron density map higher than 3σ level while monitoring the decrease of Rfree and improvement of the overall stereochemistry of the protein structure. The last two residues could not be built because of the lack of features of these residues in the electron density map. The statistics of the structure refinement is presented in Table 1. The structural figures were generated using PyMOL version 1.340. The secondary structural elements were assigned using Dictionary of Secondary Structure of Proteins (DSSP) server41.
Cell viability study
Sulforhodamine B (SRB) assay was performed to evaluate cell viability with L-929, N1H3, HeLa, KB and MCF-7 cell lines. The cells were seeded into 96-well plates at densities of 1 × 104 cells per well and incubated for 24 h. Different concentrations of the samples were added to the cells and incubated for 24 h at 37 °C in the atmosphere of 5% CO2. Thereafter, the cells were washed thrice with phosphate buffer saline (PBS) and processed for SRB assay to determine the cell viability. For this, the cells were fixed with a solution of 50% trichloroacetic acid and stained with 0.4% SRB dissolved in 1% acetic acid. Cell-bound dye was extracted with 10 mM Tris buffer solution at pH 10.5 and then the absorbance was measured at 560 nm using a plate reader. The cell viability was calculated as the ratio of the absorbance of the sample to the control, and was expressed in %.
Apoptosis study using caspase 3/7 dye
MCF-7 cells were seeded on cover slips in a 6 well plate at a density of 104 cells/mL in DMEM containing 10% FBS and a 0.1% antibiotic solution and incubated for 24 h at 37 °C and 5% CO2 for adherence. Once MCF-7 cells become fully confluent, these were treated with 15 µM of La3+-B-α-LA prepared in DMEM without phenol red (treatment media) and further incubated for 24 h. After incubation, the medium was removed from each well and the cells were washed carefully with PBS. 10 μL of ready to use caspase 3/7 dye prepared in 1 mL of PBS was added to the cells. The cells were further incubated at 37 °C for 15 min. After incubation, the cover-slip was removed from each well and mounted on glass slide using glycerol as mounting medium. Imaging was carried out using FLoid® Cell Imaging Station (Life Technologies) using 20X objective.
References
Frezza, M. et al. Novel Metals and Metal Complexes as Platforms for Cancer Therapy. Curr. Pharm. Des. 6, 1813–1825 (2010).
Casbarra, A. et al. Conformational analysis of HAMLET, the folding variant of human α-lactalbumin associated with apoptosis. Protein Sci. 13, 1322–1330 (2004).
Hakansson, A., Zhivotovsky, B., Orrenius, S., Sabharwal, H. & Svanborg, C. Apoptosis induced by a human milk protein. Proc. Natl. Acad. Sci. USA 92, 8064–8068 (1995).
Svanborg, C. et al. HAMLET kills tumor cells by an apoptosis-like mechanism-cellular, molecular, and therapeutic aspects. Adv. Cancer Res. 88, 1–29 (2003).
Svensson, M. et al. HAMLET- complex from human milk that induces apoptosis in tumor cells but spares healthy cells. Adv. Exp. Med. Biol. 503, 125–132 (2002).
Svensson, M., Hakansson, A., Mossberg, A. K., Linse, S. & Svanborg, C. Conversion of α-lactalbumin to a protein inducing apoptosis. Proc. Natl. Acad. Sci. USA 97, 4221–4226 (2000).
Svensson, M. et al. Molecular characterization of α-lactalbumin folding variants that induce apoptosis in tumor cells. J. Biol. Chem. 274, 6388–6396 (1999).
Chaudhuri, A. & Chattopadhyay, A. Lipid binding specificity of bovine α-lactalbumin: A multidimensional approach. Biochim. Biophys. Acta, Biomembr. 1838, 2078–2086 (2014).
Fang, B. et al. Bovine lactoferrin binds oleic acid to form an anti-tumor complex similar to HAMLET. Biochim. Biophys. Acta, Mol. Cell Biol. Lipids. 1841, 535–543 (2014).
Rath, E. M., Duff, A. P., Hakansson, A. P., Knott, R. B. & Bret Church, W. Small-angle X-ray scattering of BAMLET at pH 12: A complex of α-lactalbumin and oleic acid. Proteins: Struct., Funct., Bioinf. 82, 1400–1408 (2014).
Svensson, M. et al. α-Lactalbumin unfolding is not sufficient to cause apoptosis, but is required for the conversion to HAMLET (human α-lactalbumin made lethal to tumor cells). Protein Sci. 12, 2794–2804 (2003).
Kamijima, T. et al. Heat-treatment method for producing fatty acid-bound alpha-lactalbumin that induces tumor cell death. Biochem. Biophys. Res. Commun. 376, 211–214 (2008).
Park, Y. J., Kim, K. H., Lim, D. W. & Lee, E. K. Effects of pH and protein conformation on in-solution complexation between bovine α-lactalbumin and oleic acid: Binding trend analysis by using SPR and ITC. Process Biochem. (Oxford, U. K.) 50, 1379–1387 (2015).
Wen, H., Glomm, W. R. & Halskau, O. Cytotoxicity of bovine α-lactalbumin:oleic acid complexes correlates with the disruption of lipid membranes. Biochim. Biophys. Acta, Biomembr. 1828, 2691–2699 (2013).
Zhong, S. et al. Cytotoxicity and apoptosis induction of bovine alpha-lactalbumin-oleic acid complex in human breast cancer cells. Food Sci. Technol. Res. 21, 103–110 (2015).
Svensson, M., Mossberg, A. K., Pettersson, J., Linse, S. & Svanborg, C. Lipids as cofactors in protein folding: stereo-specific lipid-protein interactions are required to form HAMLET (human alpha-lactalbumin made lethal to tumor cells). Protein Sci. 12, 2805–2814 (2003).
Hakansson, A. P., Roche-Hakansson, H., Mossberg, A. K. & Svanborg, C. Apoptosis-like death in bacteria induced by HAMLET, a human milk lipid-protein complex. PLoS One. 6, e17717 (2011).
Lystvet, S. M. et al. Anticancer activity from gold-α-lactalbumin nanoconstructs? J. Phys. Chem. C. 117, 2230–2238 (2013).
Lystvet, S. M. et al. Tunable photophysical properties, conformation and function of nanosized protein-gold constructs. RSC Adv. 3, 482–495 (2013).
Yarramala, D. S., Doshi, S. & Rao, C. P. Green synthesis of protein protected fluorescent gold nanoclusters (AuNCs): Reducing the size of the AuNCs by partially occupying the Ca2+ site by La3+ in apo-α-lactalbumin. RSC Adv. 5, 32761–32767 (2015).
Yarramala, D. S., Baksi, A., Pradeep, T. & Rao, C. P. Green synthesis, characterization and anticancer activity of luminescent gold nanoparticles capped with apo-α-lactalbumin. ACS Sustainable Chem. Eng. 5, 6064–6069 (2017).
Lystvet, S. M., Volden, S., Halskau, O. & Glomm, W. R. Immobilization onto gold nanoparticles alters α-lactalbumin interaction with pure and mixed phospholipid monolayers. Soft Matter. 7, 11501–11509 (2011).
Mahanta, S. & Paul, S. Bovine α-lactalbumin functionalized graphene oxide nano-sheet exhibits enhanced biocompatibility: A rational strategy for graphene-based targeted cancer therapy. Colloids Surf B. 134, 178–187 (2015).
Ge, J., Lei, J. & Zare, R. N. Protein-inorganic hybrid nanoflowers. Nat. Nanotechnol. 7, 428–432 (2012).
Koley, P., Sakurai, M. & Aono, M. Controlled Fabrication of Silk Protein Sericin Mediated Hierarchical Hybrid Flowers and Their Excellent Adsorption Capability of Heavy Metal Ions of Pb(II), Cd(II) and Hg(II). ACS Appl. Mater. Interfaces. 8, 2380–2392 (2016).
Yilmaz, E., Ocsoy, I., Ozdemir, N. & Soylak, M. Bovine serum albumin-Cu(II) hybrid nanoflowers: An effective adsorbent for solid phase extraction and slurry sampling flame atomic absorption spectrometric analysis of cadmium and lead in water, hair, food and cigarette samples. Anal. Chim. Acta. 906, 110–117 (2016).
Chrysina, E. D., Brew, K. & Acharya, K. R. Crystal structures of apo- and holo-bovine α-lactalbumin at 2.2-Å resolution reveal an effect of calcium on inter-lobe interactions. J. Biol. Chem. 275, 37021–37029 (2000).
Acharya, K. R., Stuart, D. I., Walker, N. P., Lewis, M. & Phillips, D. C. Refined structure of Baboon α-Lactalbumin at l.7 A° resolution comparison with C-type Lysozyme. J. Mol. Biol. 208, 99–127 (1989).
Chandra, N., Brew, K. & Acharya, K. R. Structural evidence for the presence of a secondary calcium binding site in human alpha-lactalbumin. Biochemistry. 37, 4767–4772 (1998).
Pike, A. C. W., Brew, K. & Acharya, K. R. Crystal structures of guinea-pig, goat and bovine α-lactalbumin highlight the enhanced conformational flexibility of regions that are significant for its action in lactose synthase. Structure. 4, 691–703 (1996).
Mahanta, S. & Paul, S. Stable Self-Assembly of Bovine α-Lactalbumin Exhibits Target-Specific Antiproliferative Activity in MultipleCancer Cells. ACS Appl. Mater. Interfaces. 7, 28177–28187 (2015).
Brentnall, M., Rodriguez-Menocal, L., De, G. R. L., Cepero, E. & Boise, L. H. Caspase-9, caspase-3 and caspase-7 have distinct roles during intrinsic apoptosis. BMC Cell Biol. 14, 32 (2013).
Hoque, M., Dave, S., Gupta, P. & Saleemuddin, M. Oleic acid may be the key contributor in the BAMLET induced erythrocyte hemolysis and tumoricidal action. PLoSOne 8, e68390 (2013).
Kabsch, W. XDS. Acta Crystallogr D Biol Crystallogr. 66, 125–132 (2010).
Winn, M. D. et al. Overview of the CCP4 suite and current developments. Acta Crystallogr., Sect. D: Biol. Crystallogr. 67, 235–242 (2011).
McCoy, A. J. et al. Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 (2007).
Matthews, B. W. Solvent content of protein crystals. J. Mol. Biol. 33, 491–497 (1968).
Murshudov, G. N. et al. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr., Sect. D: Biol. Crystallogr. 67, 355–367 (2011).
Emsley, P. & Cowtan, K. Coot: model-building tools for molecular graphics. Acta Crystallogr., Sect. D: Biol. Crystallogr. 60, 2126–2132 (2004).
DeLano, W. L. Pymol: An open source molecular graphic tool. CCP4 Newsletter On Protein Crystallography. 40, 82–92 (2002).
Kabsch, W. & Sander, C. Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers. 22, 2577–2637 (1983).
Acknowledgements
CPR acknowledges financial support from the DST/SERB {EMR/2014/000985} and for J. C. Bose National Fellowship {SB/S2/JCB-066/2015} and IIT Bombay for Institute Chair Professorship. P.B. acknowledges financial support from the Ramalingaswami Re-entry Fellowship, DBT. D.S.Y. acknowledges UGC for research fellowship both at JRF and SRF. We acknowledge SAIF, IRCC, IIT Bombay for all the instrumental facilities. PPK acknowledges the funding for the microscope from DBT(BT/PR3871/MED/30/830/2012).
Author information
Authors and Affiliations
Contributions
D.S.Y. and C.P.R. conceived the idea of the study, designed and executed the experiments. D.S.Y. performed the experiments. Cell culture experiments were performed by D.S.R., S.D. and D.S.Y.; P.P.K. contributed the reagents/ analysis tools for the same. Crystallization of the protein complex was performed by P.P. X-ray diffraction data collection, processing and structure solution were performed by P.P. and P.B. D.S.Y., C.P.R., P.B. and P.P. analyzed the data. D.S.Y., C.P.R., P.P. and P.B. contributed to the preparation of the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yarramala, D.S., Prakash, P., Ranade, D.S. et al. Cytotoxicity of apo bovine α-lactalbumin complexed with La3+ on cancer cells supported by its high resolution crystal structure. Sci Rep 9, 1780 (2019). https://doi.org/10.1038/s41598-018-38024-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-38024-1
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
