
Abstract
The importance of neutrophils in the pathogenesis of autoimmune rheumatic diseases, such as systemic lupus erythematosus (SLE) and rheumatoid arthritis (RA), is increasingly recognised. Generation of reactive oxygen species (ROS) and release of neutrophil extracellular traps (NETs) by activated neutrophils are both thought to contribute to pathology; although the underlying mechanisms, particularly the effects of IgG autoantibodies upon neutrophil function, are not fully understood. Therefore, we determined whether purified IgG from patients with SLE or RA have differential effects upon neutrophil activation and function. We found that SLE- and RA-IgG both bound human neutrophils but differentially regulated neutrophil function. RA- and SLE-IgG both increased PMA-induced β1 integrin-mediated adhesion to fibronectin, whilst only SLE-IgG enhanced αMβ2 integrin-mediated adhesion to fibrinogen. Interestingly, only SLE-IgG modulated neutrophil adhesion to endothelial cells. Both SLE- and RA-IgG increased ROS generation and DNA externalisation by unstimulated neutrophils. Only SLE-IgG however, drove DNA externalisation following neutrophil activation. Co-culture of neutrophils with resting endothelium prevented IgG-mediated increase of extracellular DNA, but this inhibition was overcome for SLE-IgG when the endothelium was stimulated with TNF-α. This differential pattern of neutrophil activation has implications for understanding SLE and RA pathogenesis and may highlight avenues for development of novel therapeutic strategies.
Similar content being viewed by others


Introduction
Systemic lupus erythematosus (SLE) and rheumatoid arthritis (RA) are both autoimmune rheumatic diseases (ARDs) that share features of endothelial dysfunction, aberrant leukocyte activation and pathogenic autoantibody formation, all of which contribute to pathophysiology. Increasingly, neutrophil dysfunction has also been recognised in ARD immunopathology1,2.
Neutrophils are rapidly recruited to sites of infection or inflammation, where complex interactions between neutrophil selectins and integrins with their endothelial ligand counterparts regulate neutrophil extravasation and activation. Activated neutrophils fight infection and promote inflammation via phagocytosis, degranulation and neutrophil extracellular trap (NET) formation. NET release results in the externalisation of a meshwork of chromatin fibres decorated with antimicrobial proteins and proteases, via a process termed NETosis3. NETs ensnare bacteria and promote pathogen killing, but also expose neo-antigens and pro-inflammatory molecules. Aberrant NETosis can induce endothelial damage and dysfunction4,5, leading to increased risk of atherosclerosis and vascular thrombosis6,7. Moreover, NETs have been demonstrated to activate both leukocytes8,9,10,11 and stromal cells12, which can drive disease progression. Dysregulation of this process has been implicated in both SLE and RA13,14,15.
Neutrophils express several different classes of integrins but the most important are the β1 and β2 integrins. The β1 integrins recognise ligands in the extracellular matrix (ECM), in particular those with the Arg-Gly-Asp (RGD) motif (found in fibronectin, vitronectin and laminin). As well as recognising RGD, α4β1 has a ligand-binding site for Leu-Asp-Val-Pro (LDVP) and Ile-Asp-Ala-Pro (IDAP) found in fibronectin. In addition, α4β1 can also bind the Ile-Asp-Ser-Pro (IDSP) motif found in vascular cell adhesion molecule (VCAM)-116, a ligand that is upregulated on endothelial cells during inflammation. The β2 integrin αLβ2 binds intercellular cell adhesion molecule (ICAM)-1, a ligand that is upregulated on various cell types following inflammation. In contrast, αMβ2 is more promiscuous and recognises a range of ligands including ICAM-1 and fibrinogen, which are upregulated at sites of tissue injury and during active coagulation. Integrin-mediated adhesion, via β1 and/or β2 integrins, is vital to neutrophil activation17, leading to the production of reactive oxygen species (ROS) and NETosis18,19,20,21. Moreover, evidence indicates that integrin inhibition reduces NET release15,20.
ARDs are generally characterised by immune dysfunction, with many groups exploring the immune differences in patients with SLE and RA. Given the presence of autoantibodies, it is unsurprising to find that defects in B and T cell regulation have been described in both SLE and RA. B cells contribute to pathology not only through antigen presentation, but also by producing autoantibodies22. Studies in RA found that autoantibodies stimulate the production of pro-inflammatory cytokines22, which promote T cell, B cell and macrophage activation22,23. Greater peripheral B cell activation was observed in patients with SLE24, with cells being more sensitive to cytokine stimulation25, displaying abnormal receptor-mediated signalling26 and having a greater propensity to undergo epitope spreading27.
Th1 cells have conventionally been considered to drive RA pathology, however there is growing interest in other T cell subsets. Th17 cells secrete pro-inflammatory mediators that suppress regulatory T cell (Treg) generation28,29. Elevated Th17 and reduced Treg differentiation have been described in RA patients, which promote inflammatory cell phenotypes30. In addition, Tregs isolated from RA patients have limited suppressive activity31,32, which is attributed to low expression of cytotoxic T-lymphocyte-associated protein 4 (CTLA-4)32. This reduced Treg population, with defective suppressive capability, fails to suppress autoreactive T cells33. Aberrant T cell activation has also been linked to SLE pathology, with reports documenting abnormal TCR signalling and T cell hyper-responsiveness arising through defects in an array of signalling molecules34,35,36,37,38,39.
Macrophages also contribute to RA pathology through pro-inflammatory cytokine production, ROS generation, release of matrix-degrading enzymes, phagocytosis and antigen presentation40. Monocytes isolated from SLE patients have elevated expression of activation markers and adhesion molecules41,42,43,44. Aberrant cytokine production has also been described in SLE-derived monocytes25,45,46, which promote the production of anti-dsDNA autoantibodies by B cells47,48.
Studies identified neutrophils in the synovial fluid of RA patients49,50,51, with further reports finding increased neutrophil activation52,53,54. Neutrophils have also been implicated in RA mouse models55,56,57. RA neutrophils are prone to spontaneous NETosis, with elevated NETs being reported in RA serum that correlated with anti-citrullinated protein antibodies (ACPA) titres and markers of inflammation14. Adaptive immunity has an established role in SLE pathology, however in more recent years, neutrophils are being increasingly studied. Early studies noted that SLE sera induced neutrophil aggregation and interfered with phagocytosis and degranulation58. Uptake of circulating nucleosomes activates neutrophils59, which leads to the secretion of antibacterial proteins. Elevated bactericidal proteins have been documented in SLE serum, which correlate with autoantibody titres60 and disease activity61,62,63. NETosis releases dsDNA and inflammatory cytokines, which also correlate with anti-dsDNA antibodies titres64.
The presence of circulating autoantibodies in SLE and RA is well-established, and used to aid diagnosis, predict disease progression and monitor response to treatment. Patients with either SLE or RA commonly present with a multitude of autoantibodies, with a large heterogeneity of antigenic targets65,66, each believed to confer pathogenic effects. Early reports found that sub-fractions of SLE-IgG were able to activate cultured endothelial cells and promote adhesion of the monocytic U937 cell line67. Some groups have specifically examined the effects of IgG upon neutrophil activation, highlighting an increased propensity of patient-derived neutrophils to undergo spontaneous NETosis as well as serum IgG being able to induce NET release14,68,69,70. However, whilst these studies report IgG effects upon an end-stage functional outcome of neutrophil activation (NETosis), there does not appear to be any evidence examining involvement of β1 and β2 integrins, which occurs at an early stage of neutrophil activation.
The precise effects of purified ARD-IgG upon neutrophil integrin activation and adhesion are poorly characterised. Given the importance of neutrophil activation to ARD immunopathology, determining the effects of ARD-IgG upon neutrophil function may elucidate the mechanisms through which neutrophils contribute to disease progression. Therefore, we examined the effects of SLE- and RA-IgG on β1 and β2 integrin-mediated adhesion and activation.
Results
Patient demographics
Neutrophils were isolated from 12 healthy controls (HCs), 12 SLE patients and 7 RA patients. IgG was purified from a separate cohort of 9 HCs, 5 SLE patients and 9 RA patients. Demographics, clinical history and treatments of all subjects at the time of sample collection are listed in Table 1. All purified IgG samples tested negative for both PR3- and MPO-anti-neutrophil cytoplasmic antibody (ANCA), which excluded any effects being mediated by these traditional anti-neutrophil antibodies. Only RA-IgG samples tested positive (>5 U/ml) for ACPA. Our SLE cohort displayed presented with a variety of manifestations: 59% presented with renal involvement; 29% had arthritis; 12% had pulmonary involvement and 24% presented with photosensitive rashes. All of our RA cohort presented with articular involvement. In addition, 13% were hypertensive, 13% presented with rheumatoid nodules and 25% had pulmonary involvement. All patient fulfilled relevant classification criteria with stable disease. Patients with active disease requiring medication change at the time of sampling were excluded.
Purified SLE- and RA-IgG both enhance hydrogen peroxide production
To examine whether there were intrinsic differences between neutrophils derived from our patient and control populations, we first examined hydrogen peroxide (H2O2) generation in ex vivo SLE-, RA- and HC-derived neutrophils. These experiments demonstrated that SLE-neutrophils had significantly slower rates of H2O2 production compared to both HC- and RA-neutrophils (Fig. 1A). We also found no significant difference between HC- and RA-neutrophils in our assay. To build on these experiments, we subsequently explored the effects of purified IgG upon neutrophils isolated from healthy volunteers, to determine whether the presence of autoantibodies drive the activated neutrophil phenotype reported among patient populations. Here, we found that pre-conditioning neutrophils with SLE- or RA-IgG induced significantly higher rates of H2O2 generation (Fig. 1B), indicating that the presence of autoreactive IgG may prime neutrophils to enhance H2O2 production following stimulation. We therefore found that whilst ex vivo SLE-neutrophils displayed slower rates of H2O2 generation, treatment with ARD-IgG increased H2O2 production rates in HC-neutrophils.

SLE- and RA-IgG both bind neutrophils and prime neutrophils. (A) Neutrophils were isolated from patients with SLE (n = 12) or RA (n = 7) and healthy volunteers (n = 12), and rates of H2O2 production in response to 50 nM PMA using Amplex UltraRed. Data are presented as each point representing a patient sample tested with the mean and SEM also being displayed and were analysed by Kruskal-Wallis test (p = 0.0039) with a Dunn’s multiple comparison test. (B) H2O2 generation in control neutrophils was assessed in the presence of ARD-IgG in the presence of 50 nM PMA. Data are displayed as the mean and SEM from three independent experiments, with each point representing the average of IgG sample over the experimental repeats. Data were analysed by Kruskal-Wallis test (p < 0.0001) with a Dunn’s multiple comparison test. (C) Representative flow cytometry plots examining binding for purified IgG to human neutrophils. Isolated neutrophils were incubated with pooled IgG, composed of 5 different IgG samples in the absence or presence of FcγR blockade, and stained for CD15 and human IgG. IgG binding was determined by quantifying the amount of CD15+IgG+ cell within the neutrophil gate (D) IgG binding experiments were performed using neutrophils from 9 different healthy donors, with mean and SEM being displayed. Statistical significance was tested by a Kruskal-Wallis (p = 0.0010) test with a Dunn’s multiple comparison test. (E) Additional experiments examined pooled IgG binding in the presence of FcγR blockade and the number of CD15+IgG+ cells quantified by flow cytometry. Data are presented as the mean and SEM of nine donors (with each point representing a different neutrophil donor), which were compared to the number of CD15+IgG+ cells previously determined. Data were tested by repeated measures two-way ANOVA a Bonferroni’s multiple comparison test. ns = no significance, *p < 0.05, **p < 0.01.
ARD-IgG binds neutrophils
To determine whether purified IgG bound neutrophils, we employed a flow cytometry approach. HC neutrophils were incubated with pooled IgG (5 different IgG samples per group), chosen at random from each group, and stained for CD15 and human IgG. Neutrophils were then evaluated by flow cytometry, being selected based on their forward and side scatter properties (Fig. 1C). Gated neutrophils were subsequently analysed based on CD15 and the presence of human IgG, both in the absence and presence of FcR blockade (Fig. 1C). Greater numbers of CD15+IgG+ cells were observed with both SLE- and RA-IgG compared to HC-IgG (Fig. 1D). Building on this dataset, we also examined IgG binding in the presence of a FcR blocking reagent (Miltenyi, UK), which only reduced binding of SLE-IgG, indicating a contribution from FcγR-mediated interactions with this IgG source (Fig. 1E). Therefore, all subsequent functional studies were performed with FcγR blockade to specifically examine antigen-specific mediated effects.
ARD-IgG displays differential effects upon neutrophil adhesion and trans-endothelial migration
To evaluate whether β1 or β2 integrin-mediated adhesion were modulated by purified IgG, we evaluated neutrophil adhesion to their respective ligands, fibronectin (via β1 integrins) and fibrinogen (αMβ2 integrin) in the presence of individual SLE- (n = 5), RA- (n = 7) or HC-IgG (n = 6) samples. In the absence of phorbol 12-myristate 13-acetate (PMA), the effects of SLE- and RA-IgG upon neutrophil adhesion to both fibronectin and fibrinogen were identical to those of HC-IgG (Fig. 2A,B). In contrast, following PMA stimulation, both SLE- and RA-IgG increased neutrophil adhesion to fibronectin (Fig. 2A) compared to HC-IgG. Interestingly, only SLE-IgG significantly increased neutrophil adhesion to immobilised fibrinogen (Fig. 2B). The addition of pan-β1 integrin (P5D2) or αMβ2-specific (2LPM19c) blocking antibodies reduced PMA-induced binding to values similar to unstimulated cells (Fig. 2A,B). Thus, PMA-stimulated neutrophil adhesion to fibronectin was enhanced by both SLE and RA-IgG, but to fibrinogen by SLE-IgG only.

SLE- IgG enhances both neutrophil β1 and β2 integrin activation. BCECF-AM labelled neutrophil adhesion to immobilised integrin ligands was assessed in the presence of purified IgG to evaluate integrin activation. (A) 20 μg/ml fibronectin was immobilised and neutrophil adhesion examined in the absence or presence of 20 nM PMA and the 10 μg/ml pan-β1 integrin blocking antibody P5D2. (B) 200 μg/ml fibrinogen was immobilised and neutrophil adhesion evaluated in the absence or presence of 20 nM PMA and the 10 μg/ml αMβ2-specifc blocking antibody 2LPM19c. In both cases, data are presented as the mean and SEM from three independent experiments and analysed by two-way ANOVA with a Dunnett’s multiple comparison test. *p < 0.05, **p < 0.01.
We then examined the modulatory effects of individual IgG samples upon unstimulated and PMA-induced neutrophil adhesion to human umbilical cord endothelial cells (HUVEC). Interestingly, only SLE-IgG significantly enhanced both unstimulated and PMA-induced neutrophil adhesion to both resting and TNF-α-activated HUVEC compared to control IgG (Fig. 3A,B). In contrast, we found that neither SLE- nor RA-IgG affected basal or chemokine-induced neutrophil trans-endothelial migration compared to HC-IgG (Fig. 3C). Taken together, our data suggest that whilst RA- and SLE-IgG both enhanced PMA stimulated β1 integrin-mediated adhesion to fibronectin, only SLE-IgG significantly affected αMβ2-mediated adhesion to fibrinogen as well as cultured HUVEC.

SLE- IgG increased neutrophil adhesion to endothelial cells, but not trans-endothelial migration. BCECF-AM labelled neutrophil adhesion was assessed in the absence or presence of purified IgG and 20 nM PMA to (A) resting endothelial cells and (B) endothelial cells that had been activated with TNF-α prior to experimentation. (C) Fluorescently labelled neutrophil trans-endothelial migration was determined in the absence or presence of purified IgG and 150 ng/ml IL-8, using a trans-well cell culture system and flow cytometry. Cells were quantified by comparing enumerated cells in the lower chamber to the total cells added. In both cases, data are displayed as the mean and SEM of three independent experiments and were tested for significance using two-way ANOVA with a Dunnett’s multiple comparison test. **p < 0.01, ****p < 0.0001.
ARD-IgG increases DNA release by neutrophils
We next examined whether IgG could also modulate the release of extracellular DNA, both in the absence (spontaneous) and presence of PMA (inducible). Using immunofluorescence staining for histone H3, we noted that PMA stimulation induced DNA externalisation, characteristic of NET release (Fig. 4A). Comparative analysis of neutrophil supernatants also showed a significant increase in cell-free DNA in PMA-stimulated cells (Fig. 4B). We therefore used measurements of cell-free DNA, to quantitatively measure DNA released by neutrophils, as an indirect measure of NETosis. We then normalised our results against a no IgG control, thus examining any modulatory effects of IgG upon NETosis.

SLE- IgG increased neutrophil NETosis in the presence of endothelial cells. (A) PMA-induced NETosis was confirmed by immunofluorescence. Neutrophils were incubated on coverslips in the absence or presence of 40 nM PMA and then fixed. Coverslips were then stained for histone H3 and mounted using a DAPI-mounting medium. (B) Cell supernatants from neutrophils cultured in the absence or presence of 40 nM were analysed using the Quanti-iT PicoGreen dsDNA kit. Data are presented as the mean and SEM of three independent experiments. Statistical significance was determined by a Mann-Whitney test. (C–E) IgG-mediated extracellular DNA release was assessed in (C) the absence of an endothelial monolayer; (D) the presence of resting endothelial cells; and (E) the presence of activated endothelial cells. Neutrophils were cultured for 4 hours in the absence or presence of 40 nM PMA, after which cell supernatants were assessed for cell-free dsDNA. For endothelial cell activation, HUVEC were pre-stimulated with 10 ng/ml TNF-α for 24 hours and washed with warmed PBS prior to addition of neutrophils. Data are presented as the mean and SEM of three independent experiments and analysed using a two-way ANOVA with a Dunnet’s multiple comparison test. *p < 0.05, **p < 0.01, ****p < 0.0001.
Concurrent with published studies, we also report an increase in spontaneous DNA release by neutrophils in the presence of SLE- or RA-IgG compared to HC-IgG (Fig. 4C). Only SLE-IgG however, significantly elevated PMA-induced DNA externalisation by neutrophils in the absence of endothelial cells (Fig. 4C).
We then studied neutrophil DNA release in co-culture with either steady-state or TNF-α-activated HUVEC to model the cellular interactions that would occur with the vascular endothelium. Interestingly, co-culture of neutrophils with steady-state HUVEC abrogated the increases in both spontaneous and PMA-induced DNA release that we observed with neutrophils stimulated in the absence of endothelial cells (Fig. 4D). However, when HUVEC were activated with TNF-α prior to co-culture, SLE-IgG, but not RA-IgG, significantly increased both spontaneous and PMA-induced DNA externalisation (Fig. 4E).
Discussion
Abnormalities in neutrophil function contribute to the immunopathology of several ARDs, including SLE and RA. Whilst neutrophils isolated from patients with SLE and RA have been shown to exhibit increased propensities to release NETs, it is not known whether pathogenic autoantibodies directly engage and drive neutrophil dysfunction. We report that whole SLE- and RA-IgG fractions have differential binding to neutrophils in vitro, and further demonstrate that ARD-IgG enhance ROS production and DNA release, suggestive of enhanced NETosis. Taken together, these data suggest that ARD-IgG may act as priming agents that enhance neutrophil activation following stimulus.
Many groups have sought to identify the surface molecules implicated in IgG-mediated leukocyte dysfunction. Due to the wealth of potential autoantigens associated with both SLE and RA, identification of a singular antigen target has been met with varying degrees of success and is likely to be completely different in both disease groups. We initially assessed the role of FcγRs, given that immune complexes can interact with neutrophil FcγRs71 and induce ROS generation72. Indeed, SLE-IgG binding was reduced following FcγR blockade, indicating a degree of FcγR-mediated binding. This reduction may indicate a contribution of FcγR-mediated effects to neutrophil activation in patients with SLE. In our further experiments however, we assessed the antigen-specific effects of purified IgG in the presence of FcγR blockade, we found that both SLE- and RA-IgG were able to enhance β1 integrin activation, measured by adhesion to immobilised fibronectin, but only SLE-IgG significantly enhanced αMβ2-mediated adhesion to PMA stimulated neutrophils. These differences in integrin activation may highlight a subtle form of differential regulation by different ARD-IgG upon steady-state and PMA stimulated neutrophils. In this model, it is possible that the presence of ARD-IgG is able to enhance integrin affinity and/or avidity further than that seen with PMA alone. This enhancement may allow for different integrin families to either adopt a high affinity conformation or undergo integrin clustering and therefore allow high avidity interactions with integrin ligands.
Our experiments utilised immobilised fibronectin and fibrinogen, which are found in areas of tissue injury with local activation of coagulation and in regions of vascular endothelial damage where the sub-endothelial basement membrane has been exposed. Our choice of ligands has the advantage of readily allowing us to distinguish the effects of IgG on β1-mediated adhesion to ECM/VCAM-1 from the effects on αMβ2-mediated adhesion. Given the wealth of literature on ligand binding motifs, it is reasonable to extrapolate our findings to other integrin ligands for αMβ2 and α4β1, such as ICAM-1 and VCAM-1 that play a key role in neutrophil-endothelial interactions. Future work will be aimed at dissecting these ligand preferences further. It is also possible that augmented integrin-mediated binding occurs within the circulation. Fibronectin can be found incorporated into circulating NETs that can promote further neutrophil interactions73,74, circulating in plasma, as well as forming a constituent of basement membranes and ECM75. In addition, the finding of increased circulating apoptotic endothelial cells in patients with SLE76, would provide further endothelial integrin ligands to engage circulating leukocytes within the blood flow.
We found a similar pattern of differential activation with ROS generation, a process requiring αMβ277, that was increased by RA- and SLE-IgG. The requirement of αMβ2, whose affinity is commonly reduced in SLE78, may underlie our observation of reduced ROS in ex vivo SLE-derived neutrophils. Differences in treatment regimens between groups may also account for the reduced rates of H2O2 generation. If we consider both the myelosuppressive and immunosuppressive effects of mycophenolate mofetil and prednisolone, as well as their half-lives (17.6 and 2.6 hours respectively)79,80, which all SLE patients were prescribed, it is possible that the activity of freshly isolated neutrophils may still be pharmacologically suppressed. Alternatively, reduced H2O2 production may be a result of metabolic exhaustion arising from systemic activation prior to cell isolation.
Integrin activation is central to leukocyte migration, therefore we examined whether ARD-IgG affected neutrophil adhesion to, and migration across, the endothelium. Interestingly, SLE-IgG, but not RA-IgG, significantly augmented neutrophil adhesion to HUVEC, while migration across an IL-8 gradient was unaffected by ARD-IgG. This observation may suggest that the primary effects of ARD-IgG centre on the firm adhesion stage of leukocyte extravasation, as opposed to enhancing both adhesion and trans-endothelial migration. Interestingly, we also noted differences in unstimulated adhesion, with no significant differences observed to immobilised fibronectin or fibrinogen but increased baseline adhesion to HUVEC in the presence of SLE-IgG. This difference may be explained by the presence of numerous additional adhesion molecules on the endothelial monolayer, which are absent in immobilised integrin ligand. For examples, endothelial cells would also express ICAM-1 and ICAM-2, which are ligands for both αLβ2 and αMβ2 on the neutrophil. These differences highlight the complex interactions occurring between integrins and adhesion molecules under physiological conditions.
Endothelial co-culture modulated the ARD-IgG effects on NETosis. In particular, the presence of a quiescent endothelium abrogated the effects of SLE- and RA-IgG on DNA externalisation observed in the absence of endothelial cells. Stimulation of the endothelium however, restored elevated SLE-IgG induced NETosis, implicating an endothelial contribution. These results may indicate that the presence of ARD-IgG may enhance neutrophil interactions with their external environment and promote neutrophil activation to regions of endothelial activation and dysfunction. A limitation to these experiments is the use of cell-free DNA as an indirect measurement of NETosis. Whilst our approach allowed us to quantitatively compare DNA externalisation to assess the effects upon NETosis, we lose the ability to specifically assess for markers widely accepted to be associated with NET structure, such as citrullinated histones and MPO. The development of a quantitative NET standard to allow the precise measurements of DNA-protein constructs accepted as NETs would resolve this issue.
We administered FcR blockade in our experiments to focus upon the antigen-specific nature of our observations. To further study this interaction additional experiments could be performed using F(ab′)2 fragments to ensure cellular effects were mediated solely through antigen-specific binding. Differences in IgG post-translational modifications of the Fc region may also contribute to the physiological effects of IgG. In particular, modifications such as glycosylation and sialylation are believed to mediate cellular effects of IgG81,82,83,84,85, and may potentially account for differences between ARD- and HC-IgG. Future experiments however, are required to explore the contribution of any differences in post-translational modifications between IgG groups and were beyond the scope of this study.
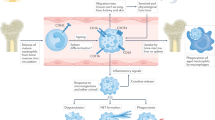
These different patterns of IgG-mediated integrin activation and neutrophil function may help elucidate the differential mechanisms driving vascular damage and dysfunction in these different ARDs. In conclusion, we found a differential pattern of autoantibody binding and integrin activation in steady-state and stimulated neutrophils exposed to purified whole IgG from patients with SLE or RA, which was modulated by the activation state of the endothelium (Fig. 5). Further work is now underway to explore the translational implications of our findings to highlight novel therapeutic targets.

Schematic of overall findings. SLE- and RA-IgG displayed differential effects upon HC-neutrophils in our experiments. SLE-IgG enhanced both β1 and β2 integrin-mediated adhesion and also adhesion to endothelial cells. In contrast, RA-IgG only increased β2 integrin-mediated adhesion. We observed higher levels of NETosis both in absence and presence of activated endothelial cells with the addition of SLE-IgG, whilst RA-IgG only enhanced NETosis in the absence of an endothelial monolayer.
Materials and Methods
Patients
Whole blood and serum were obtained by informed consent from 33 patients fulfilling classification criteria; 16 RA86 and 17 SLE87, as well as 21 HCs. Ethical approval was given by the UK National Research Ethics Committee and NHS Permission was granted for the study to be undertaken at University College London Hospitals NHS Foundation Trust (IRAS ID: 39531; REC Ref: 13/LO/0900; CSP number 39531) and all experimental procedures were performed in accordance with the Declaration of Helsinki and compliant with the principles of Good Clinical Practice.
IgG purification
Whole IgG was purified by passing patient or HC serum through protein G agarose spin columns (Thermo Scientific, UK) and eluted using 0.1 M glycine (Sigma, UK). Eluted IgG was concentrated, washed and dialysed into endotoxin-free PBS by sequential centrifugations for 20 minutes at 2050 g using centrifugal filter units (Merck Millipore, Ireland). Concentrated IgG was then passed through endotoxin removal columns (Thermo Scientific, UK) and confirmed to be endotoxin-free by EndoLISA (Hyglos, Germany) (<0.06 endotoxin units/ml).
Autoimmune rheumatic disease-associated IgG binding characterisation
Titres of ACPA in purified IgG samples were quantified using the EDIA anti-CCP-2 kit (Euro Diagnostica, Sweden). Titres of PR3-ANCA and MPO-ANCA in purified IgG were determined using the WIESLAB ANCA screen kit (Euro Diagnostica, Sweden). Quantification of anti-nuclear antibodies (ANA), anti-double stranded DNA antibodies (anti-dsDNA) and rheumatoid factor (RF) were all performed in the University College London Hospital laboratory.
Neutrophil isolation
Citrated whole blood was obtained by venepuncture and red blood cells removed by dextran sedimentation for 45 minutes. Neutrophils were isolated from the cell-enriched plasma by Percoll PLUS (Sigma, UK) gradient centrifugation at 700 g for 30 minutes and resuspended in phenol-free RPMI (Thermo Scientific, UK) supplemented with 10% FCS (Thermo Scientific, UK) and 2mM L-glutamine (Lonza, Switzerland). Isolated neutrophils were counted using a 0.4% Trypan Blue solution and diluted to 2 × 106 neutrophils/ml to ensure the same number of viable cells were used between groups and experiments. For experiments examining the effects of IgG, cells were treated with a FcR blocking reagent (Miltenyi, UK) (2 μl per 1 × 106 neutrophils) for 20 minutes prior to addition of 200 μg/ml purified IgG.
Endothelial cell culture
HUVEC at passage 2 (P2) (Lonza, Switzerland) were cultured in endothelial growth media 2 (Lonza, Switzerland) with 10% FCS (Thermo Scientific, UK) and 2mM L-glutamine (Lonza, Switzerland) to P5 for all experimental procedures. For endothelial activation, HUVEC were treated with 10 ng/ml TNF-α (R&D Systems, UK) for 24 hours and then washed with warmed HBSS prior to experimentation.
IgG binding
Neutrophils were cultured with 200 μg/ml IgG for 2 hours, fixed in 2% paraformaldehyde (PFA) (Sigma, UK) for 15 minutes, and stained using PE-conjugated anti-CD15 (clone W6D3) and FITC-conjugated anti-human IgG (clone G18-145) (BD Biosciences, UK) for 30 minutes at room temperature in the dark. Stained cells were washed twice in a sodium HEPES buffer (20 mM HEPES, 140 mM NaCl, 2 mg/ml glucose, 0.3% BSA) and analysed using a FACSVerse (BD Biosciences). Resulting data was analysed using FlowJo (TreeStar Inc., UK). IgG binding was calculated as the percentage of CD15+ neutrophils that were also positive for human IgG.
Neutrophil adhesion
Fibronectin (Sigma, UK) (for β1 integrin engagement) and fibrinogen (Sigma, UK) (for αMβ2 integrin engagement) were immobilized in 96-well black MaxiSorp microplates (Thermo Scientific, UK) and blocked with 2% fish-skin gelatin (Sigma, UK), to reduce non-specific neutrophil binding (Fig. S1). HUVEC were cultured in 96-well black tissue culture plates (Thermo Scientific, UK). HC-neutrophils were labelled with 2.5 μM 2′,7′-bis-(2-carboxyethyl)-5-(and-6)-carboxyfluoresceinacetoxymethyl ester (BCECF-AM) (Life Technologies, UK) for 30 minutes at 37 °C. Stained cells were washed twice in the sodium HEPES buffer by centrifugation at 300 g for 5 minutes and resuspended to 1 × 107 neutrophils/ml. 5 × 105 neutrophils were added to wells containing 20 nM PMA and 200 μg/ml IgG for 30 minutes at 37 °C. Fluorescence was measured using a Tecan GENios Spectra FLUOR plate reader (Tecan UK Ltd., UK). Plates were washed and read again. Adhesion was calculated by comparing the fluorescence of washed wells to initial fluorescence.
Neutrophil trans-endothelial migration
HUVEC were grown to confluence on transwell inserts (Millipore, UK). HC-neutrophils were isolated, washed with M199 media (Invitrogen, UK) labelled with a 0.5 µM CellTracker (Invitrogen, UK). Labelled cells were washed with M199 media by centrifugation at 300 g for 5 minutes and resuspended to 5.5 × 106 neutrophils/ml in M199 media supplemented with 1% FCS. Transwells were washed with M199 and 1.1 × 106 neutrophils added to the upper chamber. Transmigration was assessed both in the absence or presence of 150 ng/ml IL-8 in the lower chamber for 90 minutes. Migration was evaluated by flow cytometry, using CountBright absolute counting beads (Invitrogen, UK) to enumerate the number of cells in the lower chamber. Percent transmigration was calculated by comparing the number of cells in the lower chamber and the number of neutrophils added to the upper chamber.
Hydrogen peroxide generation
2 × 106 HC-neutrophils were incubated with 200 μg/ml IgG for 1 hour at 37 °C before addition of 0.5U/ml HRP (Sigma, UK) and 60 nM Amplex UltraRed (Invitrogen, UK). 4 × 105 cells were added to each test well of a 96-well black microplate (Thermo Scientific, UK) and fluorescence measured by FLUOstar Omega microplate reader (BMG Labetech, Germany). H2O2 generation in response to 50 nM PMA was recorded over time and rates (expressed in nM/sec) determined using Omega Mars Analysis software (BMG Labtech, Germany). In separate experiments, H2O2 generation was assessed in patient- and HC-derived neutrophils, however in the absence of autologous IgG.
Immunofluorescence staining for neutrophil extracellular traps
Glass coverslips (Fischer, UK) were sterilized with and coated with 200 μg/ml fibrinogen (Sigma, UK) at 4 °C. 5 × 105 neutrophils were then added to coverslips and stimulated for 4 hours, after which they were fixed for 15 minutes with 4% PFA at room temperature. Coverslips were blocked in a blocking solution (10% goat serum/1% BSA/2 mM EDTA/HBSS/0.1% Tween-2) overnight at 4 °C, then incubated with 1 µg/ml anti-histone H3 antibody (ab1791, Abcam, UK) and then 2 µg/ml Alexa Fluor 488-conjugated goat anti-rabbit IgG secondary antibody (Life Technologies, UK), which were diluted in blocking solution, for one hour each at room temperature. After incubations, coverslips were washed with HBSS twice, mounted and sealed on microscope slides with ProLong Gold antifade mountant with DAPI (Invitrogen, UK). Slides were subsequently visualized using a Zeiss Axio Imager.A1 inverted fluorescence microscope (Zeiss, Germany) and images analysed using Image J.
Extracellular DNA quantification
HC-neutrophils were pre-treated with IgG for 1 hour, after which NETosis was induced by stimulation with 40 nM PMA for 4 hours. Neutrophil supernatants were obtained by centrifugation at 300 g for 5 minutes and then assessed for extracellular DNA using the Quanti-iT PicoGreen dsDNA kit (Invitrogen, UK). Results were then normalised to a no IgG control, thus examining any modulatory effects of IgG.
Statistical analysis
Data were evaluated using GraphPad Prism. Data were first tested for normality using a Kolmogorov-Smirnov test. In experimental data sets only comparing two groups, a Mann-Whitney test was performed. Where multiple groups were being compared, a Kruskal-Wallis test with a Dunn’s multiple comparison test was used. In data sets with two variables, data were assessed by two-way ANOVA with a Dunnet’s multiple comparison test. A p value below 0.05 was considered significant.
References
Nemeth, T. & Mocsai, A. The role of neutrophils in autoimmune diseases. Immunology letters 143, 9–19, https://doi.org/10.1016/j.imlet.2012.01.013 (2012).
Jorch, S. K. & Kubes, P. An emerging role for neutrophil extracellular traps in noninfectious disease. Nature medicine 23, 279–287, https://doi.org/10.1038/nm.4294 (2017).
Brinkmann, V. et al. Neutrophil extracellular traps kill bacteria. Science 303, 1532–1535, https://doi.org/10.1126/science.1092385 (2004).
Gupta, A. K. et al. Activated endothelial cells induce neutrophil extracellular traps and are susceptible to NETosis-mediated cell death. FEBS letters 584, 3193–3197, https://doi.org/10.1016/j.febslet.2010.06.006 (2010).
Carmona-Rivera, C., Zhao, W., Yalavarthi, S. & Kaplan, M. J. Neutrophil extracellular traps induce endothelial dysfunction in systemic lupus erythematosus through the activation of matrix metalloproteinase-2. Annals of the rheumatic diseases 74, 1417–1424, https://doi.org/10.1136/annrheumdis-2013-204837 (2015).
Doring, Y. et al. Auto-antigenic protein-DNA complexes stimulate plasmacytoid dendritic cells to promote atherosclerosis. Circulation 125, 1673–1683, https://doi.org/10.1161/CIRCULATIONAHA.111.046755 (2012).
Borissoff, J. I. et al. Elevated levels of circulating DNA and chromatin are independently associated with severe coronary atherosclerosis and a prothrombotic state. Arteriosclerosis, thrombosis, and vascular biology 33, 2032–2040, https://doi.org/10.1161/ATVBAHA.113.301627 (2013).
Lande, R. et al. Neutrophils activate plasmacytoid dendritic cells by releasing self-DNA-peptide complexes in systemic lupus erythematosus. Science translational medicine 3, 73ra19, https://doi.org/10.1126/scitranslmed.3001180 (2011).
Sangaletti, S. et al. Neutrophil extracellular traps mediate transfer of cytoplasmic neutrophil antigens to myeloid dendritic cells toward ANCA induction and associated autoimmunity. Blood 120, 3007–3018, https://doi.org/10.1182/blood-2012-03-416156 (2012).
Nakazawa, D. et al. The responses of macrophages in interaction with neutrophils that undergo NETosis. Journal of autoimmunity 67, 19–28, https://doi.org/10.1016/j.jaut.2015.08.018 (2016).
Papadaki, G. et al. Neutrophil extracellular traps exacerbate Th1-mediated autoimmune responses in rheumatoid arthritis by promoting DC maturation. European journal of immunology 46, 2542–2554, https://doi.org/10.1002/eji.201646542 (2016).
Chrysanthopoulou, A. et al. Neutrophil extracellular traps promote differentiation and function of fibroblasts. The Journal of pathology 233, 294–307, https://doi.org/10.1002/path.4359 (2014).
Knight, J. S., Carmona-Rivera, C. & Kaplan, M. J. Proteins derived from neutrophil extracellular traps may serve as self-antigens and mediate organ damage in autoimmune diseases. Frontiers in immunology 3, 380, https://doi.org/10.3389/fimmu.2012.00380 (2012).
Sur Chowdhury, C. et al. Enhanced neutrophil extracellular trap generation in rheumatoid arthritis: analysis of underlying signal transduction pathways and potential diagnostic utility. Arthritis research & therapy 16, R122, https://doi.org/10.1186/ar4579 (2014).
Yalavarthi, S. et al. Release of neutrophil extracellular traps by neutrophils stimulated with antiphospholipid antibodies: a newly identified mechanism of thrombosis in the antiphospholipid syndrome. Arthritis Rheumatol 67, 2990–3003, https://doi.org/10.1002/art.39247 (2015).
Clements, J. M. et al. Identification of a key integrin-binding sequence in VCAM-1 homologous to the LDV active site in fibronectin. J Cell Sci 107(Pt 8), 2127–2135 (1994).
Rossaint, J. et al. Synchronized integrin engagement and chemokine activation is crucial in neutrophil extracellular trap-mediated sterile inflammation. Blood 123, 2573–2584, https://doi.org/10.1182/blood-2013-07-516484 (2014).
Raftery, M. J. et al. beta2 integrin mediates hantavirus-induced release of neutrophil extracellular traps. The Journal of experimental medicine 211, 1485–1497, https://doi.org/10.1084/jem.20131092 (2014).
Gillenius, E. & Urban, C. F. The adhesive protein invasin of Yersinia pseudotuberculosis induces neutrophil extracellular traps via beta1 integrins. Microbes and infection 17, 327–336, https://doi.org/10.1016/j.micinf.2014.12.014 (2015).
Neeli, I., Dwivedi, N., Khan, S. & Radic, M. Regulation of extracellular chromatin release from neutrophils. Journal of innate immunity 1, 194–201, https://doi.org/10.1159/000206974 (2009).
Lavigne, L. M. et al. Integrin engagement mediates the human polymorphonuclear leukocyte response to a fungal pathogen-associated molecular pattern. J Immunol 178, 7276–7282 (2007).
Smolen, J. S., Aletaha, D., Koeller, M., Weisman, M. H. & Emery, P. New therapies for treatment of rheumatoid arthritis. Lancet 370, 1861–1874, https://doi.org/10.1016/S0140-6736(07)60784-3 (2007).
Smolen, J. S. & Steiner, G. Therapeutic strategies for rheumatoid arthritis. Nature reviews. Drug discovery 2, 473–488, https://doi.org/10.1038/nrd1109 (2003).
Klinman, D. M., Shirai, A., Ishigatsubo, Y., Conover, J. & Steinberg, A. D. Quantitation of IgM- and IgG-secreting B cells in the peripheral blood of patients with systemic lupus erythematosus. Arthritis and rheumatism 34, 1404–1410 (1991).
Linker-Israeli, M. et al. Elevated levels of endogenous IL-6 in systemic lupus erythematosus. A putative role in pathogenesis. J Immunol 147, 117–123 (1991).
Liossis, S. N., Kovacs, B., Dennis, G., Kammer, G. M. & Tsokos, G. C. B cells from patients with systemic lupus erythematosus display abnormal antigen receptor-mediated early signal transduction events. The Journal of clinical investigation 98, 2549–2557, https://doi.org/10.1172/JCI119073 (1996).
Monneaux, F. & Muller, S. Epitope spreading in systemic lupus erythematosus: identification of triggering peptide sequences. Arthritis and rheumatism 46, 1430–1438, https://doi.org/10.1002/art.10263 (2002).
Chabaud, M., Fossiez, F., Taupin, J. L. & Miossec, P. Enhancing effect of IL-17 on IL-1-induced IL-6 and leukemia inhibitory factor production by rheumatoid arthritis synoviocytes and its regulation by Th2 cytokines. J Immunol 161, 409–414 (1998).
Miossec, P., Korn, T. & Kuchroo, V. K. Interleukin-17 and type 17 helper T cells. The New England journal of medicine 361, 888–898, https://doi.org/10.1056/NEJMra0707449 (2009).
Miao, J. et al. Frequencies of circulating IL-17-producing CD4+CD161+ T cells and CD4+CD161+ T cells correlate with disease activity in rheumatoid arthritis. Modern rheumatology/the Japan Rheumatism Association 24, 265–570, https://doi.org/10.3109/14397595.2013.854070 (2014).
Behrens, F. et al. Imbalance in distribution of functional autologous regulatory T cells in rheumatoid arthritis. Annals of the rheumatic diseases 66, 1151–1156, https://doi.org/10.1136/ard.2006.068320 (2007).
Cribbs, A. P. et al. Treg cell function in rheumatoid arthritis is compromised by ctla-4 promoter methylation resulting in a failure to activate the indoleamine 2,3-dioxygenase pathway. Arthritis Rheumatol 66, 2344–2354, https://doi.org/10.1002/art.38715 (2014).
Rapetti, L., Chavele, K. M., Evans, C. M. & Ehrenstein, M. R. B cell resistance to Fas-mediated apoptosis contributes to their ineffective control by regulatory T cells in rheumatoid arthritis. Annals of the rheumatic diseases 74, 294–302, https://doi.org/10.1136/annrheumdis-2013-204049 (2015).
Cedeno, S. et al. Defective activity of ERK-1 and ERK-2 mitogen-activated protein kinases in peripheral blood T lymphocytes from patients with systemic lupus erythematosus: potential role of altered coupling of Ras guanine nucleotide exchange factor hSos to adapter protein Grb2 in lupus T cells. Clin Immunol 106, 41–49 (2003).
Krishnan, S. et al. Differential expression and molecular associations of Syk in systemic lupus erythematosus T cells. J Immunol 181, 8145–8152 (2008).
Jury, E. C. et al. Abnormal CTLA-4 function in T cells from patients with systemic lupus erythematosus. European journal of immunology 40, 569–578, https://doi.org/10.1002/eji.200939781 (2010).
Tanaka, S. et al. Graded attenuation of TCR signaling elicits distinct autoimmune diseases by altering thymic T cell selection and regulatory T cell function. J Immunol 185, 2295–2305, https://doi.org/10.4049/jimmunol.1000848 (2010).
Moulton, V. R. & Tsokos, G. C. Abnormalities of T cell signaling in systemic lupus erythematosus. Arthritis research & therapy 13, 207, https://doi.org/10.1186/ar3251 (2011).
Takeuchi, T., Suzuki, K., Kondo, T., Yoshimoto, K. & Tsuzaka, K. CD3 zeta defects in systemic lupus erythematosus. Annals of the rheumatic diseases 71(Suppl 2), i78–81, https://doi.org/10.1136/annrheumdis-2011-200641 (2012).
Haringman, J. J. et al. Synovial tissue macrophages: a sensitive biomarker for response to treatment in patients with rheumatoid arthritis. Annals of the rheumatic diseases 64, 834–838, https://doi.org/10.1136/ard.2004.029751 (2005).
Funauchi, M., Ohno, M., Minoda, M. & Horiuchi, A. Abnormal expression of intercellular adhesion molecule-1 on peripheral blood mononuclear cells from patients with systemic lupus erythematosus. Journal of clinical & laboratory immunology 40, 115–124 (1993).
Nockher, W. A., Wigand, R., Schoeppe, W. & Scherberich, J. E. Elevated levels of soluble CD14 in serum of patients with systemic lupus erythematosus. Clinical and experimental immunology 96, 15–19 (1994).
Egerer, K. et al. Increased serum soluble CD14, ICAM-1 and E-selectin correlate with disease activity and prognosis in systemic lupus erythematosus. Lupus 9, 614–621 (2000).
Jin, O. et al. Lymphocyte apoptosis and macrophage function: correlation with disease activity in systemic lupus erythematosus. Clinical rheumatology 24, 107–110, https://doi.org/10.1007/s10067-004-0972-x (2005).
Llorente, L. et al. Spontaneous production of interleukin-10 by B lymphocytes and monocytes in systemic lupus erythematosus. European cytokine network 4, 421–427 (1993).
Llorente, L. et al. In vivo production of interleukin-10 by non-T cells in rheumatoid arthritis, Sjogren’s syndrome, and systemic lupus erythematosus. A potential mechanism of B lymphocyte hyperactivity and autoimmunity. Arthritis and rheumatism 37, 1647–1655 (1994).
Llorente, L. et al. Role of interleukin 10 in the B lymphocyte hyperactivity and autoantibody production of human systemic lupus erythematosus. The Journal of experimental medicine 181, 839–844 (1995).
Kanda, N., Tsuchida, T. & Tamaki, K. Estrogen enhancement of anti-double-stranded DNA antibody and immunoglobulin G production in peripheral blood mononuclear cells from patients with systemic lupus erythematosus. Arthritis and rheumatism 42, 328–337, https://doi.org/10.1002/1529-0131(199902)42:2328::AID-ANR163.0.CO;2-# (1999).
Dularay, B., Elson, C. J. & Dieppe, P. A. Enhanced oxidative response of polymorphonuclear leukocytes from synovial fluids of patients with rheumatoid arthritis. Autoimmunity 1, 159–169 (1988).
Dularay, B., Badesha, J. S., Dieppe, P. A. & Elson, C. J. Oxidative response of polymorphonuclear leucocytes to synovial fluids from patients with rheumatoid arthritis. Annals of the rheumatic diseases 49, 661–664 (1990).
den Broeder, A. A. et al. Neutrophil migration and production of reactive oxygen species during treatment with a fully human anti-tumor necrosis factor-alpha monoclonal antibody in patients with rheumatoid arthritis. The Journal of rheumatology 30, 232–237 (2003).
Barnhart, M. I., Riddle, J. M. & Bluhm, G. B. Immunocytology in arthritic joints. Annals of the rheumatic diseases 26, 281–296 (1967).
Hughes, J. R., Erhardt, C. C. & Clement, M. Neutrophilic dermatosis in association with rheumatoid arthritis. Clinical and experimental dermatology 20, 168–170 (1995).
Belcher, C., Doherty, M. & Crouch, S. P. Synovial fluid neutrophil function in RA: the effect of pregnancy associated proteins. Annals of the rheumatic diseases 61, 379–380 (2002).
Griffiths, R. J. et al. Leukotriene B4 plays a critical role in the progression of collagen-induced arthritis. Proceedings of the National Academy of Sciences of the United States of America 92, 517–521 (1995).
Wipke, B. T. & Allen, P. M. Essential role of neutrophils in the initiation and progression of a murine model of rheumatoid arthritis. J Immunol 167, 1601–1608 (2001).
Gal, I. et al. Visualization and in situ analysis of leukocyte trafficking into the ankle joint in a systemic murine model of rheumatoid arthritis. Arthritis and rheumatism 52, 3269–3278, https://doi.org/10.1002/art.21532 (2005).
Abramson, S. B., Given, W. P., Edelson, H. S. & Weissmann, G. Neutrophil aggregation induced by sera from patients with active systemic lupus erythematosus. Arthritis and rheumatism 26, 630–636 (1983).
Ronnefarth, V. M. et al. TLR2/TLR4-independent neutrophil activation and recruitment upon endocytosis of nucleosomes reveals a new pathway of innate immunity in systemic lupus erythematosus. J Immunol 177, 7740–7749 (2006).
Bakkaloglu, A. et al. Antineutrophil cytoplasmic antibodies in childhood systemic lupus erythematosus. Clinical rheumatology 17, 265–267 (1998).
Sthoeger, Z. M., Bezalel, S., Chapnik, N., Asher, I. & Froy, O. High alpha-defensin levels in patients with systemic lupus erythematosus. Immunology 127, 116–122, https://doi.org/10.1111/j.1365-2567.2008.02997.x (2009).
Vordenbaumen, S. et al. Elevated levels of human beta-defensin 2 and human neutrophil peptides in systemic lupus erythematosus. Lupus 19, 1648–1653, https://doi.org/10.1177/0961203310377089 (2010).
Ma, C. Y. et al. Elevated plasma level of HMGB1 is associated with disease activity and combined alterations with IFN-alpha and TNF-alpha in systemic lupus erythematosus. Rheumatology international 32, 395–402, https://doi.org/10.1007/s00296-010-1636-6 (2012).
Villanueva, E. et al. Netting neutrophils induce endothelial damage, infiltrate tissues, and expose immunostimulatory molecules in systemic lupus erythematosus. J Immunol 187, 538–552, https://doi.org/10.4049/jimmunol.1100450 (2011).
Bridges, S. L. Update on autoantibodies in rheumatoid arthritis. Curr Rheumatol Rep 6, 343–350 (2004).
Olsen, N. J. & Karp, D. R. Autoantibodies and SLE: the threshold for disease. Nat Rev Rheumatol 10, 181–186, https://doi.org/10.1038/nrrheum.2013.184 (2014).
Del Papa, N. et al. Anti-endothelial cell IgG fractions from systemic lupus erythematosus patients bind to human endothelial cells and induce a pro-adhesive and a pro-inflammatory phenotype in vitro. Lupus 8, 423–429 (1999).
Garcia-Romo, G. S. et al. Netting neutrophils are major inducers of type I IFN production in pediatric systemic lupus erythematosus. Science translational medicine 3, 73ra20, https://doi.org/10.1126/scitranslmed.3001201 (2011).
Khandpur, R. et al. NETs are a source of citrullinated autoantigens and stimulate inflammatory responses in rheumatoid arthritis. Science translational medicine 5, 178ra140, https://doi.org/10.1126/scitranslmed.3005580 (2013).
Knight, J. S. et al. Peptidylarginine deiminase inhibition is immunomodulatory and vasculoprotective in murine lupus. The Journal of clinical investigation 123, 2981–2993, https://doi.org/10.1172/JCI67390 (2013).
Fossati, G., Bucknall, R. C. & Edwards, S. W. Insoluble and soluble immune complexes activate neutrophils by distinct activation mechanisms: changes in functional responses induced by priming with cytokines. Annals of the rheumatic diseases 61, 13–19 (2002).
Robinson, J., Watson, F., Bucknall, R. C. & Edwards, S. W. Activation of neutrophil reactive-oxidant production by synovial fluid from patients with inflammatory joint disease. Soluble and insoluble immunoglobulin aggregates activate different pathways in primed and unprimed cells. The Biochemical journal 286(Pt 2), 345–351 (1992).
Monti, M. et al. Integrin-dependent cell adhesion to neutrophil extracellular traps through engagement of fibronectin in neutrophil-like cells. PLoS One 12, e0171362, https://doi.org/10.1371/journal.pone.0171362 (2017).
Monti, M. et al. Neutrophil Extracellular Traps as an Adhesion Substrate for Different Tumor Cells Expressing RGD-Binding Integrins. Int J Mol Sci 19, https://doi.org/10.3390/ijms19082350 (2018).
Stenman, S. & Vaheri, A. Distribution of a major connective tissue protein, fibronectin, in normal human tissues. J Exp Med 147, 1054–1064 (1978).
Rajagopalan, S. et al. Endothelial cell apoptosis in systemic lupus erythematosus: a common pathway for abnormal vascular function and thrombosis propensity. Blood 103, 3677–3683, https://doi.org/10.1182/blood-2003-09-3198 (2004).
Wu, S. Y. et al. Cell Intrinsic Galectin-3 Attenuates Neutrophil ROS-Dependent Killing of Candida by Modulating CR3 Downstream Syk Activation. Frontiers in immunology 8, 48, https://doi.org/10.3389/fimmu.2017.00048 (2017).
Fagerholm, S. C., MacPherson, M., James, M. J., Sevier-Guy, C. & Lau, C. S. The CD11b-integrin (ITGAM) and systemic lupus erythematosus. Lupus 22, 657–663, https://doi.org/10.1177/0961203313491851 (2013).
Bullingham, R. E., Nicholls, A. J. & Kamm, B. R. Clinical pharmacokinetics of mycophenolate mofetil. Clin Pharmacokinet 34, 429–455, https://doi.org/10.2165/00003088-199834060-00002 (1998).
Sagcal-Gironella, A. C. et al. Pharmacokinetics of prednisolone at steady state in young patients with systemic lupus erythematosus on prednisone therapy: an open-label, single-dose study. Clin Ther 33, 1524–1536, https://doi.org/10.1016/j.clinthera.2011.09.015 (2011).
Nandakumar, K. S. et al. Endoglycosidase treatment abrogates IgG arthritogenicity: importance of IgG glycosylation in arthritis. European journal of immunology 37, 2973–2982, https://doi.org/10.1002/eji.200737581 (2007).
Nimmerjahn, F., Anthony, R. M. & Ravetch, J. V. Agalactosylated IgG antibodies depend on cellular Fc receptors for in vivo activity. Proc Natl Acad Sci USA 104, 8433–8437, https://doi.org/10.1073/pnas.0702936104 (2007).
Lux, A., Yu, X., Scanlan, C. N. & Nimmerjahn, F. Impact of immune complex size and glycosylation on IgG binding to human FcgammaRs. J Immunol 190, 4315–4323, https://doi.org/10.4049/jimmunol.1200501 (2013).
Harre, U. et al. Glycosylation of immunoglobulin G determines osteoclast differentiation and bone loss. Nat Commun 6, 6651, https://doi.org/10.1038/ncomms7651 (2015).
Ohmi, Y. et al. Sialylation converts arthritogenic IgG into inhibitors of collagen-induced arthritis. Nat Commun 7, 11205, https://doi.org/10.1038/ncomms11205 (2016).
Aletaha, D. et al. 2010 Rheumatoid arthritis classification criteria: an American College of Rheumatology/European League Against Rheumatism collaborative initiative. Arthritis and rheumatism 62, 2569–2581, https://doi.org/10.1002/art.27584 (2010).
Tan, E. M. et al. The1982 revised criteria for the classification of systemic lupus erythematosus. Arthritis and rheumatism 25, 1271–1277 (1982).
Acknowledgements
This work was performed as part of a UCL Impact Studentship to AAK with further support from the Rosetrees Trust (ref: M136) and Breathing Matters and undertaken at UCLH/UCL who received a proportion of funding from the Department of Health’s NIHR Biomedical Research Centres funding scheme. JCP is funded by a MRC New Investigator Research Grant. We would like to acknowledge Dr Andrew Smith and Professor Tony Segal for their help with the neutrophil hydrogen peroxide generation assay and are grateful to Professors Nancy Hogg (The Francis Crick Institute) and Ian Dransfield (University of Edinburgh) for helpful discussions.
Author information
Authors and Affiliations
Contributions
A.A.K. designed and performed the research, analysed the data and contributed to writing the manuscript; C.P. contributed to the study design and critically revised the manuscript; V.M.R. contributed to the study design and critically revised the manuscript; J.C.P. and I.P.G. conceived the study, designed the experiments, were involved in data analysis, patient recruitment and characterization, and wrote the manuscript. All authors read, revised and approved the content of the final version of the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Khawaja, A.A., Pericleous, C., Ripoll, V.M. et al. Autoimmune rheumatic disease IgG has differential effects upon neutrophil integrin activation that is modulated by the endothelium. Sci Rep 9, 1283 (2019). https://doi.org/10.1038/s41598-018-37852-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-37852-5
This article is cited by
-
Targeting integrin pathways: mechanisms and advances in therapy
Signal Transduction and Targeted Therapy (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
