
Abstract
Metabolism changes extensively during the normal proliferation and differentiation of mammalian cells, and in cancer and inflammatory diseases. Since changes in the metabolic network reflect interactions between genetic, epigenetic and environmental changes, it is helpful to study the flow of label from isotopically labelled precursors into other metabolites rather than static metabolite levels. For this Nuclear Magnetic Resonance (NMR) spectroscopy is an attractive technique as it can quantify site-specific label incorporation. However, for applications using human cells and cell lines, the challenge is to optimize the process to maximize sensitivity and reproducibility. Here we present a new framework to analyze metabolism in mammalian cell lines and primary cells, covering the workflow from the preparation of cells to the acquisition and analysis of NMR spectra. We have applied this new approach in hematological and liver cancer cell lines and confirm the feasibility of tracer-based metabolism in primary liver cells.
Similar content being viewed by others

Introduction
The metabolism of a cell changes as a net downstream response to the cellular environment. This is triggered by external stimuli influencing the cell cycle and the balance between differentiation, proliferation and apoptosis, which all contribute to changes in metabolic networks. Furthermore, genetic and epigenetic changes also modify metabolism and the metabolic response to extrinsic factors. Thus, in order to better understand the regulation of metabolism, one needs to integrate analysis of regulatory protein levels with a quantitative analysis of metabolite levels.
Since the seminal findings by Otto Warburg of increased glycolysis leading to lactic acid production in cancer cells, it has become increasingly common to use metabolomic approaches to quantify levels of key mediators within cells1. However, quantification of metabolite concentration provides only a static picture of metabolic processes that are in constant flux. In order to elucidate metabolic mechanisms in cells one needs to employ metabolic flux analysis, or at least tracer-based metabolism. The advantage of tracer-based analyses is that changes in metabolites can be assigned to particular mechanisms. For tracer-based analyses, different isotopically labelled precursors have been used as starting points to determine the intermediates and products of metabolism in cells. Typically-used isotopes include 13C and 15N, both are amenable to NMR examination. A tracer-based metabolic analysis can assign a product to one particular or multiple pathways, or even describe the contribution of different pathways which is often not possible based on static metabolite concentrations. A typical example is the consumption and/or production of glutamic acid in cancer cells, which can involve production from glycolysis and the Krebs cycle or from glutamine2. Tracer-based methods can also distinguish between different entry mechanisms into the Krebs cycle, to determine contributions of pyruvate dehydrogenase activity vs the anaplerotic pathways using pyruvate carboxylase or glutaminolysis. In the past 13C-labelled precursors such as glucose, glutamine, glutamic acid, pyruvate, acetate, aspartate, glycerol, serine and fatty acids3 have been used for this purpose.
The two predominantly used technologies in this context have been mass spectrometry (MS) and NMR spectroscopy. MS has a significant advantage over NMR in sensitivity. However, its information content is limited to mass increments, which can often only be interpreted in the context of predefined models4,5. With the mechanistic details we now see unraveled in current biology, it is becoming increasingly clear that it is desirable to add an analytical level that can also detect site-specific label incorporation in small molecules.
First applications of NMR for tracer-based metabolism reach back into the 1970s6,7,8, with significant progress in the 1990s when Szyperski and Wüthrich9,10,11 introduced 1H-13C-HSQC spectra for such analyses. Chikayama has used HSQC spectra and other spectra to assign metabolites in plants and silkworm larvae12. Several seminal publications have established roadmaps for tracer-based metabolism using mass spectrometry, in particular in the context of metabolic flux analysis13,14.
Here we present a workflow for efficiently using NMR in the context of tracer-based metabolism using mammalian cell lines or even primary cells under physiologically relevant conditions. This includes methods of preparing cells, along with NMR methods suitable for such analyses. We also discuss possible precursors that can be used to decipher different pathways. Furthermore, we show applications in cancer cell lines and in primary liver cells.
Results
The tracer-based metabolism framework
Figure 1 shows the workflow that we propose for NMR tracer-based metabolism. The different stages of this process will be discussed below, with an emphasis on steps taken to increase reproducibility between samples, to maximize sensitivity per unit time in the NMR experiments, and to obtain validated results.

Workflow for NMR tracer-based metabolism. Cell cultures are grown in a medium containing 13C or 15N- labelled precursors. Typically, 10–20 million cells are required. Adherent cells are removed from the flask surface using a scraper, thereby avoiding the use of trypsin which generates unwanted metabolites. Cells in suspension (e.g. hematological cancer cells) are harvested by centrifugation (See Materials and Methods for detailed protocols). Metabolites are obtained using a methanol/chloroform/water system where the polar extract in the upper phase is easily collected. Samples are vacuum dried and re-suspended in NMR buffer before being transferred to an NMR tube. Different NMR spectra are acquired, typically including a 1D-1H-NOESY and a 2D-1H,13C-HSQC, often also 2D-1H,1H-TOCSY spectra. For a complete isotopomer analysis, a combination of NMR and GC-MS may be used, which can be analyzed using the MetaboLab software24. From this analysis site specific label incorporations can be derived19. The spectrometer in this Figure is taken from WikiCommons, CC BY-SA 3.0 (https://creativecommons.org/licenses/by-sa/3.0/) https://commons.m.wikimedia.org/wiki/File:Nuclear_magnetic_resonance_(NMR).png.
NMR Sample Preparation
Reproducible metabolite extraction from cell lines or primary cells is not trivial as metabolism is not static and metabolite concentrations change quickly, even during the extraction process. High reproducibility is of particular importance for comparing labeled and unlabeled samples. Cells must be grown within a controlled environment, with identical cell numbers, and typically a final cell number of 10–20 million cells must be obtained in order to acquire 1H,13C-HSQC and other NMR spectra in a reasonable time frame. Highly reproducible growth conditions are required and the amount of media must be minimized as labeled precursors are expensive. The overall procedure is summarized in the flowchart depicted in Fig. 2 with further details provided in Materials and Methods. To achieve optimal reproducibility, it is essential that cold solvents are used for the extraction to stop all metabolic processes15. We chose to lyse the cells using cold methanol which has the advantage of immediately quenching metabolism, followed by chloroform extraction to obtain and separate the organic and polar phase. Alternatively, others have used liquid nitrogen to quench metabolism or to lyse cells by sonication. The latter has the drawback of long incubation times, requiring enzyme inhibitors to stop protein degradation, but allows the fractionation of cell compartments16,17. Media for tracer-based metabolism can either be prepared from scratch or media with one or several metabolites missing can be prepared or purchased. For any quantitative analysis it is important to replace the entire pool of one or several metabolites with its isotope labelled equivalent.

Flowchart summarizing the preparation of cells for tracer-based metabolism NMR analyses for adherent cell lines (left) and for cells growing in suspension (right).
When this protocol is followed rigorously we obtain 1D-NMR spectra with excellent reproducibility, as shown in Fig. 3a for a series of hematological cancer cell lines. The variability between intensities within replicates should be between 1 and 3% as relevant changes in metabolism can be small. Data of this quality enables excellent separation of different cell-lines by principal component analysis (PCA) (Fig. 3b). Without this level of reproducibility, it is impossible to compare labelled and unlabeled spectra in a quantitative analysis, and comparing metabolism between different samples also becomes questionable.

Increasing the power of NMR metabolomics analysis by increasing sample reproducibility, and by enhancing sensitivity with modern probe technology. (a) Spectra of four hematological cell lines (black: U266 and green: UM3, both myeloma cell lines, blue: Sudhl6 diffuse large B-cell lymphoma, red: Glor Burkitt’s lymphoma cell line) with 6 replicates per cell line to illustrate the required level of reproducibility. (b) Principal Component Analysis plot of spectra shown in (a) with 95% confidence intervals showing a clear separation between classes of cells and excellent clustering within the individual cell lines. (c,d) Comparison of 1D spectra obtained from two equal mass samples, either sucrose (c) or hepatocarcinoma cell line (HuH7) extracts (d) using a 1.7 mm micro-cryoprobe (blue), a 5 mm cryoprobe (black) and a 5 mm room temperature (RT) probe (red). These panels illustrate a factor 5–7 sensitivity gain from the RT to the cryoprobe, and another factor of 5–7 for the micro-cryoprobe. Overall the gain for equal sample masses from the RT to the micro-cryoprobe can be up to 40 fold.
Improvements in NMR technology
Tracer-based NMR analysis is considerably facilitated by huge signal-to-noise improvements arising from the introduction of micro-cryoprobe technology by the Bruker company. This is illustrated by comparing spectra from equal mass samples acquired with a 5.0 mm room temperature (RT) probe, a 5.0 mm cryoprobe and a 1.7 mm micro-cryoprobe (Fig. 3c,d). For this assessment, sucrose samples were prepared to a final concentration of 1 mM when using the micro-cryoprobe, i.e. 12 µg of sucrose in 35 µL for the micro-cryoprobe and in 600 µL for the other probes. We observed essentially no signal after 32 scans in the RT probe and a S/N ratio gain of at least 10 from using the 1.7 mm cryoprobe instead of the 5 mm cryoprobe. The same trend is illustrated for cell extracts from hepatocarcinoma cell line (HuH7, Fig. 3d). Two samples were prepared from the same flask. To ensure that equal cell masses were used, we vacuum-dried equal volumes from the polar layer of the methanol/chloroform/water extraction and dissolved these samples in different volumes as explained previously. Here the RT probe shows very low-intensity signals for 128 scans, whereas cryoprobes show a huge improvement with a further signal enhancement factor of 4–5 for the micro-cryoprobe.
Overall sensitivity improves by a factor of at least 40 with this probe technology for both samples. Importantly, the salt tolerance of this type of probe is much better than that of conventional cryoprobes but shimming becomes more challenging due to the large length/width ratio and the temperature gradient along the sample length of the NMR tube.
Tailoring HSQC experiments for tracer-based studies
It is primarily this advancement in technology that enables the use of 2D-1H,13C-HSQC spectra from cell extracts within a reasonable time period of 4 to 5 hours. HSQC spectra represent one of several choices for this type of analysis. By correlating proton (1H) and 13C (or 15N) chemical shifts, spectral overlap is considerably reduced compared with homonuclear two-dimensional (2D) experiments, such as 1H-1H-TOCSY. HSQC spectra offer at least two options for the measurement of label incorporation: (i) Measuring intensities from metabolite spin systems, which rise with the amount of label incorporation. This approach requires an unlabeled reference spectrum, and to derive absolute concentrations, it is also necessary to calibrate for different metabolites owing to variable coupling constants (see18 and Materials and Methods). (ii) Analysis of the complex multiplet patterns in the indirect 13C-dimension arising from scalar couplings (1JCC couplings) between adjacent 13C-atoms. This enables the determination of label incorporations in adjacent atoms as shown recently19.
Using signal intensities in HSQC spectra as analytical tools to quantify site-specific label incorporation ratios in metabolites requires appropriate scaling. This is challenging as algorithms typically used for 1D spectra fail because intensity depends on label incorporation levels, as well as concentration (The intensity of HSQC signals is proportional to IHSQC ∼ Γ · n · x, where Γ is the pulse sequence specific transfer function, n the number of spins, and x the label incorporation). In our workflow we have used the total spectral area (TSA) or Probabilistic Quotient Normalization (PQN)20 in the corresponding 1D spectrum of the same sample to scale the HSQC (see Materials and Methods for further details). These methods fail in the presence of systematic changes affecting many metabolites. In such cases it is sometimes possible to use intensities of selected metabolites, e.g, the sum of all branched chain amino acids, as a reference. For well-resolved resonances the accuracy of label incorporations may be cross-checked using 13C-edited and filtered spectra.
To resolve JCC coupling constants high-resolution HSQC spectra are needed, typically acquired with 16384 increments but only 2 scans. To acquire such spectra, we use non-uniform sampling (NUS) with a 25% sampling rate. Such spectra can be acquired in approximately 4.5 h. The multiplet patterns obtained this way are virtually indistinguishable from those with regular sampling (Supplementary Fig. 2). Spectra acquired using NUS can be processed using different approaches21,22 and the application for small molecules has recently been demonstrated23.
It is important to realize that signals in HSQC spectra arise solely from protons attached to 13C. In contrast to 1D-1H and 1H, 1H-TOCSY spectra, a 1H-12C pair does not lead to any signal in an HSQC spectrum. In mass spectrometry terms, it is only the m ≥ +1 molecules that can be observed. In other words, the interpretation of HSQC spectra is complicated by the fact that the amount of 12C for a particular atom in a metabolite cannot be measured directly. In principle, one can estimate the amount of unlabeled sample from intensities in the natural abundance spectrum; the total amount of carbon (13C + 12C) must then be 100 times the observed 13C. For a labeled sample grown under identical conditions, the amount of the metabolite will be same, although different isotopomers may be present. The level of label incorporation can be inferred by comparing signal intensities in the labelled and natural abundance reference spectra.
Although signal intensities arising from the 1.1% natural abundance 13C in most metabolites can be quite low, which limits quantification, comparing relative intensities between related labelled samples can help overcome this problem. In this paper we demonstrate how intensities from both unlabeled and labeled HSQC metabolites can be used to determine the 13C-incorporation in specific positions.
The value of using HSQC spectra for isotopomer analysis arises from the quantitative information in the multiplet structure in the incremented 13C-dimension, as originally suggested by Szyperski10. In a mixture of isotopomers for the central carbon of a 3-carbon fragment, there is a superimposition of resonances arising from isolated 13C singlets, two possible 13C-13C doublets, and the possibility of doublets of doublets when all three carbons are labelled (Supplementary Fig. 4). The MetaboLab software24 has a module that simulates line shapes and deconvolutes contributions from different isotopomers in slices taken from HSQC spectra19.
For such HSQC spectra, the choice of HSQC pulse sequence is important. The echo-antiecho-selected HSQC25 is commonly used as it provides excellent solvent and artefact suppression, but leads to complex line shapes in the presence of 2JCC couplings (Fig. 4a,b). These arise from scalar coupling evolution during the time when the selective encoding gradient is applied. While such signals can be simulated using a quantum mechanical approach19, pure in-phase absorption type signals can be obtained when using HSQC sequences without echo-antiecho encoding (Fig. 4c,d). Such spectra are best acquired in D2O due to reduced solvent suppression efficiency. Simulation of lineshapes in the incremented dimension can be used for isotopomer analysis19. Examples of such simulations are shown in Fig. 4a–d for lactic and for glutamic acid in samples derived from HuH7 cells (blue experimental, red simulated spectra). This approach is particularly robust as it does not depend on signal intensities. In-phase spectra acquired in D2O offer a fast and reliable assessment of metabolic mechanisms without any need of simulations.

Sections from 1H-13C HSQC spectra of polar cell extracts illustrating spectral improvements obtained using acquisition mode of the indirect directions States rather than Echo-AntiEcho in HSQCs with NUS. (a–d) Show slices and spectral regions for L-lactic acid resonances from 1H,13C-HSQC spectra for HuH7 cells grown for 12 hours in a medium containing [U-13C] glucose. Echo-antiecho-1H,13C-HSQC spectra acquired for samples in a mixed 90% H2O/10% D2O NMR buffer (panels a and b) and conventional 1H,13C-HSQC spectra acquired for samples in a D2O buffer (panels c and d). The high resolution in the 13C dimension obtained with 25% NUS sampling over 8k complex points is illustrated in panels e and f where the different couplings obtained from the PC-derived 13C2–13C3 and PDH-derived 13C2-13C1 moieties are clearly resolved in the L-aspartic acid (e) and L-malic acid (f) H2C2 resonances. (g) shows the alanine H2/C2 resonance at 3.78/53.2ppm from GLOR cells labelled with [U-13C]glucose and [15N]glutamine. The complexity of the 15N-undecoupled (blue) spectrum arises from the superimposition of resonances from mainly [13C-]alanine with a small proportion of [15N,13C]alanine. While JC2C3 and JC1C2 produce a well-resolved doublet of doublets. For [15N,13C]alanine, these 4 resonances are further split by the small JC2N2 coupling. The resulting spectrum is further complicated by a 14N/15N isotope shift that causes overlap of one of the double components of [13C,15N]alanine with the resonance from [13C]alanine. (f): AXP (ADP and AMP) H1/C1 resonances at 6.15/89.7ppm from [15N,13C]glucose and [15N]glutamine labelled GLOR cells showing a complex pattern based on a doublet due to the large 1JC1C2 coupling in the undecoupled (blue) spectrum. Additional resonances are seen because a proportion of the C1 is directly attached to a 15N in the adenine ring. This complexity is reduced by 15N decoupling in the red spectrum but the remaining peaks appear broadened by the 14N/15N isotope shift45 on the directly-attached 13C.
Additional information can also be inferred from NMR proton observed NMR spectra which show 13C and 12C- linked signals, such as 1D-1H or 2D-1H-TOCSY. This approach has been successfully employed by Fan and coworkers26. The limitation here lies in the overlap of signals in metabolite mixtures derived from mammalian cells. From TOCSY spectra it is usually possible to obtain label incorporations for several metabolites as shown for lactic acid and threonine. The same is possible for some other metabolites including alanine, glutamic acid, aspartate, aspartate derived UDP-pyrimidine resonances, although signal overlap is a problem for less abundant metabolites (Supporting Fig. 3). Another option is the use of 13C-filtered 1D-1H-spectra27 and possibly 13C-filtered TOCSY spectra.
Insights from tracer-based analyses
Several metabolic pathways from central carbon metabolism can be explored using the aforementioned methods, as summarized in Fig. 5. These include glycolysis, the oxidative and non-oxidative branch pentose phosphate pathway (PPP), Krebs cycle metabolism, the relative importance of pyruvate dehydrogenase and pyruvate carboxylase28 in feeding pyruvate-derived carbons into the Krebs cycle, glutaminolysis in the Krebs cycle2 nucleotide synthesis, serine/glycine, creatine synthesis and one carbon metabolism (Supplementary Table 1). This list is likely to lengthen with the use of additional labeling schemes.

Label incorporation schemes for NMR.
It has previously been shown that [1,2-13C]glucose and [3-13C]glutamine represent excellent choices for such analyses29,30 In many cases it is useful to have multiple carbons labeled in the same molecule as the preservation of adjacent labels bears considerable information. For example in [1,2-13C]glucose labelled samples, the size of the coupling constant in aspartate and malate can be used to determine the balance between pyruvate carboxylase (PC) and pyruvate dehydrogenase (PDH) activity28, as the two entry mechanisms for the TCA cycle produce different labelling patterns. In the case of PC C2&3 of aspartate and malate become labelled with a smaller coupling constant compared to coupling to the COO groups in the case of PDH activity (C1&2 or C3&4). An example is depicted in Fig. 4e,f which is similar to what we have previously observed, although now with considerably improved spectral quality. In haematological cancer cells we often observe that aspartate is primarily formed from PC, and in one case (in K562 cells) we were able to follow the path of label incorporation via carbamoyl-aspartate, dihydroorotate and orotate into pyrimidines2.
Lactate label incorporation can be used to identify PPP activity with [1,2-13C]glucose as a precursor because the oxidative PPP removes C1 from glucose and this alters label incorporation in lactic acid and in riboses28. We also found that uridine diphosphate (UDP) can be used to determine relative amounts of oxidative vs non-oxidative PPP branch activities2. Pyruvate is well-known as an agent for fast metabolic fluxes using Dynamic Nuclear Polarization (DNP) MRI. In this process the 13C1 is polarized as it has the longest T1 relaxation time. For HSQC analysis the C2,3 are more informative. Interestingly we also observed serine and glycine formation in U266 multiple myeloma cell lines. Serine is a valuable precursor as it gets catabolized into glycine and formate, and both become building blocks for pyrimidines.
Dual labeling
Another option with NMR is to label multiple precursors in a way that metabolite signals will show couplings arising from label incorporation in adjacent positions with isotopes derived from two different labelled precursors. For example we observe alanine with JC2C3 and JC2N coupling arising from labelling with [U-13C]glucose and [15N]glutamine (Fig. 4g). This double labelling strategy is particularly useful to look at nucleotide synthesis (Supplementary Fig. 4). For example, the C1 of the ribose of adenosine nucleotides shows coupling to the directly attached 15N of the adenine ring from labelling with [U-13C]glucose and [15N]glutamine (Fig. 4h). For double labelling, we have explored several combinations: [U-13C]glucose and [15N]glutamine, and [1,2-13C]glucose and [3-13C]glutamine. These experiments facilitate the assessment of the contributions of glycolysis and glutaminolysis in feeding the Krebs cycle. Likewise, one-carbon metabolism can be probed by [13C,15N]serine and [15N]glutamine. A recent report shows the utility of combining [1,2-13C]acetate and [1,6-13C]glucose31.
Proof of concept studies using human cells
Carbon-13 enrichment was explored using various labeled precursors for different human cell lines, including the adherent hepatocellular epithelial cell line HuH7, and two hematological cancer cell lines, U266, a multiple myeloma cell line, and GLOR, a Burkitt’s lymphoma cell line. The choice of GLOR and U266 cell lines permitted comparison of a more glycolytic cell line and a cell line with stronger Krebs cycle activity. For GLOR and U266, we compare glucose and glutamine labeling, however HuH7 cells do not seem to consume glutamine over the period of this experiment. Moreover, we looked at serine and pyruvate as precursors for hematological cell lines.
Figure 6a,b compare label incorporations using [U-13C]glucose and [3-13C]glutamine, respectively, as precursors. GLOR show a typical Warburg effect with greater lactate and alanine production and glutaminolysis to supply the Krebs cycle, driven by c-Myc overexpression. Only the HuH7 hepatocytes produced labelled aspartate and showed labelling in aspartate-derived pyrimidine nucleotides from glucose.

Label incorporations in extracts from different cell types. The labelled precursors used were [U-13C]glucose (a), [3-13C]glutamine (b), [3-13C]pyruvate (c) and [2-13C]acetate (d) and [15N,13C]serine (e). In a–c & e, bars denote label incorporations in the U266, GLOR and HuH7 cell-lines respectively (additional information in Supplementary Fig. 5).
Diverse insights into different metabolic pathways are provided by using different labelled precursors as exemplified below:
-
Using [3-13C]pyruvate as a labelled precursor unexpectedly produced [3-13C]serine in both GLOR and U266 cell-lines, although in different amounts (Fig. 6c) providing unique information about gluconeogenesis arising from the activity of pyruvate carboxylase and PEP carboxykinase which convert pyruvate to phosphenolpyruvate, which is then converted to 3-phosphoglycerate and subsequently to serine. PEP carboxykinase is a gluconeogenesis enzyme that was not expected to be active in either of these hematological cancer cell lines.
-
Acetate is one of the most rapidly metabolized precursors with detectable label incorporation into glutamate in Kasumi cells after only 5 minutes (Fig. 6d) and may therefore even be suitable for DNP-MRI.
-
Using [U-13C,15N]serine as precursor in the hematological cell-lines, AXP was labelled at the C8 of the adenine ring (Fig. 6e), which is derived from formyl-tetrahydrofolate, illustrating the use of serine in supplying one-carbon metabolism. Purines and pyrimidines represent excellent reporter molecules in nucleobases and when ligated to riboses in nucleotides.
-
Using [U-13C,15N]serine as precursor also led to labelling of creatine (Fig. 7), whose production is altered in many cancers.
Figure 7 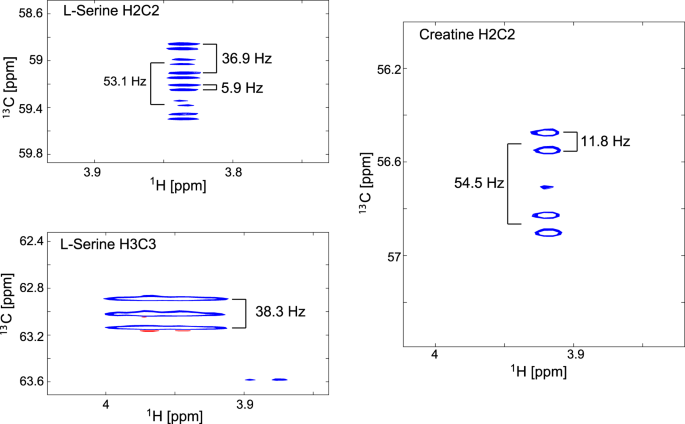
Coupling patterns at the C2/H2 resonances of serine and creatine in [U-13C,15N]serine labelled U266 cells from 1H-13C-HSQCs. The serine C2 resonance is predominantly a doublet of doublets of doublets because of JC1C2 coupling (53.1 Hz), JC2C3 coupling (36.9 Hz) and JC2N2 coupling (5.9 Hz). The C2 resonance of creatine is a doublet of doublets as the glycine-derived moiety gives rise to a JC1C2 coupling (54.5 Hz) and JC2N2 coupling (11.8 Hz).

Tracer-based experiments on primary patient tissue
Tracer-based metabolism in primary liver samples has great potential for diagnostic purposes in translational medicine, particularly in the context of understanding glucose and lipid homeostasis in fatty liver disease. Primary human hepatocytes were prepared from liver tissue, as previously described32. The harvested primary hepatocytes were metabolically active allowing metabolic studies to measure label incorporations. This is demonstrated in Fig. 8 for a flux time of 4.5 hours with [U-13C]glucose. Here we observe label incorporation in alanine, lactic acid, glutamic acid and glutathione. This is in partial agreement with Winnike et al.33 although their analysis was limited to 1D-1H-NMR spectra.

Feasibility of tracer-based analysis from donor liver samples. Overlaid sections from 1H-13C -HSQC of polar extracts from hepatocytes isolated from a donor liver sample. Black and blue resonances arise from samples exposed for 4.5h to media containing unlabeled glucose and [U-13C]glucose, respectively. Panels show the spectral regions for L-alanine C3 (a), L-lactic acid C3 (b), L-glycine C2 (c), L-glutamic acid C3 (d), phosphorylcholine C1 (e) and L-glutathione C3 (f). Phosphorylcholine was not enriched and is included as a scaling reference but the other metabolites were enriched by [U-13C]Glucose.
Discussion
Although NMR has been employed for tracer-based metabolic studies since the 1970s, it is only recent developments in NMR technology that provide the sensitivity required to analyze extracts from mammalian cells. Tracer-based analyses offers considerable opportunities in the context of metabolism in various diseases including cancer. Different NMR approaches include 13C-observed spectra, 1D-1H spectra where signals arising from 13C-coupled protons can be detected, and 2D-1H,1H-TOCSY spectra. Observing 13C directly is very insensitive, even when using 13C-observe probes, although the recent development of a much more sensitive 13C-observe micro-cryoprobe, which uses high-temperature superconducting receiver coils, may provide additional advantages by observing 13C-couplings in the directly observed dimension34. The use of proton observed 1D spectra is prone to large errors arising from signal overlap and differences between samples. Indeed errors reported in a recent publication were as large as 20–30%35. This is further complicated by the complex line shapes arising from proton-proton and longer-range proton-carbon couplings.
TOCSY spectra have helped to overcome some of these problems (Supplementary Fig. 3) and have been used widely for metabolic analyses26,36. One big disadvantage of TOCSY spectra is the long acquisition times, in particular for increased resolution, which are needed to resolve overlapping metabolite signals. This can potentially be overcome with fast TOCSY methods37.
Here we propose to base analysis mainly on HSQC spectra and describe a framework to work with reasonable numbers of mammalian cells. For such analyses it is imperative to use the most sensitive probes, at this stage the best option is the micro-cryoprobe available from Bruker. Here we have used a commercially available micro-cryoprobe at 14.1 Tesla (600 MHz proton frequency). We expect further improvements from such probe developments using high-temperature superconducting receiver coils34 and by using such probes at much higher field strengths, especially as 28.2 Tesla magnets are on the horizon38.
Tracer-based metabolism has started to make an impact in the analysis of metabolic pathways, some of which can only be probed using isotope-labelled tracers. This approach is relevant for future drug discovery in the field of metabolism, and also for elucidating the mechanistic differences between healthy and diseased states in cell lines and, as demonstrated here, potentially also in patient-derived tissue. There have already been reports showing the effectiveness of tracer-based metabolism in vivo39. In particular, Mashimo et al. showed the simultaneous oxidation of [1,2-13C]acetate and [1,6-13C]glucose in diverse brain tumours31. This illustrated the power of using multiple labelled precursors which give distinct labelling patterns in target metabolites. Likewise, [1,2-13C]glucose and [3-13C]glutamine have the potential to determine the balance between glycolysis and glutaminolysis in supplying the TCA cycle. There is great potential for in vivo approaches in metabolically active organs such as the liver and kidney. Such tracer-based studies can be closely linked to DNP enhanced chemical shift MR imaging which can observe the same type of reactions in vivo40,41, although only within a shorter time frame.
In summary, we present a framework for tracer-based analysis of mammalian cells using an NMR approach that provides robust data for site-specific label incorporations. This type of data can be used to elucidate metabolic mechanisms. With serial sampling, NMR should well complement the well-established MS for metabolic flux analyses. This approach shows that NMR is now suitable for working with limited numbers of mammalian cells.
Materials and Methods
Cell cultures
Detailed extraction protocol for adherent HuH7 cells
Cell culture: The hepatocellular carcinoma cell line, HuH7, was available in house. Cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 1% penicillin-streptomycin and 10% (v/v) heat-inactivated fetal bovine serum (FBS), then grown in a humidified atmosphere of 5% CO2 at 37 °C in 150 cm2 Corning flasks. The cells were routinely subcultured every 3 days (when they reached 80% confluency) by using trypsin to detach the cells from the flask. Briefly, the spent media was discarded, the flask was washed with 10 ml phosphate-buffered saline (PBS) and the PBS was discarded. Then, 5 ml of trypsin was added to the flask and incubated for 5 minutes. After microscopic assessment to confirm the cells were detached, 5 ml of media was added to the flask to inactivate the trypsin. In triplicate, 2 ml of this mixture was then inoculated into 18 ml of media and the flasks were incubated again at 37 °C.
Isotope Labelling: For tracer-based metabolic analysis the cells were transferred to a modified DMEM media lacking the metabolic precursor that was intended to be labelled, and completed with labelled precursors. The cells were then cultured with tracer for a further 6 hours prior to intracellular metabolite extraction. This was sufficient time to give an average cell yield of 15 × 106 cells/ml, the required cell density to provide a strong signal for NMR analysis.
Intracellular Metabolite extraction: PBS and purified water were kept at −4 °C, and the methanol and chloroform at −20 °C prior to use. For the quenching process, the spent medium was aspirated and each flask was washed twice with cold phosphate-buffered saline (PBS). Then, intracellular aqueous metabolites and lipids were extracted using a dual phase extraction procedure adapted from Teng et al.42. Briefly, 1200 µ of cold methanol (HPLC grade) was added to stop metabolic activity, and the cells were scraped off the flask. Afterwards, the suspension was transferred into a labelled vial and vortexed for 30 s. Chloroform (400 µl + 400 µL) and purified water (600 µl) were then added to each sample, each addition being followed by 30 s vortexing, and the samples were left to rest on ice for 10 min. After centrifuging at 4000 RPM for 10 min, the upper aqueous phase (~500 µ) and the lower organic phase (~200 µl) were then carefully transferred into new vials. The upper (polar) phase was dried under vacuum while the lower organic phase was stored at −80 °C until further analysis.
Detailed extraction protocol for haematological cancer cells grown in suspension
Cell culture: Cells were cultured in Roswell Park Memorial Institute (RPMI) 1640 media supplemented with 1% penicillin-streptomycin and 10% (v/v) heat-inactivated fetal bovine serum at 37 °C. Cells were maintained in an exponential proliferation at density 0.5 × 106 cells/ml.
Isotope Labelling: After cell counting, 15 × 106 cells were transferred to a universal tube and centrifuged at 1500 rpm for 5 min at room temperature. The supernatant was discarded and the cells were resuspended in 15 ml media prepared with labelled components. After 6 hours incubation, cells were counted and 10 × 106 cells were centrifuged at 1500 rpm for 5 min at room temperature. 5 ml of the supernatant (media) was taken for media analysis and the remaining media was discarded.
Intracellular metabolite extraction: Methanol was pre-chilled on dry ice and MilliQ water and chloroform were pre-chilled on wet ice. Cells were washed with 1 ml PBS: The cells were transferred to glass vials in 1 ml PBS and then centrifuged at 14000 rpm for 10 seconds. The use of glass vials is essential as chloroform will extract small molecules from most plastic materials. The PBS was removed and 400 μl methanol pre-chilled on dry ice was added. The cell pellet was resuspended thoroughly by vortexing for 10 secs and transferred to a glass vial. After adding 325 μl H2O and 400 μl chloroform, the sample was vortexed for 40 secs. The mixture was placed on ice for 10 mins to allow phase separation and then the vials were centrifuged at 4000 rpm for 10 mins at 4 °C in a swingout rotor. After allowing the vials to stand at room temperature for 5 mins, 400μl of the upper (polar) layer and 200μl of the lower (non-polar) layer were transferred to glass vials. Care was taken throughout to avoid carryover of proteins. The upper (polar) phase was dried under vacuum while the lower organic phase was stored at −80 °C until further analysis.
Preparation of primary hepatocytes: Hepatocytes were isolated from marginal human donor livers unsuitable for transplantation surgery under an ethically approved study (Local Research Ethics Committee: reference number 06/Q702/61), with informed patient consent. An encapsulated wedge of tissue was flushed with PBS to remove blood and and initial dissociation buffer was used to loosen cell junctions (10 mM HEPES, 0.5 mM EGTA). After another wash with PBS, tissue was dissociated using a combined enzymatic digestion buffer (0.5% w/v Collagenase A, 0.25% w/v Protease, 0.125% w/v Hyaluronidase and 0.05% w/v Deoxyribonuclease units/mg) as previously described32. Following this the liver was mechanically disaggregated in media (DMEM + 10%FCS). The cell suspension was sieved and centrifuged at 50 g to pellet hepatocytes. These were counted using trypan blue and plated out onto collagen-coated plastic.
NMR spectra
NMR sample preparation
Polar extracts were resuspended in 50 µl metabolomics buffer (100 mM sodium phosphate buffer, 0.5 mM TMSP, 10% or 100% D2O, pH 7) with vortexing. For samples in 10% D2O, 40 µl of supernatant was transferred to champagne vials. 35 µl supernatant was then transferred to 1.7 mm NMR tubes using a Bruker Biospin liquid handler. For samples in 100% D2O buffer, 35 µl supernatant was transferred manually to 1.7 mm NMR tubes. Samples were kept at 4 °C prior to NMR.
NMR data acquisition
All spectra were acquired at 300 K on a Bruker 600 MHz spectrometer with a TCI 1.7 mm z-PFG cryogenic probe using a cooled Bruker SampleJet autosampler. In all experiments, the 1H carrier was on the water frequency and the 1H 90° pulse was calibrated at a power of 0.256 W.
For the 1H 1D spectra, the standard Bruker pulse sequence noesygppr1d for a 1D NOESY with water pre-saturation was used. Key parameters were as follows: spectral width 11.97ppm/7183.9 Hz; complex points, TD 32768; interscan delay, d1 4 s; short NOE mixing time, d8 10 ms; number of scans, ns = 128; dummy scans, ds = 8. Total experiment time was 14 minutes.
For 1H,13C-HSQC spectra in water, the pulse sequence used was based on the Bruker standard pulse program hsqcetgpsp, which uses echo/anti-echo TPPI gradient selection. For the 1H,13C-HSQC spectra in D2O, the pulse sequence used was based on the Bruker standard pulse program, hsqcgphprsp. In both cases additional gradient pulses were added to improve water suppression in H2O and to help suppress T1 noise in D2O and soft 180°-pulses were used for 13C. For the 1H observe dimension, a spectral width of 7812.5 Hz/13.02ppm with 1024 complex points was used. For the 13C indirect dimension, 2048 complex points were acquired with a spectral width of 24154.6/160.1ppm. Spectra were acquired with 2 scans and an interscan delay of 1.5 s giving a total experiment time of ca. 4 h. All spectra were processed using NMRLab in MATLAB (The Mathworks Inc). Cosine-squared window functions were applied to both dimensions. For the NUS spectra, the 25% NUS list used was calculated for 8192 complex points using Wagner’s schedule generator43 with a tolerance of 0.01 and other parameters with default values.
NMR data processing
1D 1H NMR spectra were processed using the NMRlab and Metabolab programs within MATLAB. The free induction decay (FID) signals were multiplied by a 0.3 Hz exponential window function and zero-filled to 32 K points prior to Fourier transformation. Spectra were phased, referenced to TMSP δ 0.00 ppm, baseline corrected and the water region and regions devoid of signal at the edges of the spectrum were excluded. Finally, the spectra were scaled to a constant total spectral area (TSA-scaling). Resonances were assigned using Chenomx (Alberta, Canada, 2015), and by consulting the NMR metabolic profiling human metabolome database (HMDB).
2D HSQC spectra were processed using NMRLab in MATLAB. Cosine-squared window functions were applied to both dimensions and spectra were phased manually. Calibration was carried out manually using L-lactic acid as a reference peak (δ 1.31/22.9 ppm). Peak identification used MetaboLab24 with reference to HMDB. For 2D NUS-HSQC, data processing was initially performed using NMRPipe44 with the Hyberts extension for processing NUS spectra43 and subsequent analysis was then performed with NMRLab as described above for regular HSQC spectra.
Intensity scaling of HSQC spectra
To directly derive concentrations from HSQC spectra one needs to consider that JCHx coupling constants vary between different compounds. For echo-anti-echo HSQC sequences using a refocussed INEPT (hsqcetgpsp) a complex transfer function is obtained which also depends on the multiplicity x of the CHx group. This becomes considerably simpler for an HSQC that does not use a refocussed INEPT. Nevertheless, the intensity depends on the transfer function. If one wants to derive absolute concentrations from HSQC spectra one has to calibrate for each spin system unless the exact JCHx coupling constant is known.
Here we chose not to derive exact concentrations, as this would also require the use of longer recycling times, and relative intensities between labelled and unlabelled, or between different labelled compounds will not be affected by either the transfer function, or T1 relaxation effects. Moreover, the use of JCC couplings is also independent of these factors.
Relative intensities were processed as follows. Theoretically, the percentage label incorporation can be calculated from: Ilab/Ictl where Ilab is the intensity of the resonance from the labeled sample, and Ictl is the intensity of the resonance from the reference unlabeled sample.
It is often impossible to prepare differently labelled but otherwise identical samples because the overall amount of sample depends on cell number. Using a chemical shift referencing standard (usually TMS, TMSP or DSS) is completely unsuitable for intensity referencing because it is added after the sample has been prepared. Therefore, we propose another method to scale HSQC spectra. A common approach to scale one-dimensional spectra is to calculate the total spectral area (TSA) which reflects the total metabolite concentration. The same approach is not possible for HSQC spectra as intensities depend on concentration and label incorporation. However, we can use TSA scaling factors from the one-dimensional spectra to scale the associated HSQCs. Therefore, the expression for the percentage label incorporation becomes (Ilab * Sctl)/(Ictl * Slab), where Sctl and Slab are the scaling factors in the corresponding 1D NOESY spectra for the control and labelled samples. This approach has the advantage that the correction factor uses all the metabolite intensities rather than just one reference metabolite. This method was used to compare intensities between HSQC spectra to obtain label incorporations.
References
Markley, J. L. et al. The future of NMR-based metabolomics. Curr. Opin. Biotechnol. 43, 34–40 (2017).
Carrigan, J. B. et al. Tracer-Based Metabolic NMR-Based Flux Analysis in a Leukaemia Cell Line. ChemPlusChem 81, 453–459 (2016).
Lane, A. N. & Fan, T. W.-M. NMR-based Stable Isotope Resolved Metabolomics in systems biochemistry. Arch. Biochem. Biophys. 628, 123–131 (2017).
Wiechert, W. 13C Metabolic Flux Analysis. Metab. Eng. 3, 195–206 (2001).
Dai, Z. & Locasale, J. W. Understanding metabolism with flux analysis: From theory to application. Metab. Eng. 43, 94–102 (2017).
Eakin, R. T., Morgan, L. O., Gregg, C. T. & Matwiyoff, N. A. Carbon-13 nuclear magnetic resonance spectroscopy of living cells and their metabolism of a specifically labeled 13C substrate. FEBS Lett. 28, 259–264 (1972).
Kainosho, M., Ajisaka, K. & Nakazawa, H. In situ analysis of the microbial fermentation process by natural abundance 13C and 31P NMR spectroscopy. Production of adenosine-5′-triphosphate from adenosine. FEBS Lett. 80, 385–389 (1977).
Ugurbil, K., Brown, T. R., Den Hollander, J. A., Glynn, P. & Shulman, R. G. High-resolution 13C nuclear magnetic resonance studies of glucose metabolism in Escherichia coli. Proc. Natl. Acad. Sci. 75, 3742–3746 (1978).
Szyperski, T. Biosynthetically Directed Fractional 13C-labeling of Proteinogenic Amino Acids. Eur. J. Biochem. 232, 433–448 (1995).
Szyperski, T., Bailey, J. E. & Wüthrich, K. Detecting and dissecting metabolic fluxes using biosynthetic fractional 13C labeling and two-dimensional NMR spectroscopy. Trends Biotechnol. 14, 453–459 (1996).
Szyperski, T., Glaser, R. W., Hochuli, M. & Fiaux, J. Bioreaction Network Topology and Metabolic Flux Ratio Analysis by Biosynthetic Fractional 13C Labeling and Two-Dimensional NMR Spectroscopy. Metab. Eng. 1, 189–197 (1999).
Chikayama, E. et al. Systematic NMR Analysis of Stable Isotope Labeled Metabolite Mixtures in Plant and Animal Systems: Coarse Grained Views of Metabolic Pathways. PLoS ONE 3, e3805 (2008).
Cascante, M. & Marin, S. Metabolomics and fluxomics approaches. Essays Biochem. 45, 67–82 (2008).
Buescher, J. M. et al. A roadmap for interpreting 13 C metabolite labeling patterns from cells. Curr. Opin. Biotechnol. 34, 189–201 (2015).
Beckonert, O. et al. Metabolic profiling, metabolomic and metabonomic procedures for NMR spectroscopy of urine, plasma, serum and tissue extracts. Nat. Protoc. 2, 2692–2703 (2007).
Bertini, I., Hu, X. & Luchinat, C. Global metabolomics characterization of bacteria: pre-analytical treatments and profiling. Metabolomics 10, 241–249 (2014).
Bernacchioni, C. et al. NMR metabolomics highlights sphingosine kinase-1 as a new molecular switch in the orchestration of aberrant metabolic phenotype in cancer cells. Mol. Oncol. 11, 517–533 (2017).
Wan, Q., Wang, Y. & Tang, H. Quantitative 13C Traces of Glucose Fate in Hepatitis B Virus-Infected Hepatocytes. Anal. Chem. 89, 3293–3299 (2017).
Chong, M. et al. Combined Analysis of NMR and MS Spectra (CANMS). Angew. Chem. 129, 4204–4208 (2017).
Dieterle, F., Ross, A., Schlotterbeck, G. & Senn, H. Probabilistic Quotient Normalization as Robust Method to Account for Dilution of Complex Biological Mixtures. Application in 1H NMR Metabonomics. Anal. Chem. 78, 4281–4290 (2006).
Hyberts, S. G., Arthanari, H., Robson, S. A. & Wagner, G. Perspectives in magnetic resonance: NMR in the post-FFT era. J. Magn. Reson. 241, 60–73 (2014).
Kazimierczuk, K. & Orekhov, V. Non-uniform sampling: post-Fourier era of NMR data collection and processing: Non-uniform sampling. Magn. Reson. Chem. 53, 921–926 (2015).
Lee, S. et al. Carbon Isotopomer Analysis with Non-Unifom Sampling HSQC NMR for Cell Extract and Live Cell Metabolomics Studies. Anal. Chem. 89, 1078–1085 (2017).
Ludwig, C. & Günther, U. L. MetaboLab-advanced NMR data processing and analysis for metabolomics. BMC Bioinformatics 12, 366 (2011).
Schleucher, J. et al. A general enhancement scheme in heteronuclear multidimensional NMR employing pulsed field gradients. J. Biomol. NMR 4, 301–306 (1994).
Fan, T. W.-M. & Lane, A. N. Structure-based profiling of metabolites and isotopomers by NMR. Prog. Nucl. Magn. Reson. Spectrosc. 52, 69–117 (2008).
Smith, T. B. et al. High-Speed Tracer Analysis of Metabolism (HS-TrAM). Wellcome Open Res. 3, 5 (2018).
Reed, M. A. C., Ludwig, C., Bunce, C. M., Khanim, F. L. & Günther, U. L. Malonate as a ROS product is associated with pyruvate carboxylase activity in acute myeloid leukaemia cells. Cancer Metab. 4 (2016).
Metallo, C. M., Walther, J. L. & Stephanopoulos, G. Evaluation of 13C isotopic tracers for metabolic flux analysis in mammalian cells. J. Biotechnol. 144, 167–174 (2009).
Walther, J. L., Metallo, C. M., Zhang, J. & Stephanopoulos, G. Optimization of 13C isotopic tracers for metabolic flux analysis in mammalian cells. Metab. Eng. 14, 162–171 (2012).
Mashimo, T. et al. Acetate Is a Bioenergetic Substrate for Human Glioblastoma and Brain Metastases. Cell 159, 1603–1614 (2014).
Bhogal, R. H. et al. Isolation of Primary Human Hepatocytes from Normal and Diseased Liver Tissue: A One Hundred Liver Experience. PLoS ONE 6, e18222 (2011).
Winnike, J. H. et al. Stable isotope resolved metabolomics of primary human hepatocytes reveals a stressed phenotype. Metabolomics 8, 34–49 (2012).
Ramaswamy, V. et al. Development of a 13C-optimized 1.5-mm high temperature superconducting NMR probe. J. Magn. Reson. 235, 58–65 (2013).
Vinaixa, M. et al. Positional Enrichment by Proton Analysis (PEPA): A One-Dimensional 1H-NMR Approach for 13C Stable Isotope Tracer Studies in Metabolomics. Angew. Chem. Int. Ed. 56, 3531–3535 (2017).
Bingol, K., Bruschweiler-Li, L., Li, D.-W. & Brüschweiler, R. Customized Metabolomics Database for the Analysis of NMR 1H–1H-TOCSY and 13C–1H-HSQC-TOCSY Spectra of Complex Mixtures. Anal. Chem. 86, 5494–5501 (2014).
Hansen, A. L., Li, D., Wang, C. & Brüschweiler, R. Absolute Minimal Sampling of Homonuclear 2D NMR TOCSY Spectra for High-Throughput Applications of Complex Mixtures. Angew. Chem. Int. Ed. 56, 8149–8152 (2017).
Schwalbe, H. Editorial: New 1.2 GHz NMR Spectrometers- New Horizons? Angew. Chem. Int. Ed. 56, 10252–10253 (2017).
Fan, T. W. et al. Altered regulation of metabolic pathways in human lung cancer discerned by 13C stable isotope-resolved metabolomics (SIRM). Mol. Cancer 8, 41 (2009).
Brindle, K. M. Imaging Metabolism with Hyperpolarized 13C-Labeled Cell Substrates. J. Am. Chem. Soc. 137, 6418–6427 (2015).
Sriram, R., Kurhanewicz, J. & Vigneron, D. B. Hyperpolarized Carbon-13 MRI and MRS Studies. In eMagRes (John Wiley & Sons, Ltd, 2007), https://doi.org/10.1002/9780470034590.emrstm1253.
Teng, Q., Huang, W., Collette, T. W., Ekman, D. R. & Tan, C. A direct cell quenching method for cell-culture based metabolomics. Metabolomics 5, 199 (2008).
Hyberts, S. G., Milbradt, A. G., Wagner, A. B., Arthanari, H. & Wagner, G. Application of iterative soft thresholding for fast reconstruction of NMR data non-uniformly sampled with multidimensional Poisson Gap scheduling. J. Biomol. NMR 52, 315–327 (2012).
Delaglio, F. et al. NMRPipe: A multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6, 277–293 (1995).
Lapidot, A. & Gopher, A. Quantitation of metabolic compartmentation in hyperammonemic brain by natural abundance 13C-NMR detection of 13C-15N coupling patterns and isotopic shifts. FEBS J. 243, 597–604 (1997).
Acknowledgements
Much of the work presented is based on the FP7 Metaflux project (FP7-PEOPLE-ITN-2010–264780). We thank the EC for supporting ZE and RS within the EUROPOL-ITN project (H2020-MSCA-ITN-2014-642773), PhenoMeNal for supporting software development (H2020-EINFRA-2014-2-654241), and BBSRC for supporting JR in the context of a BBSRC CASE studentship. We are also grateful to the Wellcome Trust for supporting access to NMR instruments at the Henry Wellcome Building for Biomolecular NMR in Birmingham (grant number 208400/Z/17/Z). We are also grateful for access to NMR instrumentation at the BMRZ NMR centre in Frankfurt with support from INext, and thank Christian Richter for technical assistance. This report includes independent research funded by the NIHR Birmingham Biomedical Research Centre at the University Hospitals Birmingham NHS Foundation Trust and the University of Birmingham. The views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR or the Department of Health.
Author information
Authors and Affiliations
Contributions
R.S., Z.E., J.R. carried out the experiments; U.G. & M.A.C.R. designed the experiments. P.F.L. and F.K. contributed towards the design of lab-based experiments. U.L.G., M.A.C.R., R.S., Z.E. wrote the manuscript, U.G., M.A.C.R., P.F.L., Z.E., R.S. contributed to proof reading.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Saborano, R., Eraslan, Z., Roberts, J. et al. A framework for tracer-based metabolism in mammalian cells by NMR. Sci Rep 9, 2520 (2019). https://doi.org/10.1038/s41598-018-37525-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-37525-3
This article is cited by
-
Low-field NMR with multilayer Halbach magnet and NMR selective excitation
Scientific Reports (2023)
-
Isolated murine skeletal muscles utilize pyruvate over glucose for oxidation
Metabolomics (2022)
-
Discriminating aspects of global metabolism of neonatal cardiomyocytes from wild type and KO-CSRP3 rats using proton magnetic resonance spectroscopy of culture media samples
In Vitro Cellular & Developmental Biology - Animal (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.

.png){kind=link}