
Abstract
Understanding the gut microbiota in metabolic disorders, including type 2 diabetes mellitus (T2DM), is now gaining importance due to its potential role in disease risk and progression. We previously established a zebrafish model of T2DM, which shows glucose intolerance with insulin resistance and responds to anti-diabetic drugs. In this study, we analysed the gut microbiota of T2DM zebrafish by deep sequencing the 16S rRNA V3-V4 hypervariable regions, and imputed a functional profile using predictive metagenomic tools. While control and T2DM zebrafish were fed with the same kind of feed, the gut microbiota in T2DM group was less diverse than that of the control. Predictive metagenomics profiling using PICRUSt revealed functional alternation of the KEGG pathways in T2DM zebrafish. Several amino acid metabolism pathways (arginine, proline, and phenylalanine) were downregulated in the T2DM group, similar to what has been previously reported in humans. In summary, we profiled the gut microbiome in T2DM zebrafish, which revealed functional similarities in gut bacterial environments between these zebrafish and T2DM affected humans. T2DM zebrafish can become an alternative model organism to study host-bacterial interactions in human obesity and related diseases.
Similar content being viewed by others


Introduction
Worldwide prevalence of obesity continues to increase at an alarming rate, and more than 415 million people were estimated to be suffering from type 2 diabetes mellitus (T2DM) in 20151. T2DM is a metabolic disorder, which is primarily caused by obesity-induced insulin resistance, and more than 90% of people with T2DM are overweight or obese. In addition, obesity and T2DM are associated with specific changes in the gut microbiota composition. For example, many studies have found that T2DM patients have a reduced abundance of butyrate-producing species, leading to a low-grade inflammation in the gut2. This has been reported in people of different races, ethnicities and after controlling for the effect of anti-diabetic drugs on the gut microbiome3.
Over these recent decades, Zebrafish has emerged as a pre-eminent vertebrate model organism for biomedical research. The number of microbiota related studies in zebrafish are still limited but growing, in developmental and physiological microbiology4,5,6,7,8, colitis models9,10,11, effects of antibiotics12 and immune responses13,14,15,16,17. Among the studies on zebrafish microbiota in obesity, Semova I, et al. were the first reported that diet-induced alteration of gut microbiota composition influenced fat absorption18. After that, Felcinelli S, et al. demonstrated that Lactobacillus rhamnosus, a probiotic bacterium beneficial to humans, could modulate gut microbiota composition in zebrafish19 and attenuate obese phenotypes20. However, there are no reports that have evaluated the possibility of using zebrafish gut microbiota as a model for obesity and related metabolic disorders.
We previously created a diet-induced T2DM model of zebrafish (T2DM zebrafish), which shows glucose intolerance and insulin resistance with common obese phenotypes (body weight increase, visceral adiposity, hepatic steatosis and dyslipidaemia)21. This zebrafish model also shares common pathological transcriptome pathways associated with human T2DM, and shows improvement with anti-diabetic medications (metformin and glibenclamide). We believe that this model could be suitable for therapeutic target identification and chemical screening. To further evaluate the possibility of utilizing T2DM zebrafish as a model for human gut microbiota in T2DM, we conducted deep sequencing of 16S rRNA and compared T2DM-induced microbiota alternations between zebrafish and humans. Furthermore, we predicted the metabolic pathways associated with the microbiota of T2DM zebrafish using Phylogenetic Investigation of Communities by Reconstruction of Unobserved States (PICRUSt)22, based on the 16S rRNA sequence datasets.
Methods
T2DM zebrafish
All zebrafish procedures were approved by the Ethics Committee of Mie University and were performed according to the Japanese animal welfare regulation ‘Act on Welfare and Management of Animals’ (Ministry of the Environment, Japan) and complying with international guidelines. Zebrafish (AB strain; the Zebrafish International Resource Center, Eugene, OR, USA) were maintained in our facility according to standard guidelines. Each adult male zebrafish (6 months old) was assigned to either overfeed or a control group, under a single fish per 2L tank condition. To induce T2DM, zebrafish were overfed with Otohime B2 for 4 weeks, based on our previous study21. Body metrics (body weight, total length, standard length and height at anterior of anal fin [HAA], and body mass index [BMI])23,24, and fasting blood glucose25,26 were measured at the end of the feeding experiment.
Gut bacterial DNA isolation
Zebrafish were sacrificed 2 h after the feeding experiment, with ice water. Each fish was then surface sterilized using 70% ethanol, and the intestinal contents were collected by gently squeezing the intestine with forceps. DNA was isolated with the PowerSoil DNA Isolation Kit (MoBio Laboratories, Carlsbad, CA, USA) according to the manufacturer’s instructions.
16S rRNA sequencing of intestinal content
Illumina MiSeq paired-end sequencing of the hypervariable V3-V4 regions of the 16S rRNA was performed at Bioengineering Lab. Co., Ltd., Kanagawa, Japan. A two-step, tailed PCR approach was used according to the protocol for 16S metagenomic sequencing library preparation (Illumina, San Diego, CA, USA). Both the V3 and V4 regions of the 16S ribosomal RNA were amplified with primers containing the Illumina overhang adaptor (Forward primer 5′ ACA CTC TTT CCC TAC ACG ACG CTC TTC CGA TCT CCT ACG GGN GGC WGC AG; Reverse primer 5′ GTG ACT GGA GTT CAG ACG TGT GCT CTT CCG ATC TGA CTA CHV GGG TAT CTA ATC C). Index PCR was performed with Index 1 and Index 2 Primers from the Nextera XT Index Kit (Illumina), using 2 μL of amplicon derived from the previous PCR. The indexed libraries were cleaned and analysed with Bioanalyzer, using a high sensitivity DNA kit (Agilent Technologies, Wilmington, DE). The prepared libraries were used for paired-end sequencing using MiSeq v3 reagents and 2 × 300-bp reads on the MiSeq (Illumina).
Determination of colony forming units in the tank water
Heterotrophic bacteria in tank water were analysed by spread plating method on R2A-agar (Difco Laboratories, Sparks, MD, USA). Briefly, R2A-agar plates were inoculated with 0.1 ml of the samples and 0.1 ml of the 10−1 dilutions in triplicates, and spread with a sterile glass rod. All plates were incubated for 14 days at 25 °C to reach a plateau, and then the numbers of colonies were counted.
Analysis of bacterial composition in 16S rRNA datasets
The paired-end reads of the 16S rRNA amplicons were assembled using QIIME (version 1.9.1)27, with the script multiple_join_paired_ends.py. Further processing of paired-end reads, including quality filtering based on a quality score of >19, removal of mismatched barcodes and sequences below length thresholds (130 bases), was performed. Quality filtered reads were assigned to Operational Taxonomic Units (OTUs) (97% identity) using de novo OTU picking and taxonomic assignment using the UCLUST algorithm28 against the Greengenes (version 13.8)29 reference using the script pick_de_novo_otus.py.
Measurement of fructose and BCAA
Twelve hours after starvation, zebrafish blood was collected as described in our previous study25,30. Contents of the gut were collected and homogenized in D-PBS at five-times the original volume. Then, the supernatants were obtained by centrifugation at 15,000 g for 10 min at 4 °C. Plasma and gut fructose levels were quantified using PicoProbe Fructose Fluorometric Assay Kit (Biovision, Mountain View, CA, USA), according to manufacturer’s instruction. Branched chain amino acids were quantified using Branched Chain Amino Acid Assay Kit (Abcam, Cambridge, MA, USA), according to manufacturer’s instruction.
Measurement of butyrate
Samples for LC-MS/MS analysis were prepared using a previously reported method31. In brief, 10 µL of sample containing 2.0 nmol heptanoic acid as internal standard was saponified by mixing with 25 µL of 0.3 M KOH-EtOH and heating at 80 °C for 45 min. Then, derivatization was performed by adding 100 µL 2-NPH·HCl and EDC·HCl, and incubating at 60 °C for 20 min. Fatty acid NPH derivatives were obtained using potassium phosphate buffer and diethyl ether extraction, followed by vacuum drying of the diethyl ether layer. The dried residue was dissolved in 100 µL of methanol before injection. LC-MS/MS analysis was performed using a Surveyor HPLC system and a TSQ Quantum Access MAX mass spectrometer with a heated electrospray ionization (H-ESI) probe (Thermo Fisher Scientific, Waltham, MA, USA). LC was carried out on an Ascentis Express Phenyl-Hexyl column (5 cm × 2.1 mm I.D., 2.7 µm; Supelco, Bellefonte, PA, USA) at 45 °C. The injection volume was set at 5.0 µL. The mobile phase consisted of 5 mM aqueous ammonium acetate (A), isopropanol (B), and methanol (C) at a flow rate of 200 µL/min. The following gradient elution was applied: 0.0−0.5 min 65% A and 35% C; 0.5–1.0 min 30% A, 20% B, 50% C; 1.0−4.5 min 5% A, 30% B, 65% C; 4.5–5.0 min 33% B, 67% C; this ratio was kept up to 8.0 min; 8.0–10.0 min, ratios were returned to initial gradient for re-equilibration. The selected reaction monitoring (SRM) under negative mode was utilized for MS detection, and the main parameters were optimized. Spray voltage was set at 3000 V. Nitrogen was used as the sheath gas and the auxiliary gas (set at 50 psi and 10 psi, respectively). The vaporizer temperature and the capillary temperature was set at 350 °C and 200 °C, respectively. Collision gas (argon) pressure was set at 1.8 mTorr. Quantification was performed using Thermo Xcalibur 2.1 software (Thermo Fisher Scientific).
Diversity analysis
Diversity analyses were performed on QIIME using the default parameters. α-diversity (microbial diversity within samples) using Chao1 (a richness estimator based on OTU frequency counts) was analysed based on sample sizes, normalized using the minimum number of sequences obtained among samples. β-diversity (microbial diversity between samples) across the samples, was calculated based on unweighted UniFrac32 distance matrices using the script beta_diversity_through_plots.py. A Principal Coordinate Analysis (PCoA) plot was constructed showing the overall dissimilarity of bacterial communities in different groups.
Metabolic reconstruction of 16S rRNA datasets
Phylogenetic Investigation of Communities by Reconstruction of Unobserved States (PICRUSt)22 was used to predict the functional gene content in the faecal microbiota based on the taxonomy obtained from the Greengenes reference database (http://greengenes.lbl.gov/cgi-bin/nph-index.cgi)29.
Statistical analysis
Comparison of OTU percentages between groups was performed using the Mann-Whitney U-test, adjusted for multiple comparisons, using GraphPad Prism version 7 (GraphPad Software, San Diego, CA, USA). Data from the two groups (body metrics and plasma or faecal parameters) were analysed using a non-parametric two-sample t test. All error bars indicate SEM.
Results and Discussion
Hyperglycaemic-zebrafish show different composition of gut microbiota
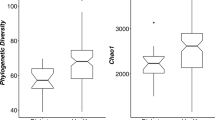
We created T2DM zebrafish by overfeeding them for 4-weeks (about 408 calories per fish per day) as previously described21. This resulted in a significant (p < 0.01) increase in body weight (Fig. 1A; 0.31 ± 0.02 g in controls vs. 0.45 ± 0.02 g in T2DM), HAA (Table S1; 5.50 ± 0.19 mm in controls vs. 6.60 ± 0.22 mm in T2DM) and BMI (Table S1; 0.28 ± 0.01 kg/m2 in controls vs. 0.34 ± 0.01 kg/m2 in T2DM), as a consequence of diet-induced obesity. Controls were fed 68 calories per fish per day as per normal feeding procedures. The fasting blood glucose was doubled (p < 0.01) in the T2DM group compared with the controls (Fig. 1B; 33.8 ± 4.7 mg/dL in control vs. 60.4 ± 4.9 mg/dL in T2DM group), consistent to our previous study21. We then constructed and sequenced 16S rRNA amplicon libraries from the intestinal content samples. The numbers of read tags in each sample were almost the same at about 120-thousand (Fig. S1), corresponded to 1,226 predicted species of the microbiota. Principle coordinate analysis (PCoA; Fig. S2) and jackknifed tree constructed (Fig. S3) with unweighted UniFrac32 revealed that bacterial composition in T2DM zebrafish was different from that of the control group. The dynamic alteration in bacterial compositions between control and T2DM zebrafish indicates that overfeeding affected the microbiota in the zebrafish gut, even though these fish were fed with the same kind of fish feed (the feeding volume of the T2DM group was five-times that of the controls). Bacterial load in the breeding water of the overfed group doubled to that of the control group after feeding experiments (Fig. S4). Because the UV-irradiated breeding water of these tanks was obtained from the same source, the environmental bacterial burden, which was induced by overfeeding and overfed-zebrafish, could not be eliminated by the regular water exchange rate (2.8 ± 0.1 min/exchange). In addition, the zebrafish fed on their faeces, which resulted in enhanced alternation of their gut microbiota in this study. The α-diversity estimates are methods used for describing the number of types of organisms in a single sample, and are useful for examining patterns of dominance, rarity, and community complexity. We evaluated the α-diversity by using the Chao1 index and found that the T2DM group showed lower (p < 0.05) α-diversities (Taxa richness) of operational taxonomic units (OTUs) compared to those of the controls (Fig. 1C). This result indicates that overfeeding reduced bacterial diversity in the zebrafish gut. Because a lower bacterial diversity has also been described in association with obesity and insulin resistance in human studies33,34, the T2DM zebrafish shares common microbiota responses with human T2DM patients in their disease state.

Microbiome in T2DM zebrafish. (A) Body weight gain after 4-weeks of feeding experiment. The T2DM (overfed) group had increased body weight compared to the control group. **p < 0.01, n = 10. (B) Fasting blood glucose (FBG) after the 4-week feeding experiment. The T2DM group exhibited higher FBG compared to the control group. **p < 0.01, n = 5. (C) α-diversity analysis. Whiskers in the boxplot represent minimum and maximum α-diversity values within each group. **p < 0.01, n = 4. (D,E) Median relative abundance of dominant bacteria at phylum level (D) and class level (E).
Comparison of gut microbiota alteration between zebrafish and human T2DM patients
Proteobacteria phylum dominated the zebrafish gut microbiome (Figs 1D and S5), consistent with a previous study5. The second most dominant phylum was Bacteroidetes, which showed a tendency (p < 0.2) to reduce in abundance in T2DM zebrafish. Gaulke CA, et al. previously reported that the main phyla in adult zebrafish gut were Proteobacteria and Fusobacteria12, and there were very few Bacteroidetes. Our result was slightly different from their result. This may be attributed to the difference in feeding, Gaulke CA, et al. fed their zebrafish with processed fish feed containing algae as an omnivorous diet, while we mainly fed our zebrafish Artemia and not algae-containing processed feed. As bacterial communities are known to be different in different fish facilities5, we hypothesized that the difference in feed may have resulted in the difference in compositions of Bacteroidetes and Fusobacteria. In addition, Bacteroidetes are widely distributed in sea water, and our fish feed was made of krill and sea fish, which could be sources of Bacteroidetes in our zebrafish gut. Common zebrafish gut bacteria, Cetobacterium sp., was detected in our control group and its percentage of total reads (0.9%) is similar to the previous study5. Interestingly, Cetobacterium disappeared from the T2DM group, probably due to the reduction of bacterial diversity (Fig. 1C).
At the class level, bacterial compositions between controls and T2DM zebrafish was also different (Figs 1E and S6). Gamma-proteobacteria, Beta-proteobacteria and Flavobacteriia dominated in the control zebrafish. Gamma-proteobacteria class was slightly increased (p < 0.1) and the Beta-proteobacteria class was slightly decreased (p < 0.1) in T2DM zebrafish (Fig. 2A). In the Gamma-proteobacteria class, order Aeromonadales was significantly (p < 0.05) increased in T2DM (26.3% in controls vs. 62.8% in T2DM; Fig. 2B). The majority of Aeromonadales belonged to the Aeromonadaceae family, as reported in a previous zebrafish study35, and Aeromonadaceae was also increased in human T2DM patients36,37, consistent with the result of our T2DM zebrafish. In addition, even while occurring in a small proportions in the microbiome (less than 2%), Alteromonadales and Vibrionales orders in the Gamma-proteobacteria class were significantly (p < 0.01 and p < 0.05, respectively) reduced in T2DM compared to the controls (Fig. S7), reflecting the reduction of α-diversity in T2DM zebrafish (Fig. 1C). In the Beta-proteobacteria class, orders Neisseriales and Burkholderiales were decreased (p < 0.1 and p < 0.05, respectively) in T2DM (Fig. 2C). Burkholderiales included Oxalobacter and related genera, which is unusual since they use oxalic acid as their carbon source in the human colon, to prevent the formation of kidney stones38. Because kidney stone formation, T2DM and obesity are all highly related to the gut microbiome39, our result indicates a possibility that reduction in Burkholderiales may promote kidney stone formation in T2DM zebrafish and humans.

Difference in bacterial compositions between T2DM zebrafish and control. (A) In the Proteobacteria phylum, Gamma-proteobacteria and Beta-proteobacteria were increased and decreased in T2DM zebrafish compared with the control group, respectively n = 4. (B) In the Gamma-proteobacteria class, Aeromonadales were increased in T2DM zebrafish compared to the control group n = 4. (C) In the Beta-proteobacteria class, Neisseriales and Burkholderiales were decreased in T2DM zebrafish compared to the control group n = 4. (D) The Bacteroidetes-to-Firmicutes (B/F) ratio in T2DM and control zebrafish. n = 4.
Several studies in mice and humans have provided evidence that an increase in body weight is associated with a larger proportion of Firmicutes and fewer Bacteroidetes33,40,41. Even though Firmicutes were not the main bacteria in our zebrafish, the Bacteroidetes-to-Firmicutes ratio in T2DM zebrafish tended (p < 0.2) to be lower than that of the control group (Fig. 2D). This was caused by the decrease of the Bacteroidetes phylum (p < 0.2, Fig. 1D), mainly the Flavobacteriia class of the Bacteroidetes phylum (18.7% in controls vs. 0.7% in T2DM; p < 0.1, Fig. S8). Because Firmicutes generate more harvestable energy than Bacteroidetes, it seems reasonable that obese humans and rodents have relatively more Firmicutes42. However, we did not detect many Firmicutes in the zebrafish gut (less than 1%), which implies that other phyla compensate for the decrease of Bacteroidetes to produce enough energy. In fact, Semova I et al. reported that Firmicutes were rich (about 30%) only in zebrafish embryos (6 day-post-fertilization), and not in adults (less than 1%)18, suggesting that other phyla compensate for the lack of Firmicutes in adult fish. Akkermansia muciniphila, which improves insulin resistance in humans43,44, was not detected in our zebrafish.
Metabolic functional pathways in T2DM zebrafish microbiome
To understand the T2DM-induced metabolic alterations in gut microbiota, bacterial metagenomes were predicted by PICRUSt using 16S rRNA sequencing as previously reported45. Predicted proteins in each bacterium were classified into KEGG ortholog entities, which resulted in the identification of 6909 entities across all samples. Of these, we selected 965 ortholog entities that were increased (>2) or decreased (<0.5) in T2DM zebrafish compared to the control group, and used them to construct pathways using KEGG Mapper46. Our analysis revealed 55 reference pathways that contained more than five ortholog entities each (Table S2). Apart from Glycolysis/Gluconeogenesis (KO00010, contains 10 entities) and the Citrate cycle pathway (KO00020, contains nine entities), several pathways of amino acid metabolisms are listed below. The pathway with the most entities, amino sugar and nucleotide sugar metabolism (KO00529, contains 26 entities), is particularly well-studied in bacteria because these molecules are required for the synthesis of glycoconjugates on the surfaces of these organisms47. The dysregulation of this pathway is caused by hyperglycaemia in T2DM zebrafish because of the excessive sugars inside and/or outside the gut epithelium. In addition, the fructose and mannose metabolism (KO00051, contains 17 entities) pathway were also dysregulated in T2DM zebrafish (Fig. S9), consistent with a human T2DM study48. Fructose is closely related to glucose, with growing evidence of its contribution to several metabolic disorders, including obesity and T2DM49,50. Excessive fructose in the gut results in bacterial fermentation, which leads to formation of short-chain fatty acids (SCFAs)51. Several types of SCFAs have been reported to improve T2DM features52, while the end products were associated with arteriosclerosis and colon cancer53. Thus, the health promoting function of SCFAs can be still disputed and needs to be reconsidered. We also focused on the metabolic pathways of arginine and proline (KO00330, contains 22 entities; Fig. 3A) and phenylalanine (KO00360, contains 20 entities; Fig. 3B) in T2DM zebrafish. Downregulation of arginine and proline metabolism (Fig. 3A; red entities) results in the decrease of glutamic acid via a reduction of glutamate-5-semialdehyde. Arginine and its metabolites promote insulin secretion54,55 and improve insulin resistance in an obese mice56,57 and humans58. Regarding phenylalanine (Fig. 3B), various metabolome analysis revealed that its levels are altered in blood59, plasma60, blood cells, and urine61 in human T2DM. The pathway for butyrate (butanoate) metabolism (KO00650, 17 entities), a downstream pathway of arginine and proline metabolism, was also downregulated (Fig. 3C).

Alteration of microbial KEGG metabolic pathways in T2DM zebrafish. (A–C) Several KEGG metabolic pathways were downregulated in the microbiome of T2DM zebrafish compared with those of the control group. Arginine and proline metabolism (A), phenylalanine metabolism (B) and butyrate (butanoate) metabolism (C). Red and green indicates downregulation and upregulation, respectively. KEGG pathway maps (ko00330, ko00360, ko00650) are adapted here from http://www.kegg.jp/kegg/kegg1.html. The KEGG database has been described previously46.
To validate the predicted functional content of the microbiome in T2DM zebrafish, we measured plasma and faecal levels of fructose. Both plasma and faecal fructose levels increased in T2DM zebrafish compared to the controls (Fig. 4A,B), in accordance with the switch from fructose to mannose metabolism (Fig. S9), similar to that in rodents and humans62,63. We also measured the plasma and faecal concentration of butyrate (Total FA 4:0). As shown in Fig. 4C,D, only faecal butyrate levels were significantly (p < 0.05) reduced in T2DM zebrafish compared to the controls, in accordance with the downregulation of butyrate metabolism pathway (Fig. 3C). Dietary supplementation of butyrate improves insulin sensitivity and increases energy expenditure in mice64. In addition, butyrate producing intestinal bacteria seems to play an important role in blood glucose regulation and lipid metabolism, as shown by faecal transplantation studies in humans65,66. Because branch-chain amino acids (BCAA) have been shown to be associated with insulin resistance and T2DM in humans67,68, we also measured plasma and faecal BCAA in the zebrafish. Similar to T2DM studies in humans, T2DM zebrafish showed higher BCAA concentration in their plasma (Fig. 4E) and faeces (Fig. 4F). Although faecal transplantation in zebrafish has not been reported yet, probably because of the lack of germ-free adult zebrafish69, we expect normal microbiota transfer into T2DM zebrafish gut to improve these dysregulations and insulin resistance. Our results indicate that microbiota in T2DM zebrafish adversely affects its health via inhibition of these amino acid metabolisms.

Alteration of fructose, butyrate and BCAA in plasma and faeces of T2DM zebrafish. (A,B) Plasma (A) and faecal (B) fructose levels in zebrafish. (C,D) Plasma (C) and faecal (D) butyrate levels in zebrafish. (E,F) Plasma (E) and faecal (F) branched-chain amino acids (BCAA) in zebrafish. *p < 0.05, **p < 0.01, n = 4–5.
Summary
In this study, we performed 16S rRNA sequencing and functional metagenomics profiling of T2DM zebrafish gut microbiome. The main class in the normal zebrafish gut was Gamma-proteobacteria, Beta-proteobacteria and Flavobacteriia. In addition, Beta-proteobacteria and Flavobacteriia were decreased in T2DM. As for bacterial metabolic functions predicted using PICRUSt and KEGG, several pathways of amino acid metabolism were dysregulated in T2DM, in addition to sugar metabolism. The alternation of bacterial compositions and metabolic functions are similar to human T2DM, indicating that T2DM zebrafish can become a suitable alternative model organism to study host-bacterial interaction in human obesity and its-related diseases.
References
da Rocha Fernandes, J. et al. IDF Diabetes Atlas estimates of 2014 global health expenditures on diabetes. Diabetes Res Clin Pract 117, 48–54, https://doi.org/10.1016/j.diabres.2016.04.016 (2016).
Komaroff, A. L. The Microbiome and Risk for Obesity and Diabetes. JAMA 317, 355–356, https://doi.org/10.1001/jama.2016.20099 (2017).
Forslund, K. et al. Disentangling type 2 diabetes and metformin treatment signatures in the human gut microbiota. Nature 528, 262–266, https://doi.org/10.1038/nature15766 (2015).
Cantas, L., Sorby, J. R., Alestrom, P. & Sorum, H. Culturable gut microbiota diversity in zebrafish. Zebrafish 9, 26–37, https://doi.org/10.1089/zeb.2011.0712 (2012).
Roeselers, G. et al. Evidence for a core gut microbiota in the zebrafish. ISME J 5, 1595–1608, https://doi.org/10.1038/ismej.2011.38 (2011).
Rurangwa, E. et al. Impact of a novel protein meal on the gastrointestinal microbiota and the host transcriptome of larval zebrafish Danio rerio. Front Physiol 6, 133, https://doi.org/10.3389/fphys.2015.00133 (2015).
Rawls, J. F., Samuel, B. S. & Gordon, J. I. Gnotobiotic zebrafish reveal evolutionarily conserved responses to the gut microbiota. Proc Natl Acad Sci USA 101, 4596–4601, https://doi.org/10.1073/pnas.0400706101 (2004).
Bates, J. M. et al. Distinct signals from the microbiota promote different aspects of zebrafish gut differentiation. Dev Biol 297, 374–386, https://doi.org/10.1016/j.ydbio.2006.05.006 (2006).
He, Q., Wang, L., Wang, F. & Li, Q. Role of gut microbiota in a zebrafish model with chemically induced enterocolitis involving toll-like receptor signaling pathways. Zebrafish 11, 255–264, https://doi.org/10.1089/zeb.2013.0917 (2014).
Brugman, S. et al. Oxazolone-induced enterocolitis in zebrafish depends on the composition of the intestinal microbiota. Gastroenterology 137, 1757–1767 e1751, https://doi.org/10.1053/j.gastro.2009.07.069 (2009).
Oehlers, S. H. et al. A chemical enterocolitis model in zebrafish larvae that is dependent on microbiota and responsive to pharmacological agents. Dev Dyn 240, 288–298, https://doi.org/10.1002/dvdy.22519 (2011).
Gaulke, C. A., Barton, C. L., Proffitt, S., Tanguay, R. L. & Sharpton, T. J. Triclosan Exposure Is Associated with Rapid Restructuring of the Microbiome in Adult Zebrafish. PLoS One 11, e0154632, https://doi.org/10.1371/journal.pone.0154632 (2016).
Udayangani, R. M. C. et al. Metagenomics analysis of gut microbiota and immune modulation in zebrafish (Danio rerio) fed chitosan silver nanocomposites. Fish Shellfish Immunol 66, 173–184, https://doi.org/10.1016/j.fsi.2017.05.018 (2017).
Bates, J. M., Akerlund, J., Mittge, E. & Guillemin, K. Intestinal alkaline phosphatase detoxifies lipopolysaccharide and prevents inflammation in zebrafish in response to the gut microbiota. Cell Host Microbe 2, 371–382, https://doi.org/10.1016/j.chom.2007.10.010 (2007).
Liu, Y. et al. Influence of Endogenous and Exogenous Estrogenic Endocrine on Intestinal Microbiota in Zebrafish. PLoS One 11, e0163895, https://doi.org/10.1371/journal.pone.0163895 (2016).
Stagaman, K., Burns, A. R., Guillemin, K. & Bohannan, B. J. The role of adaptive immunity as an ecological filter on the gut microbiota in zebrafish. ISME J 11, 1630–1639, https://doi.org/10.1038/ismej.2017.28 (2017).
Merrifield, D. L. et al. Ingestion of metal-nanoparticle contaminated food disrupts endogenous microbiota in zebrafish (Danio rerio). Environ Pollut 174, 157–163, https://doi.org/10.1016/j.envpol.2012.11.017 (2013).
Semova, I. et al. Microbiota regulate intestinal absorption and metabolism of fatty acids in the zebrafish. Cell Host Microbe 12, 277–288, https://doi.org/10.1016/j.chom.2012.08.003 (2012).
Falcinelli, S. et al. Lactobacillus rhamnosus lowers zebrafish lipid content by changing gut microbiota and host transcription of genes involved in lipid metabolism. Sci Rep 5, 9336, https://doi.org/10.1038/srep09336 (2015).
Falcinelli, S. et al. Dietary lipid content reorganizes gut microbiota and probiotic L. rhamnosus attenuates obesity and enhances catabolic hormonal milieu in zebrafish. Sci Rep 7, 5512, https://doi.org/10.1038/s41598-017-05147-w (2017).
Zang, L., Shimada, Y. & Nishimura, N. Development of a Novel Zebrafish Model for Type 2 Diabetes Mellitus. Sci Rep 7, 1461, https://doi.org/10.1038/s41598-017-01432-w (2017).
Langille, M. G. I. et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nature Biotechnology 31, 814, https://doi.org/10.1038/nbt.2676 (2013).
Oka, T. et al. Diet-induced obesity in zebrafish shares common pathophysiological pathways with mammalian obesity. BMC Physiol 10, 21, https://doi.org/10.1186/1472-6793-10-21 (2010).
Parichy, D. M., Elizondo, M. R., Mills, M. G., Gordon, T. N. & Engeszer, R. E. Normal table of postembryonic zebrafish development: staging by externally visible anatomy of the living fish. Dev Dyn 238, 2975–3015, https://doi.org/10.1002/dvdy.22113 (2009).
Zang, L., Shimada, Y., Nishimura, Y., Tanaka, T. & Nishimura, N. Repeated Blood Collection for Blood Tests in Adult Zebrafish. J Vis Exp, e53272, https://doi.org/10.3791/53272 (2015).
Zang, L., Shimada, Y., Nishimura, Y., Tanaka, T. & Nishimura, N. A novel, reliable method for repeated blood collection from aquarium fish. Zebrafish 10, 425–432, https://doi.org/10.1089/zeb.2012.0862 (2013).
Caporaso, J. G. et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7, 335–336, https://doi.org/10.1038/nmeth.f.303 (2010).
Edgar, R. C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461, https://doi.org/10.1093/bioinformatics/btq461 (2010).
DeSantis, T. Z. et al. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Applied and Environmental Microbiology 72, 5069–5072, https://doi.org/10.1128/aem.03006-05 (2006).
Zang, L., Shimada, Y., Nishimura, Y., Tanaka, T. & Nishimura, N. A Novel, Reliable Method for Repeated Blood Collection from Aquarium Fish. Zebrafish, https://doi.org/10.1089/zeb.2012.0862 (2013).
Chen, Z. et al. ANNALS EXPRESS: Determination of Total, Free, and Esterified Short-Chain Fatty Acid in Human Serum by LC-MS/MS. Ann Clin Biochem, 4563218801393, https://doi.org/10.1177/0004563218801393 (2018).
Lozupone, C. & Knight, R. UniFrac: a new phylogenetic method for comparing microbial communities. Appl Environ Microbiol 71, 8228–8235, https://doi.org/10.1128/AEM.71.12.8228-8235.2005 (2005).
Turnbaugh, P. J. et al. A core gut microbiome in obese and lean twins. Nature 457, 480–484, https://doi.org/10.1038/nature07540 (2009).
Le Chatelier, E. et al. Richness of human gut microbiome correlates with metabolic markers. Nature 500, 541–546, https://doi.org/10.1038/nature12506 (2013).
Stephens, W. Z. et al. The composition of the zebrafish intestinal microbial community varies across development. ISME J 10, 644–654, https://doi.org/10.1038/ismej.2015.140 (2016).
Larsen, N. et al. Gut microbiota in human adults with type 2 diabetes differs from non-diabetic adults. PLoS One 5, e9085, https://doi.org/10.1371/journal.pone.0009085 (2010).
Wang, Y. et al. Gut microbiome analysis of type 2 diabetic patients from the Chinese minority ethnic groups the Uygurs and Kazaks. PLoS One 12, e0172774, https://doi.org/10.1371/journal.pone.0172774 (2017).
Jalanka-Tuovinen, J. et al. Intestinal microbiota in healthy adults: temporal analysis reveals individual and common core and relation to intestinal symptoms. PLoS One 6, e23035, https://doi.org/10.1371/journal.pone.0023035 (2011).
Stern, J. M. et al. Evidence for a distinct gut microbiome in kidney stone formers compared to non-stone formers. Urolithiasis 44, 399–407, https://doi.org/10.1007/s00240-016-0882-9 (2016).
Backhed, F. et al. The gut microbiota as an environmental factor that regulates fat storage. Proc Natl Acad Sci USA 101, 15718–15723, https://doi.org/10.1073/pnas.0407076101 (2004).
Ley, R. E. et al. Obesity alters gut microbial ecology. Proc Natl Acad Sci USA 102, 11070–11075, https://doi.org/10.1073/pnas.0504978102 (2005).
Turnbaugh, P. J. et al. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 444, 1027–1031, https://doi.org/10.1038/nature05414 (2006).
Zhang, X. et al. Human gut microbiota changes reveal the progression of glucose intolerance. PLoS One 8, e71108, https://doi.org/10.1371/journal.pone.0071108 (2013).
Cani, P. D. & de Vos, W. M. Next-Generation Beneficial Microbes: The Case of. Front Microbiol 8, 1765, https://doi.org/10.3389/fmicb.2017.01765 (2017).
Mukherjee, A. et al. Bioinformatic Approaches Including Predictive Metagenomic Profiling Reveal Characteristics of Bacterial Response to Petroleum Hydrocarbon Contamination in Diverse Environments. Sci Rep 7, 1108, https://doi.org/10.1038/s41598-017-01126-3 (2017).
Kanehisa, M., Goto, S., Sato, Y., Furumichi, M. & Tanabe, M. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res 40, D109–114, https://doi.org/10.1093/nar/gkr988 (2012).
Samuel, G. & Reeves, P. Biosynthesis of O-antigens: genes and pathways involved in nucleotide sugar precursor synthesis and O-antigen assembly. Carbohydr Res 338, 2503–2519 (2003).
Karlsson, F. H. et al. Gut metagenome in European women with normal, impaired and diabetic glucose control. Nature 498, 99–103, https://doi.org/10.1038/nature12198 (2013).
Johnson, R. J. et al. Potential role of sugar (fructose) in the epidemic of hypertension, obesity and the metabolic syndrome, diabetes, kidney disease, and cardiovascular disease. Am J Clin Nutr 86, 899–906 (2007).
Kolderup, A. & Svihus, B. F. Metabolism and Relation to Atherosclerosis, Type 2 Diabetes, and Obesity. J Nutr Metab 2015, 823081, https://doi.org/10.1155/2015/823081 (2015).
Wong, J. M., de Souza, R., Kendall, C. W., Emam, A. & Jenkins, D. J. Colonic health: fermentation and short chain fatty acids. J Clin Gastroenterol 40, 235–243 (2006).
Puddu, A., Sanguineti, R., Montecucco, F. & Viviani, G. L. Evidence for the gut microbiota short-chain fatty acids as key pathophysiological molecules improving diabetes. Mediators Inflamm 2014, 162021, https://doi.org/10.1155/2014/162021 (2014).
Wang, Z. et al. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature 472, 57–63, https://doi.org/10.1038/nature09922 (2011).
Malaisse, W. J. et al. Stimulus-secretion coupling of arginine-induced insulin release. Metabolism of L-arginine and L-ornithine in pancreatic islets. Biochim Biophys Acta 1013, 133–143 (1989).
Schmidt, H. H., Warner, T. D., Ishii, K., Sheng, H. & Murad, F. Insulin secretion from pancreatic B cells caused by L-arginine-derived nitrogen oxides. Science 255, 721–723 (1992).
Carvalho, D. S. et al. L-Arginine supplementation improves insulin sensitivity and beta cell function in the offspring of diabetic rats through AKT and PDX-1 activation. Eur J Pharmacol 791, 780–787, https://doi.org/10.1016/j.ejphar.2016.10.001 (2016).
Fu, W. J. et al. Dietary L-arginine supplementation reduces fat mass in Zucker diabetic fatty rats. J Nutr 135, 714–721 (2005).
Monti, L. D. et al. Decreased diabetes risk over 9 year after 18-month oral L-arginine treatment in middle-aged subjects with impaired glucose tolerance and metabolic syndrome (extension evaluation of L-arginine study). Eur J Nutr, https://doi.org/10.1007/s00394-017-1548-2 (2017).
Mook-Kanamori, D. O. et al. Type 2 diabetes is associated with postprandial amino acid measures. Arch Biochem Biophys 589, 138–144, https://doi.org/10.1016/j.abb.2015.08.003 (2016).
Rahimi, N. et al. Amino acid profiling in the gestational diabetes mellitus. J Diabetes Metab Disord 16, 13, https://doi.org/10.1186/s40200-016-0283-1 (2017).
Tam, Z. Y. et al. Metabolite profiling in identifying metabolic biomarkers in older people with late-onset type 2 diabetes mellitus. Sci Rep 7, 4392, https://doi.org/10.1038/s41598-017-01735-y (2017).
Kawasaki, T., Akanuma, H. & Yamanouchi, T. Increased fructose concentrations in blood and urine in patients with diabetes. Diabetes Care 25, 353–357 (2002).
Kawasaki, T. et al. Postprandial plasma fructose level is associated with retinopathy in patients with type 2 diabetes. Metabolism 53, 583–588 (2004).
Gao, Z. et al. Butyrate improves insulin sensitivity and increases energy expenditure in mice. Diabetes 58, 1509–1517, https://doi.org/10.2337/db08-1637 (2009).
Udayappan, S. D., Hartstra, A. V., Dallinga-Thie, G. M. & Nieuwdorp, M. Intestinal microbiota and faecal transplantation as treatment modality for insulin resistance and type 2 diabetes mellitus. Clin Exp Immunol 177, 24–29, https://doi.org/10.1111/cei.12293 (2014).
Vrieze, A. et al. Transfer of intestinal microbiota from lean donors increases insulin sensitivity in individuals with metabolic syndrome. Gastroenterology 143, 913–916.e917, https://doi.org/10.1053/j.gastro.2012.06.031 (2012).
Pedersen, H. K. et al. Human gut microbes impact host serum metabolome and insulin sensitivity. Nature 535, 376–381, https://doi.org/10.1038/nature18646 (2016).
Wang, T. J. et al. Metabolite profiles and the risk of developing diabetes. Nat Med 17, 448–453, https://doi.org/10.1038/nm.2307 (2011).
Melancon, E. et al. Best practices for germ-free derivation and gnotobiotic zebrafish husbandry. Methods Cell Biol 138, 61–100, https://doi.org/10.1016/bs.mcb.2016.11.005 (2017).
Acknowledgements
This work was supported in part by Grants-in-Aid for Scientific Research from the Japan Science and Technology Agency and Okasan-Kato Foundation. The authors thank Ms. Takako Taguchi and Ms. Azusa Kato for their secretarial assistance. We also thank Editage (www.editage.jp) for English language editing.
Author information
Authors and Affiliations
Contributions
Y.S. and N.N. designed and conceived the presented study. F.O., L.Z., H.N. and Y.S. carried out the zebrafish experiments. Z.C., Z.G., H.C. and S.H. performed LC-MS analysis. F.O. performed the bioinformatics analysis. Y.S. wrote manuscript with support from F.O. and T.A.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Okazaki, F., Zang, L., Nakayama, H. et al. Microbiome Alteration in Type 2 Diabetes Mellitus Model of Zebrafish. Sci Rep 9, 867 (2019). https://doi.org/10.1038/s41598-018-37242-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-37242-x
This article is cited by
-
Graphene oxide elicits microbiome-dependent type 2 immune responses via the aryl hydrocarbon receptor
Nature Nanotechnology (2023)
-
Effect of crude polysaccharide from seaweed, Dictyopteris divaricata (CDDP) on gut microbiota restoration and anti-diabetic activity in streptozotocin (STZ)-induced T1DM mice
Gut Pathogens (2022)
-
Molecular and culture-based assessment of the microbiome in a zebrafish (Danio rerio) housing system during set-up and equilibration
Animal Microbiome (2021)
-
Rhamnan sulphate from green algae Monostroma nitidum improves constipation with gut microbiome alteration in double-blind placebo-controlled trial
Scientific Reports (2021)
-
Using Zebrafish to Investigate Interactions Between Xenobiotics and Microbiota
Current Pharmacology Reports (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
