
Abstract
We have previously shown that high fat diet (HFD) for 2 weeks increases airway hyperresponsiveness (AHR) to methacholine challenge in C57BL/6J mice in association with an increase in IL-1β levels in lung tissue. We hypothesize that obesity increases AHR via the IL-1β mechanism, which can be prevented by caloric restriction and IL-1β blockade. In this study, we fed C57BL/6J mice for 8 weeks with several hypercaloric diets, including HFD, HFD supplemented with fructose, high trans-fat diet (HTFD) supplemented with fructose, either ad libitum or restricting their food intake to match body weight to the mice on a chow diet (CD). We also assessed the effect of the IL-1β receptor blocker anakinra. All mice showed the same total respiratory resistance at baseline. All obese mice showed higher AHR at 30 mg/ml of methacholine compared to CD and food restricted groups, regardless of the diet. Obese mice showed significant increases in lung IL-1 β mRNA expression, but not the protein, compared to CD and food restricted mice. Anakinra abolished an increase in AHR. We conclude that obesity leads to the airway hyperresponsiveness preventable by caloric restriction and IL-1β blockade.
Similar content being viewed by others

Introduction
The prevalence of obesity is increasing worldwide. The epidemic of obesity can be attributed to increased consumption of high caloric food, sedentary life style and genetic factors. Obesity has been linked to multiple comorbidities including insulin resistance and type 2 diabetes, cardiovascular disorders, cancer and systemic low-grade inflammation1.
Asthma is a chronic inflammation of the bronchi leading to airway hyperresponsiveness (AHR), the major functional outcome of this disease. The relationship between obesity and asthma is well established2,3,4. Obese asthma poses a significant public health problem due to poor understanding of the pathogenesis and a lack of effective treatment. In the mouse model, IL-17, IL-1β and the NLRP3 inflammasome have been implicated in the pathogenesis of obesity-induced airway hyperresponsiveness5. Additionally, a high fat diet (HFD) has been linked to asthma, independent of obesity, although the underlying mechanism is not clear. Moreover, weight loss by caloric restriction improved disease control and quality of life in asthmatics6, but mechanisms are also insufficiently understood.
We have previously shown that HFD leads to AHR in mice after 2 weeks of feeding in association with IL-1 β upregulation in lung tissue, whereas other pro-inflammatory cytokines were unchanged7. However, it was unclear whether a small weight gain or dietary fat led to hyperresponsiveness and whether IL-1β played a causal role. In addition, emerging epidemiology literature suggests that a diet high in fructose is associated with asthma8. We hypothesized that obesity rather than a diet leads to AHR via the IL-1β mechanism and that caloric restriction on the same diet will abolish both AHR and pulmonary inflammation. We investigated our hypothesis in two experiments. In the first experiment, C57BL/6J mice were fed with several hypercaloric diets including HFD, HFD supplemented with 30% fructose added to drinking water, and high trans-fat diet (HTFD) supplemented with 30% fructose added to drinking water for 8 weeks, either ad libitum [HFD(O), HFD + HFr(O), and HTFD + HFr(O) groups, respectively] or food restricted to match their weight to the control group on a chow diet [HFD(R), HFD + HFr(R), and HTFD + HFr(R) groups, respectively], and subsequently AHR and pulmonary inflammation were measured. In the second experiment, C57BL/6J mice were fed with a HFD ad libitum for 8 weeks and treated with an IL-1β receptor blocker or placebo during the last 2 weeks of the experiment followed by the same measurements.
Results
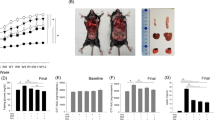
In the first experiment, mice in all groups gained significant amount of weight over the period of 8 weeks as compared to their initial weight. As expected, the hypercoloric ad libitum groups gained more weight compared to the chow (CD) group and the restricted HFD, HFD + HFr, HTFD + HFr groups (p < 0.001, Table 1). There was no weight difference between HFD(R), HFD + HFr(R), HTFD + + HFr(R) and CD group across the 8-week time course (Fig. 1). There was no difference in lung volumes measured by water displacement between the groups (Table 2).

Weight trajectory of chow diet (CD), HFD (high fat diet), HFD + high fructose, HTFD + high fructose ad libitum (O) and restricted (R) groups over the period of 8 weeks. *,**,***Denote that the final weight was significantly different as compared to the chow diet (CD), *p < 0.05, **p < 0.01, ***p < 0.001.
Total respiratory resistance (Rrs) at baseline was identical in all seven groups, 1.00 ± 0.06 cmH2O.s/mL, 1.00 ± 0.02 cmH2O.s/mL, 1.00 ± 0.03 cmH2O.s/mL, 1.00 ± 0.02 cmH2O.s/mL, 1.00 ± 0.05 cmH2O.s/mL, 1.00 ± 0.04 cmH2O.s/mL, and 1.00 ± 0.02 cmH2O.s/mL in the CD, HFD(O), HFD + HFr(O), HTFD + HFr(O), HFD(R), HFD + HFr(R), HTFD + HFr(R) groups, respectively. AHR assessed by methacholine challenge showed no difference between the groups at 3 mg/ml dose of methacholine. However, when the methacholine dose was increased to 30 mg/ml, HFD(O), HFD + HFr(O) and HTFD + HFr(O) group exhibited a robust increase in airway reactivity (Fig. 2). Specifically, at this dose of methacholine, HFD(O), HFD + HFr(O) and HTFD + HFr(O) mice showed 7.2 ± 0.4, 6.6 ± 0.3 and 6.3 ± 0.4 fold increase in AHR from baseline vs 4.3 ± 0.3, 3.9 ± 1.0, 4.4 ± 0.6, and 4.7 ± 0.5 fold in CD, HFD(R), HFD + HFr(R) and HTFD + HFr(R) groups, respectively (p < 0.05).

HFD, HFD + high fructose and HTFD + high fructose ad libitum (O) groups have increased total resistance of the respiratory system (Rrs) in response to methacholine as compared to chow diet group (*p < 0.05, ***p < 0.001). The Rrs values were normalized to baseline (no significant difference between groups at baseline). No difference in Rrs was observed between CD and caloric restricted groups.
Metabolic measurements showed that the HFD(O) and HFD + HFr(O) groups had higher fasting blood glucose levels compared to CD (p < 0.05, Table 1). Fasting blood glucose was lower in HTFD + HFr(R) group compared to CD group but not different between CD, HFD(R) and HFD + HFr(R) group (Table 1). Serum insulin and leptin levels were significantly higher in HFD(O), HFD + HFr(O) groups compared to the CD group. There was no difference in adiponectin levels across the groups (Table 1). Serum free fatty acids (FFAs) were higher in HFD + HFr(R) and HTFD + HFr(R) group than CD group. There was no difference in serum FFAs between HFD(O), HFD + HFr(O), HTFD + HFr(O) and CD groups. The HFD(R) group had lower triglycerides as compared to CD group and HFD (O) group. HFD(O), HFD + HFr(O) and HTFD + HFr(O) groups showed a greater than 2-fold increase in the bronchoalveolar lavage (BAL) cellularity with the predominance of macrophages (Table 2). Neither eosinophils nor basophils were observed in BAL, and the proportion of neutrophils were less than 1% in all groups. Lung triglycerides were higher in HFD + HFr(O) than CD group, but the values were similar between HFD(O), HTFD + HFr(O), caloric restricted groups and CD group (Table 2). There was no difference in lung FFAs across the groups (Table 2). Lung TNF-α and IL-6 were not significantly different between CD, HFD(O) and HFD(R) group (Suppl. Fig. 1). Such pro-inflammatory cytokines as IL-4, IL-5, IL-13, IL-17, IL-18, IL-21, IL-23 were not detected. There were a 3.1 ± 1.0-fold, 4.7 ± 1.6-fold and 8.7 ± 5.2-fold increases in IL-1 β mRNA levels in the lung tissue of HFD(O), HFD + HFr(O) and HTFD + HFr(O) mice compared to the CD group. In contrast, IL-1 β mRNA expression in lung tissue of the HFD(R), HFD + HFr(R), HTFD + HFr(R) groups were not elevated (Fig. 3A). However, IL-1 β protein levels in lung lysates were not significantly different across all the obese and caloric restricted groups, either (Fig. 3B). Flow cytometry also did not show any difference in cell-specific IL-1β production from pulmonary macrophages, neutrophils or lymphocytes between the groups (Suppl. Fig. 2). IL-1 β protein secretion by pulmonary macrophages was similarly not different between the CD, HFD(O) and HFD(R) groups (Fig. 3C). IL-1β mRNA levels in epididymal fat and inguinal fat tissues were not increased in mice fed on HFD ad libitum as compared to CD group (Table 2).

(A) Interleukin-1 beta (IL-1β) mRNA levels in lung tissue of chow diet (CD), HFD, HFD + high fructose, HTFD + high fructose ad libitum (O) and restricted (R) groups; (B) Interleukin-1 beta (IL-1β) protein levels in lung lysates of chow diet (CD), HFD, HFD + high fructose, HTFD + high fructose ad libitum (O) and restricted (R) groups. The results were adjusted by sample protein concentration. (C) IL-1β protein secretion to the media by adherent cells isolated from the lung single cell suspension in chow diet (CD), high fat diet ad libitum (O) and restricted (R) group. *,**Denote that these values were significantly different as compared to the chow diet (CD), *p < 0.05, **p < 0.01.
In the second experiment we examined the effect of an IL-1β blocker anakinra on metabolic parameters and airway responsiveness in mice fed HFD. The placebo and anakinra groups gained similar amount of weight after feeding with HFD for 8 weeks (Fig. 4). Serum insulin levels were lower in the anakinra group compared to the placebo group (Table 3). There was no significant difference in fasting glucose, leptin, adiponectin, FFAs or triglyceride levels between two groups (Table 3). Anakinra had no effect on Rrs at baseline, 0.65 ± 0.03 cmH2O.s/mL vs 0.67 ± 0.03 cmH2O.s/mL in the placebo group. There was no difference between the groups at 3 mg/ml of methacholine. However, at 30 mg/mL of methacholine the AHR in the anakinra group was significantly lower compared to the placebo group (2.9 ± 0.9 vs 5.1 ± 1.4 respectively, p = 0.01) (Fig. 5). In fact, the AHR in obese mice treated with anakinra was identical to the lean CD mice and the HFDR mice from Experiment 1. The proportion of lymphocytes in BAL was lower in the anakinra group compared to the placebo group. The lung volumes, serum and lung FFAs, triglycerides and pro-inflammatory cytokines, total BAL cell count and other than lymphocyte cell content were not different between placebo and anakinra groups (Table 4).

Weight trajectory of HFD ad libitum + placebo and HFD ad libitum + anakinra group over the period of 8 weeks.

Anakinra injection decreased total resistance of the respiratory system (Rrs) in response to methacholine in HFD induced obese mice. The Rrs values were normalized to baseline (no significant difference between groups at baseline). *Denotes p < 0.05.
Discussion
The main finding of our study was that, in mice on a high fat hypercaloric diet, caloric restriction prevented the development of airway hyperresponsiveness and upregulation of IL-1β gene expression in lung parenchyma, regardless of the diet. Moreover, IL-1β receptor blockade also prevented and maybe even reversed the development of airway hyperresponsiveness in obese mice, despite persistent metabolic abnormalities.
Our study was designed to determine whether obesity per se or diet lead to asthma. A positive correlation between high fat diet intake and asthma has been previously observed9. Wood et al. suggests that the abundance of saturated fatty acids and lack of antioxidants in HFD can induce inflammation by activating toll-like receptors and hence stimulating the NF-κB inflammatory cascade10. HFD also induces the proliferation of invasive bacteria and eliminates the protective bacteria in the gut which can induce inflammation11,12. A high fructose diet has also been associated with asthma8. Our data demonstrated that two different types of HFD fed ad libitum induced pulmonary inflammation with increased macrophages in BAL, especially in the HTFD + HFr group. However, our food restriction protocol showed that food restricted mice did not develop AHR, despite being fed the same diet. These results lead to the conclusion that the airway hyperresponsiveness is a consequence of obesity rather than high fat or high fructose diets. We recently reported that HFD feeding for 2 weeks led to an increase in AHR of the similar magnitude as HFD feeding for 8 weeks, despite much more significant weight gain in a longer term experiment7. Taken together these data suggest that even mild obesity can lead to airway hyperresponsiveness.
Several physiological and immunological mechanisms have been implicated in the pathogenesis of obese asthma. Excessive adiposity can have a restrictive effect on the lung decreasing functional residual capacity and expiratory reserve volume13. Radial traction around the distal airway is decreased at low lung volume contributing to airway narrowing14,15. A reduction in initial airway caliber allows a greater increase in resistance for a given absolute reduction in smooth muscle shortening, which manifests as increased airway reactivity16. Obesity’s effect on lung and chest wall compliance also lessens the effectiveness of a bronchoprotective deep breath to dilate airways17. In addition to these mechanical effects, our current study highlights an even more important role of pulmonary inflammation in the pathogenesis of obesity-induced airway hyperresponsiveness in mice.
Our previous study showed that HFD feeding for two weeks increases AHR in association with increased IL-1 β gene expression in the lung and augmented IL-1 β secretion by pulmonary macrophages7. The current study showed that the expression of IL-1 β mRNA was increased in the mouse lungs after HFD feeding for 8 weeks and this increase was prevented by caloric restriction. The IL-1β receptor blockade also prevented both obesity-induced AHR and pulmonary inflammation, supporting the concept that up-regulation of IL-1β gene expression in the lung could be a mechanism linking obesity and asthma5. Given that obesity increases AHR early in the time course7 and that anakinra was administered only during last two weeks of the 8-week experiment, our data suggest that IL-1β receptor blockers not merely prevent, but also reverse obese asthma.
Mechanisms of obesity-induced up-regulation of IL-1β have been linked to the NLRP3 inflammasome, which was examined in detail by Umetsu et al.5. However, we did not detect an increase in adipose IL-1β in mice on a HFD. We found an increase in lung triglycerides only in the HFD + HFr (O) group, whereas the AHR was increased in all obese groups. Nevertheless, the lung is an important organ of triglyceride rich lipoprotein clearance18 and it is conceivable that particular species of fatty acids, e.g. long chain fatty acids, which were not measured specifically in our study, induced IL-1β expression in the lungs. IL-1β could increase bronchial reactivity via up-regulation of TH2/TH17 cells, which were associated with severe asthma phenotype in humans19,20.
Other potential mechanisms contributing to obesity-induced pulmonary inflammation and AHR are hyperleptinemia21, hyperglycemia22 and insulin resistance23,24, which were observed in obese mice on a HFD, but not in control chow fed mice or food restricted mice on a HFD25. Of note, the IL-1β receptor blocker anakinra has been shown to improve hyperglycemia and insulin secretion in type 2 diabetes26. In our study, anakinra lowered fasting serum insulin levels without a significant change in fasting glucose levels indicating increased insulin sensitivity. Our finding suggests that anakinra could contribute to improvement in AHR due to its off-target effects by improving glucose metabolism.
The most important novel finding of our study was that caloric restriction prevented the development of airway hyperresponsiveness. The data from several trials showed that weight loss in asthmatics through caloric restriction can lead to clinical improvement6,27,28,29. We agree and further propose that caloric restriction may be beneficial for asthma in obese individuals, possibly by suppressing the IL-1β response.
Our study had several limitations. First, we were unable to demonstrate an increase in IL-1β protein. We have previously shown that IL-1β secretion by pulmonary macrophages is dramatically increased after 2 weeks of high fat diet7. After 8 weeks of HFD and development of severe obesity, IL-1β mRNA levels remained elevated, but the IL-1β protein levels and secretion by pulmonary macrophages were no longer increased. These data may suggest that IL-1β peaked early in the time course of HFD – induced obesity. Second, there was an apparent discrepancy between the lack of increase in IL-1β protein levels and beneficial effects of the IL-1β receptor blocker on AHR. Besides possible fluctuations of IL-1β levels over the time course, anakinra could exert a therapeutic effect in the absence of an increase in IL-1β by blocking the receptor and downstream inflammation. It is also possible that anakinra had non-specific off-target beneficial effects, for instance by alleviating insulin resistance as discussed above. Third, other than IL-1β mechanisms by which obesity may increase AHR were not addressed. Recent literature suggest that long chain fatty acids may induce inflammation by altering macrophage lipid metabolism indirectly via the toll receptor 430. Finally, we did not investigate sex differences by including female mice.
Conclusions and Implications
Diet induced obesity increases airway hyperresponsiveness and the effects of obesity are preventable by caloric restriction and IL-1β blockade. Taken together our data suggest that caloric restriction should be used for prevention of obese asthma and that IL-1β blockade may be considered as an adjunct therapy.
Methods
Animals and study design
The study was approved by the Institutional Animal Care and Use Committee of the Johns Hopkins University Animal Use and Care Committee (Protocol # MO15M257) and complied with the American Physiological Society Guidelines for Animal Studies. The study consisted of two experiments. In total, 80 C57BL/6J male mice (Jackson Labs Bar Harbor, MA), 6–8 weeks old) were used in the study. Mice were housed in 4 per cage, in a temperature and humidity-controlled room with a 12/12 light/dark cycle (9 am–9 pm lights on) with access to water at all time. 64 C57BL/6J male mice were used in the first experiment. Mice were divided in 7 groups, Control (CD) (n = 16), high fat diet ad libitum group [HFD(O)](n = 8), high fat diet + high fructose ad libitum group [(HFD + HFr)(O)](n = 8), high trans-fat diet + high fructose ad libitum group [(HTFD + HFr)(O)](n = 8), and the restricted HFD [HFD(R)](n = 8), HFD + HFr [(HFD + HFr)(R)](n = 8) and HTFD + HFr [(HTFD + HFr)(R)] groups. CD group was fed with chow diet (3.0 kcal/g, 4.4% fat, 13% kcal from fat) ad libitum. HFD(O) group was fed with high fat diet (TD 03584, Teklad WI, 5.4 kcal/g, 35.2% fat, 58.4% kcal from fat,) ad libitum for 8 weeks. (HFD + HFr)(O) group was fed with high fat diet (TD 03584) and fructose (30% by wt) ad libitum for 8 weeks. (HTFD + HFr)(R) group was fed with high trans-fat diet (Research diets, D09100301, 4.49 kcal/g, 19.9% fat, 40% kcal from fat] and fructose (30% by wt) ad libitum for 8 weeks. In caloric-restricted groups, mice were provided with the same HFD, HTFD and fructose but the amount of food was restricted to match body weight to the CD group. The composition of HFD has been described previously7. Diets were refrigerated at 4–8 °C before it was added to the cages. 16 C57BL/6J male mice were used in the second experiment. The second experiment consisted of 2 groups, placebo group (n = 8) and anakinra group (n = 8). Anakinra was a gift from Sobi (Stockholm, Sweden). Both groups were fed with HFD ad libitum for 8 weeks. During last 14 days of the experiment, the anakinra group was injected subcutaneously at 50 mg/kg in 250 µl of saline daily and placebo group was injected subcutaneously with 250 µl of saline daily.
Physiological measurements
Mice were anesthetized with ketamine/xylazine i.p., tracheostomized and the total respiratory resistance (Rrs) was measured by forced oscillation technique (Flexivent, SCIREQ Québec, Canada) at baseline and after methacholine aerosol challenge at 3 and 30 mg/mL as described7,31. Blood was collected from the aorta, BAL was performed with 2 × 0.8 mL of sterile phosphate-buffered saline (PBS) through a tracheal cannula. The thorax was opened, and the right lung was tied off, dissected free and immediately frozen in liquid nitrogen and stored at −80 °C. Inguinal fat and epididymal fat tissue were collected, immediately frozen in liquid nitrogen and stored at −80 °C. The remaining left lung was inflated with formalin at 26 cmH2O pressure for 20 min, tied off and placed inflated in formalin for 2 days. Left lung volumes were measured by fluid displacement method32.
Plasma, lung and adipose tissue analysis
Triglycerides and free fatty acids (FFA) were measured in lung homogenates and plasma with kits from Wako Inc (Richmond, VA). Plasma insulin, adiponectin and leptin were measured with kits from Alpco Diagnostics (Salem, NH), Millipore (Billerica, MA) and Abcam (Cambridge, MA), respectively. Blood glucose was measured following a 4-h fast by tail-snip technique using a handheld glucometer (ACCUCHECK Aviva Plus, Roche, Indianapolis, IN). IL-1β protein levels in lung lysates were measured with an ELISA kit (R&D systems). Total RNA was extracted from lung, inguinal fat and epididymal fat tissue with a Trizol reagent (Life Technologies, Rockville, MD). cDNA was produced from total RNA using Advantage RT for PCR kit from Clontech (Palo Alto, CA). Real time PCR was performed for the cytokine panel, including interleukins (IL) 1β, 4, 5, 6, 10, 13, 17, IL-18, TNF-α, IL-21, and IL-23 with premade primers from Invitrogen (Carlsbad, CA) and Taqman probes from Applied Biosystems (Foster City, CA) using 18S as a housekeeping gene. Custom made 18S primers were forward 5′-CTCTTTCGAGGCCCTGTAATTGT-3′, reverse, 5′-AACTGCAGCAACTTTAATATACGCTATT-3′ and the probe 6FAM-AGTCCACTTTAAATCCTT. Target mRNA level was normalized to 18 s rRNA, using the formula: Target/18 s = 2Ct(18 s)–Ct(target).
Cytokine secretion and flow cytometry
In a subset of mice left lungs were harvested, minced and placed in gentle MACS Dissociator (Miltenyi Biotec), and digested using Collagenase type 1 (Worthington) and DNase I (Sigma Aldrich, St. Louis, MO) for 10 minutes at 37 °C. The lung digests were passed through a 70-μm nylon cell strainer (Becton Dickinson, Franklin Lakes, NJ), and erythrocytes were subsequently lysed using RBC Lysis Buffer (eBioscience, San Diego, CA). The cells were counted, and cells viability was assessed by Trypan Blue staining. Then, 2 × 106 of viable cells were seeded in 96 well plates in the presence of DMEM + 10% FBS + Pen/Strep 1:100 medium. Two hours later, non-adherent cells were removed and 100 ul of the medium were added to the attached cells. The cells were incubated for 24 hours at 37 °C, the media was collected, centrifuged to remove cells and debris, and IL-1β secretion was measured with an ELISA kit (R&D systems). Four hours before end of the ex vivo culture, cells were stimulated with Phorbol myristate acetate (PMA, 40 ng/ml)) and ionomycin (500 ng/ml) in the presence of Golgistop (BD Biosciences). For flow cytometry, cells were washed with FACS buffer (PBS + 0.5% BSA) and incubated with PE-Cy CD64 Ab. Then, non-specific staining of Fcγ III/II receptors was blocked with Fc Block-2.4G2 (BD Biosciences — Pharmingen) Ab. The following Abs (BD Biosciences — Pharmingen) were used for cell phenotyping: PerCp Cy 5.5-conjugated anti-CD11c, PE-CF594-conjugated anti-CD11b, APC-Cy7-conjugated anti-MHCII, BV421-conjugated anti-SigF, BV605-conjugated anti-Ly6c, BV510-conjugated anti-Ly6g, BV395-conjugated anti-CD4 and BV737-conjugated anti-CD8 and respective isotype Abs. Cells were subsequently fixated and permeabilized and stained for IL-1β (Thermo Fisher). Lymphocytes, monocytes, neutrophils, alveolar and interstitial macrophages were gated with characteristic low forward scatter/side scatter, using a FACSAria instrument and FACSDiva for data acquisition (Becton Dickinson) and Flowjo for analysis (Tree Star Inc.) as previously described33.
Statistical analysis
The statistical analysis was done using STATA version 12. All values were reported as means ± SEM. All the data in the study was checked for normality with Jarque-Bera test. Statistical analysis of normally distributed variables was determined by student t-test or one-way analysis of variance test (ANOVA) with repeated measures when appropriate. Non-normally distributed values were analyzed by Kruskal-Wallis rank test. A p-value of <0.05 was considered significant.
References
Gomez-Llorente, M. A., Romero, R., Chueca, N., Martinez-Canavate, A. & Gomez-Llorente, C. Obesity and Asthma: A Missing Link. International journal of molecular sciences 18, https://doi.org/10.3390/ijms18071490 (2017).
Camargo, C. A. et al. Prospective study of body mass index, weight change, and risk of adult-onset asthma in women. Archives of internal medicine 159, 2582–2588 (1999).
Ather, J. L., Poynter, M. E. & Dixon, A. E. Immunological characteristics and management considerations in obese patients with asthma. Expert review of clinical immunology 11, 793–803, https://doi.org/10.1586/1744666x.2015.1040394 (2015).
Periyalil, H. A., Gibson, P. G. & Wood, L. G. Immunometabolism in obese asthmatics: are we there yet? Nutrients 5, 3506–3530, https://doi.org/10.3390/nu5093506 (2013).
Kim, H. Y. et al. Interleukin-17-producing innate lymphoid cells and the NLRP3 inflammasome facilitate obesity-associated airway hyperreactivity. Nature medicine 20, 54–61, https://doi.org/10.1038/nm.3423 (2014).
Forte, G. C. et al. Diet effects in the asthma treatment: A systematic review. Critical reviews in food science and nutrition, 1–10 https://doi.org/10.1080/10408398.2017.1289893 (2017).
Fricke, K., Vieira, M. & Younas, H. High fat diet induces airway hyperresponsiveness in mice 8, 6404, https://doi.org/10.1038/s41598-018-24759-4 (2018).
DeChristopher, L. R. & Tucker, K. L. Excess free fructose, high-fructose corn syrup and adult asthma: the Framingham Offspring Cohort. The British journal of nutrition 119, 1157–1167, https://doi.org/10.1017/s0007114518000417 (2018).
Wood, L. G., Garg, M. L. & Gibson, P. G. A high-fat challenge increases airway inflammation and impairs bronchodilator recovery in asthma. The Journal of allergy and clinical immunology 127, 1133–1140, https://doi.org/10.1016/j.jaci.2011.01.036 (2011).
Wood, L. G. & Gibson, P. G. Dietary factors lead to innate immune activation in asthma. Pharmacology & therapeutics 123, 37–53, https://doi.org/10.1016/j.pharmthera.2009.03.015 (2009).
Martinez-Medina, M. et al. Western diet induces dysbiosis with increased E coli in CEABAC10 mice, alters host barrier function favouring AIEC colonisation. Gut 63, 116–124, https://doi.org/10.1136/gutjnl-2012-304119 (2014).
Agus, A. et al. Western diet induces a shift in microbiota composition enhancing susceptibility to Adherent-Invasive E. coli infection and intestinal inflammation. Scientific reports 6, 19032, https://doi.org/10.1038/srep19032 (2016).
Jones, R. L. & Nzekwu, M. M. The effects of body mass index on lung volumes. Chest 130, 827–833, https://doi.org/10.1378/chest.130.3.827 (2006).
Mead, J., Takishima, T. & Leith, D. Stress distribution in lungs: a model of pulmonary elasticity. Journal of applied physiology 28, 596–608, https://doi.org/10.1152/jappl.1970.28.5.596 (1970).
Bates, J. H. & Dixon, A. E. Potential role of the airway wall in the asthma of obesity. Journal of applied physiology (Bethesda, Md.: 1985) 118, 36–41, https://doi.org/10.1152/japplphysiol.00684.2014 (2015).
Raviv, S., Dixon, A. E., Kalhan, R., Shade, D. & Smith, L. J. Effect of obesity on asthma phenotype is dependent upon asthma severity. The Journal of asthma: official journal of the Association for the Care of Asthma 48, 98–104, https://doi.org/10.3109/02770903.2010.534220 (2011).
Kapsali, T., Permutt, S., Laube, B., Scichilone, N. & Togias, A. Potent bronchoprotective effect of deep inspiration and its absence in asthma. Journal of applied physiology (Bethesda, Md.: 1985) 89, 711–720, https://doi.org/10.1152/jappl.2000.89.2.711 (2000).
Yao, Q. et al. Effect of chronic intermittent hypoxia on triglyceride uptake in different tissues. Journal of lipid research 54, 1058–1065, https://doi.org/10.1194/jlr.M034272 (2013).
Liu, W. et al. Mechanism of TH2/TH17-predominant and neutrophilic TH2/TH17-low subtypes of asthma. The Journal of allergy and clinical immunology 139, 1548–1558.e1544, https://doi.org/10.1016/j.jaci.2016.08.032 (2017).
Peebles, R. S. Jr. Is IL-1beta inhibition the next therapeutic target in asthma? The Journal of allergy and clinical immunology 139, 1788–1789, https://doi.org/10.1016/j.jaci.2017.03.018 (2017).
Shore, S. A. et al. Effect of leptin on allergic airway responses in mice. The Journal of allergy and clinical immunology 115, 103–109, https://doi.org/10.1016/j.jaci.2004.10.007 (2005).
McKeever, T. M., Weston, P. J., Hubbard, R. & Fogarty, A. Lung function and glucose metabolism: an analysis of data from the Third National Health and Nutrition Examination Survey. American journal of epidemiology 161, 546–556, https://doi.org/10.1093/aje/kwi076 (2005).
Cardet, J. C., Ash, S., Kusa, T., Camargo, C. A. Jr. & Israel, E. Insulin resistance modifies the association between obesity and current asthma in adults. The European respiratory journal 48, 403–410, https://doi.org/10.1183/13993003.00246-2016 (2016).
Forno, E., Han, Y. Y., Muzumdar, R. H. & Celedon, J. C. Insulin resistance, metabolic syndrome, and lung function in US adolescents with and without asthma. The Journal of allergy and clinical immunology 136, 304–311.e308, https://doi.org/10.1016/j.jaci.2015.01.010 (2015).
Arai, T. et al. Effects of dietary fat energy restriction and fish oil feeding on hepatic metabolic abnormalities and insulin resistance in KK mice with high-fat diet-induced obesity. The Journal of nutritional biochemistry 24, 267–273, https://doi.org/10.1016/j.jnutbio.2012.06.002 (2013).
Larsen, C. M. et al. Interleukin-1-receptor antagonist in type 2 diabetes mellitus. The New England journal of medicine 356, 1517–1526, https://doi.org/10.1056/NEJMoa065213 (2007).
Pakhale, S., Baron, J., Dent, R., Vandemheen, K. & Aaron, S. D. Effects of weight loss on airway responsiveness in obese adults with asthma: does weight loss lead to reversibility of asthma? Chest 147, 1582–1590, https://doi.org/10.1378/chest.14-3105 (2015).
Scott, H. A. et al. Dietary restriction and exercise improve airway inflammation and clinical outcomes in overweight and obese asthma: a randomized trial. Clinical and experimental allergy: journal of the British Society for Allergy and Clinical Immunology 43, 36–49, https://doi.org/10.1111/cea.12004 (2013).
Stenius-Aarniala, B. et al. Immediate and long term effects of weight reduction in obese people with asthma: randomised controlled study. BMJ (Clinical research ed.) 320, 827–832 (2000).
Lancaster, G. I. et al. Evidence that TLR4 Is Not a Receptor for Saturated Fatty Acids but Mediates Lipid-Induced Inflammation by Reprogramming Macrophage Metabolism. Cell metabolism 27, 1096–1110.e1095, https://doi.org/10.1016/j.cmet.2018.03.014 (2018).
Soutiere, S. E. & Mitzner, W. On defining total lung capacity in the mouse. Journal of applied physiology (Bethesda, Md.: 1985) 96, 1658–1664, https://doi.org/10.1152/japplphysiol.01098.2003 (2004).
Scherle, W. A simple method for volumetry of organs in quantitative stereology. Mikroskopie 26, 57–60 (1970).
Misharin, A. V., Morales-Nebreda, L., Mutlu, G. M., Budinger, G. R. & Perlman, H. Flow cytometric analysis of macrophages and dendritic cell subsets in the mouse lung. American journal of respiratory cell and molecular biology 49, 503–510, https://doi.org/10.1165/rcmb.2013-0086MA (2013).
Acknowledgements
N.N.H. and V.Y.P. are supported by NIEHS Grant P50 ES018176 and EPA Agreements 83615201 & 83451001. V.Y.P. is also supported by NHLBI Grants R01 HL128970, HL133100, and HL138932. This publication has not been formally reviewed by EPA. The views expressed in this document are solely those of V.Y.P. and colleagues and do not necessarily reflect those of the Agency. EPA does not endorse any products or commercial services mentioned in this publication. We are grateful to Sobi (Stockholm, Sweden) for their generous gift of anakinra.
Author information
Authors and Affiliations
Contributions
H.Y., M.V., C.G., R.L., S.B., F.R.D., A.N., M.C.M., N.N.H., W.M., V.Y.P. conceived and carried out experiments. J.L., M.K.S., S.B.F., Q.Z. carried out experiments. H.Y., M.V., C.G. and R.L. carried out experiments and performed statistical analysis. All authors were involved in writing the paper and had final approval of the submitted and published versions.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Younas, H., Vieira, M., Gu, C. et al. Caloric restriction prevents the development of airway hyperresponsiveness in mice on a high fat diet. Sci Rep 9, 279 (2019). https://doi.org/10.1038/s41598-018-36651-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-36651-2
This article is cited by
-
Exercise training attenuates pulmonary inflammation and mitochondrial dysfunction in a mouse model of high-fat high-carbohydrate-induced NAFLD
BMC Medicine (2022)
-
IL-33, diet-induced obesity, and pulmonary responses to ozone
Respiratory Research (2020)
-
Caloric restriction attenuates C57BL/6 J mouse lung injury and extra-pulmonary toxicity induced by real ambient particulate matter exposure
Particle and Fibre Toxicology (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
