
Abstract
In this study, novel phosphatidylcholines containing ibuprofen or naproxen moieties were synthesized in good yields and high purities. Under the given synthesis conditions, the attached drug moieties racemized, which resulted in the formation of phospholipid diastereomers. The comperative studies of the cytotoxicity of ibuprofen, naproxen and their phosphatidylcholine derivatives against human promyelocytic leukemia HL-60, human colon carcinoma Caco-2, and porcine epithelial intestinal IPEC-J2 cells were carried out. The results of these studies indicated that phospholipids with NSAIDs at both sn-1 and sn-2 positions (15 and 16) were more toxic than ibuprofen or naproxen themselves, whereas 2-lysophosphatidylcholines (7 and 8) were less toxic against all tested cell lines. Phospholipids with NSAIDs at sn-1 and palmitic acid at sn-2 (9 and 10) were also less toxic against Caco-2 and normal cells (IPEC-J2).
Similar content being viewed by others
Introduction
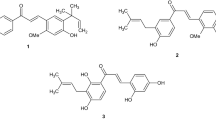
Nonsteroidal anti-inflammatory drugs (NSAIDs) exhibit anti-inflammatory, analgesic, and antipyretic effects1,2 and are currently some of the most widely used drugs in the world. NSAIDs are a large heterogeneous group of compounds and their classification is based mainly on their chemical structure. The most well-known group of NSAIDs are derivatives of 2-arylpropionic acid (profens) such as ibuprofen (1) and naproxen (2) (Fig. 1). The fact that these over-the-counter (OTC) drugs are readily available and relatively inexpensive is what makes them first-line drugs (FLD) for pain relief and inflammation treatment.

Structures of ibuprofen (1) and naproxen (2). IBU and NAP are shortcuts of ibuprofen and naproxen moieties used in the following schemes.
Only S isomers of profens consistently bind to proteins and inhibit prostaglandin synthesis3,4. In-vivo studies have shown that in some animal organisms, including humans, the inactive R enantiomer can be converted to a pharmacologically active S isomer by 2-arylpropionyl-CoA epimerase4,5. Since the synthesis of optically active drugs is costly, the production and sale of racemic mixtures has been approved. However, preparations containing drugs as (S)-isomers are also available, e.g., dexibuprofen and dexketoprofen6,7.
The mechanism of action of all NSAIDs is closely related to the inhibition of the enzyme cyclooxygenase-2 (COX-2). Unfortunately, most drugs, including ibuprofen (1) and naproxen (2), show lack of selectivity and inhibit the cyclooxygenase-1 (COX-1) enzyme, which is involved in maintaining the normal homeostasis of the body8. Therefore, long-term use of ibuprofen (1) or naproxen (2) has negative effects on the gastrointestinal tract (GI), leading to damage of the mucous membrane and consequent bleeding and perforation8,9.
Because of this GI toxicity, there is a clear need to create new generations of NSAIDs. In recent years, many NSAID derivatives have been found to exhibit similar activities but with less side effects than standard drugs. From these, noteworthy are the nitric oxide-releasing prodrugs of NSAIDs (NO-NSAIDs)10,11 and the derivatives of NSAIDs with phosphate moieties (phospho-NSAIDs)12. Moreover, it has been reported that phospho-NSAIDs are safer than normal drugs because of their reduced gastrointestinal toxicity13. In addition to their anti-inflammatory properties, it has been found that phospho-NSAIDs exhibit anticancer properties against colon and breast cancer12,14,15,16.
Among different approaches to improving drug quality, there has been a huge potential for drugs combined with phospholipids17,18,19. One reason for this is that they are non-toxic and biocompatible. Studies that were carried out on animal and human cells indicated that the mixture of NSAIDs and phosphatidylcholine exhibits stronger activity at identical concentrations and lower GI toxicity than NSAIDs alone19,20,21,22. Moreover, phospholipids with linked drugs mediate the transport of compounds and increase their bioavailability23. Our studies showed that compounds with anticancer activity linked covalently with phosphatidylcholine in the place of natural fatty acids lose their toxicity toward normal cells24,25.
In this paper, we report the synthesis of phosphatidylcholines containing ibuprofen (1) or naproxen (2) at sn-1 and/or sn-2 positions. Phosphatidylcholines with ibuprofen have previously been reported26 but herein we applied a different, shorter synthetic pathway. Additionally, the cytotoxicity of the newly synthesized phospholipid derivatives against human promyelocytic leukemia HL-60 cells, human colon carcinoma Caco-2 cells, and porcine epithelial intestinal IPEC-J2 cells was studied.
Results and Discussion
Chemistry
Phosphatidylcholines containing ibuprofen (1) or naproxen (2) moieties at the sn-1 position (7–10) were synthesized according to Fig. 2. In the first steps, the corresponding chlorides 3 and 4, and stannylene acetal (6) were obtained. Acyl chlorides resulted from the reaction of acids 1 and 2 with oxalyl chloride in the presence of catalytic amounts of anhydrous DMF. Then, according to known procedures24,27,28, the reaction of stannylene acetal (6) and acyl chloride (3 or 4) gave the corresponding 2-lysophosphatidylcholine (7 or 8) in good yields (68 and 70%, respectively). In the next step, lysophosphatidylcholines 7 and 8 were esterified with palmitic acid using the Steglich conditions to give phosphatidylcholines 9 and 10, respectively.

Synthesis of 2-lysophosphatidylcholines 7 and 8, and phosphatidylcholines 9 and 10. Reagents and conditions: (a) oxalyl chloride (3 equiv), DMF, CH2Cl2, 2 h; (b) DBTO, 2-propanol, reflux, 2 h; (c) TEA, 30 min; (d) palmitic acid, DMAP, DCC, 72 h.
The structures of lysophosphatidylcholines 7 and 8 were confirmed by NMR spectroscopy, since in the 1H and 13C NMR spectra of the two compounds, all hydrogen and carbon signals from the ibuprofen and naproxen moieties, respectively, were observed. Characteristic signals from the isobutyl group of compound 7 were observed at 0.85 and 0.86 ppm (d, J = 6.7 Hz) for the methyl groups and at 2.41 ppm (d, J = 7.2 Hz) for the methylene group. Moreover, the isobutyl methine proton was observed as a multiplet at δ = 1.81, CH3-3′ was detected as a doublet (J = 7.2 Hz) at 1.46 ppm, and the H-2′ methine proton was identified as a quartet (J = 7.2 Hz) at 3.72 ppm. Additionally, in the 1H NMR spectrum of lysophosphatidylcholine 8 a characteristic singlet of the methoxy group at δ = 3.88 was visible, and the H-2′ and H-2 methine protons overlapped and gave multiplets in the range of 3.89–3.93 ppm (Supplementary Information).
The introduction of palmitic acid into the sn-2 position was confirmed by the appearance of signals from fatty acid residue in the 1H NMR spectra of phosphatidylcholines 9 and 10. Triplets from terminal methyl groups at δ = 0.84 (J = 7.1 Hz) for 9 and at δ = 0.85 (J = 7.0 Hz) for 10 were observed. Additionally, the structures of 9 and 10 were confirmed by the shift in the methine proton H-2 signals from δ = 3.92 (7) to δ = 5.19 (9) and from δ = 3.91 (8) to δ = 5.19 (10), respectively. Moreover, phosphatidylcholines 9 and 10 were obtained as mixtures of diastereomers, which was a result of the enolization of the carboester group with contribution of H-2′ from the ibuprofen or naproxen moiety under mildly alkaline conditions29. However, in acidic solutions, NSAIDs are reportedly highly stable and do not undergo racemization30.
The presence of two diastereomers of phosphocholine 9 was confirmed by the two quartets (J = 7.2 Hz) of H-2′ observed at 3.66 and 3.69 ppm and the two doublets (J = 7.2 Hz) of CH3-3′ at 1.43 and 1.44 ppm in the 1H NMR spectrum. Double signals were also observed for CH2-1 and CH2-3, and in the 13C NMR spectrum: C-1, C-1′, C-2′, C-3′, C-1″′, C-2″′, and C-3″′. The composition of the phosphatidylcholine isomers mixture (9) was determined by integrating one of the CH2-1 protons. Assuming that there was a higher content of phosphatidylcholine with S-ibuprofen, which was the starting enantiomer in the synthesis, the quantitative composition was defined as 77% 2 R,2′S isomer and 23% 2 R,2′R isomer.
Phosphatidylcholine (10) with a naproxen moiety at the sn-1 position and palmitoyl residue at the sn-2 position was also obtained as a mixture of diastereomers. However, a pronounced advantage of the 2 R,2′S isomer was observed in this case. The intensities of the 1H NMR signals from the 2 R,2′R isomer was so low (below 5%) that the percentage composition of the mixture could not be accurately determined. The smaller degree of racemization of the S-naproxen can be explained by the electron-donating effect of the methoxy group on the enol formation process.
Phosphatidylcholines containing palmitoyl residue at the sn-1 position and ibuprofen (12) or naproxen (13) at the sn-2 position were synthesized as depicted in Fig. 3. First, 2-lysophosphatidylcholine 11 was prepared according to a known procedure by reacting stannylene acetal (6) and palmitoyl chloride31. Then, 2–lysophosphatidylcholine 11 was esterified with ibuprofen (1) or naproxen (2) using the Steglich conditions.

Synthesis of phosphatidylcholines 12 and 13. Reagents and conditions: room temperature (a) DBTO, 2-propanol, reflux, 2 h; (b) palmitoyl chloride (2 equiv), TEA, 30 min; (c) S-ibuprofen or S-naproxen (2 equiv), DMAP, DCC, 72 h.
The structures of phosphatidylcholines 12 and 13 were confirmed by NMR spectroscopy. In the 1H NMR spectrum of 12 all signals of the palmitoyl residue were visible. Signals from the isobutyl group were observed at 2.41 ppm (d, J = 7.2 Hz) for the methylene protons, at 1.81 ppm (multiplet) for the methine proton, and at 0.86 ppm (d, J = 6.7 Hz) for the methyl groups. The presence of a multiplet at 5.20 ppm for H-2 indicated the attachment of the ibuprofen molecule onto the sn-2 position. Also, in the range of 7.03–7.17 ppm multiplets of the ibuprofen aromatic protons were visible. In addition, the structure of phosphatidylcholine 13 was confirmed by the presence of the following signals: 1.53 ppm (d, J = 7.1 Hz) from CH3-3′, 3.88 ppm (singlet) from the methoxy group, 5.21 ppm (multiplet) from H-2 and multiplets in the range of 7.08–7.70 ppm from the aromatic protons (Supplementary Information).
Similar to compound 9, phosphatidylcholine 12, was obtained as a mixture of diastereomers with similar quantitative composition: 73% of the 2 R,2′S isomer and 27% of the 2 R,2′R isomer. The presence of two diastereomers of 12 was indicated by the doubled signals for C3′ (17.57 and 17.63 ppm), C-2″′ (33.25 and 33.52 ppm), and C-3″′ (24.14 and 24.77 ppm) in the 13C NMR spectrum. Interestingly, the rest of the carbon atoms from phosphatidylcholine 12 were present in the 13C NMR spectrum as single signals. In contrast to compound 12, phosphatidylcholine 13 containing naproxen at the sn-2 position was detected as one isomer (2 R,2′S).
Phosphatidylcholines containing ibuprofen or naproxen moieties at both sn-1 and sn-2 positions (15 and 16) were synthesized in a one-step synthesis from the highly pure sn-glycero-3-phosphocholine (GPC) and ibuprofen (1) or naproxen (2), respectively (Fig. 4). In order to increase the solubility of GPC, its complex with cadmium chloride (14) was prepared according to a previously published procedure24. In the synthesis of 15 and 16, the Steglich esterification of two free hydroxyl groups in the presence of a fourfold molar excess of ibuprofen (1) or naproxen (2) was applied.

Synthesis of phosphatidylcholines 15 and 16. Reagents and conditions: (a) S-ibuprofen or S-naproxen (4 equiv), DMAP, DCC, 25 °C, N2, 48 h.
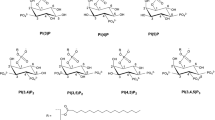
The use of mass spectrometry was highly significant in the determination of the structures of products 15 and 16, since it detected intensive signals with m/z 634.3510 for 15 and m/z 682.2760 for 16. Additionally, the structures of these compounds were confirmed by 1H, 13C, and 31P NMR spectroscopy. In the 1H NMR spectra of 15 and 16, multiplied signals of acid moieties, glycerol, and choline were observed, which suggested the presence of four diastereomers in the product mixtures. Due to the substitution of identical molecules at the sn-1 and sn-2 positions and the racemization of acids bonded to GPC, the interpretation of the spectral data was complicated and very difficult. The racemization caused a formation of four diastereomers for both 15 and 16, respectively (Fig. 5). Unfortunately, the separation of the individual isomers by a chiral HPLC column was impossible.

Diastereoisomers of phosphatidylcholines 15 and 16. Indexes “a” and “b” are assigned to the acid residues at the sn-1 and sn-2 positions, respectively.
When looking at the 1H NMR spectrum of 15 in the range of 1.29–1.43 ppm, the overlapping doublets (J = 7.2 Hz) of CH3-3′a-b could clearly be observed. Their differentiation was caused by the direct attachment of the methyl group to the chiral C-2′a-b carbon atom of the ibuprofen moiety. Moreover, the methine protons of H-2′a-b were detected as multiplets in the range of 3.43–3.69 ppm. The separation of signals was imperceptible due to the large distance from the ibuprofen isobutyl moiety to the stereogenic center. In addition, in the range of 0.84–0.88 ppm doublets (J = 6.6 Hz) from the isobutyl methyl groups were detected and at 3.11, 3.12, 3.15, and 3.16 ppm, four singlets from the choline methyl groups were detected. This indicated the formation of four diastereomers of compound 15. Two-dimensional HSQC spectroscopy was instrumental in elucidating the structure of 15. The measurements detected signals of the CH2-α (δ = 4.11 and 4.19) and CH2-β (δ = 3.49 and 3.55) protons, multiplets of the CH2-1 protons in the range of 3.98–4.42 ppm, and multiplets of the CH2-3 protons in the range of 3.84–3.96 ppm. Multiplets of the methine proton H-2 were detected at δ = 5.15 and 5.20 ppm. In the 13C NMR spectrum of phosphatidylcholine 15, signals of C-3′a-b and C-2′a-b in the ranges of 17.44–17.87 and 44.24–44.58 ppm, respectively, were observed. Multiplied signals of the glycerophosphocholine carbon atoms were detected at 58.54 and 58.60 ppm (C-α, two d, JC-P = 5.1 Hz), as well as in the ranges of 62.29–63.11 ppm (C-1 and C-3) and 65.75–65.90 (C-β). Interestingly, a single doublet (JC-P = 8.0 Hz) from the chiral C-2 carbon atom appeared at 70.21 ppm. The carbon atoms from the aromatic ring did not show any differentiation with exception of C-1″a-b, which was closest to the chiral C-2′a-b carbon atom and gave 8 signals in the range of 136.78–136.97 ppm. The signal of the ester carbon atom C-1′a-b was also multiplied eight times and appeared in the range of 173.86–174.36 ppm. In the 31P NMR spectrum, only one signal from the phosphorus atom of 15 was visible at δ = –1.02.
In the synthesis of phosphatidylcholine 16, a racemization of naproxen was also expected. The 1H NMR spectrum clearly indicated the formation of diastereomers and the multiplication of almost all signals. Signals of CH3-3′a-b appeared as eight doublets (J = 7.2 Hz) in the range of 1.23–1.49 ppm, while H-2′a-b were visible as overlapping quartets (J = 7.2 Hz) at 3.39–3.81 ppm. The signals of the methoxy group were observed at 3.86 and 3.87 ppm as two singlets. Hydrogen atoms from the glycerophosphocholine fragment were also distinctly differentiated. The CH2-1 groups of each diastereomer gave rise to two doublet of doublets (J = 12.0, 7.0 Hz) with signals at 3.98, 4.11, 4.15, and 4.16 ppm from one of the protons, and a doublet of doublets (J = 12.0, 3.0 Hz) with signals at 4.23, 4.33, 4.41, and 4.44 ppm from the second. The CH2-3 groups appeared as two multiplets in the range of 3.80–3.95 ppm. Moreover, at 2.90, 2.91, 3.01, and 3.03 ppm four singlets that were assigned to the choline methyl groups were detected. Particularly interesting was the chemical shift of the first two singlets, because these proton signals have never been observed at such a high field for phosphatidylcholines. The CH2-β methylene groups appeared on the spectrum as two multiplets at 3.08 and 3.36 ppm, and the CH2-α protons appeared as three multiplets in the range of 3.72–3.84 ppm and at 4.05 ppm. The shifts of the N(CH3)3 and CH2-β signals toward the higher field, as well as the differentiation of the CH2-α protons, may indicate the influence of the naproxen moiety from the sn-2 position on the hydrophilic choline part of the molecule. Modeling of the phosphatidylcholine 16 structure showed that this may be the result of the rotation around the C2-C3 bond and the existence of two distinctive rotamers: one with the phosphocholine moiety in syn conformation to the naproxen at the sn-2 position and the second one in the anti. In the first conformation, which was stabilized by the nucleophilic interaction of the –N+(CH3)3 group and naphthalene electrons, the choline moiety was located in the shielding cone of the magnetic field induced by the electrons of the naphthalene ring. In the second conformation, the electrons of the aromatic ring did not affect the chemical shifts of the hydrogen atoms of the choline moiety.
In the 13C NMR spectrum of phosphatidylcholine 16, a differentiation of the signals of C-3′a-b (17.22–17.66 ppm) and C-2′a-b (44.42–44.83 ppm) was observed. A doubling of the signals from the carbon atoms was observed: C-α at 58.35 (d, JC-P = 5.0 Hz) and at 58.59 ppm (d, JC-P = 4.9 Hz), C-β in the range of 65.41–65.77 ppm (two m), and C-3 at 62.96 and 63.03 ppm (two d, JC-P = 5.2 Hz) and at 63.13 and 63.18 ppm (two d, JC-P = 5.6 Hz). In the range of 62.13–62.42 ppm, differentiated signals of the C-1 carbon atom were detected. Signals of the C-1′a-b carbonyl atoms were visible at: 173.73, 173.82, 173.92, 174.09, 174.11, 174.21, and 174.25 ppm. The phosphorus atom was also differentiated, and the signals were registered at −1.01 and −1.19 ppm.
Cytotoxicity of the phosphatidylcholines containing ibuprofen and naproxen moieties
Taking into consideration the idea of the future application of novel phosphatidylcholines containing ibuprofen or naproxen moieties as anti-inflammatory drugs with reduced gastrointestinal toxicity, it is necessary to analyze the cytotoxicity of these derivatives against the sensitive and normal cells. Then, the cytotoxicity of ibuprofen, naproxen, and their phospholipid derivatives was analyzed by a WST-1 cell proliferation assay. Our earlier studies indicated that among different cell lines, human promyelocytic leukemia HL-60 cells are the most sensitive to modified phosphatidylcholines24,25,32. Therefore, HL-60 cells, gastrointestinal origin - human colon cancer Caco-2 cells and normal porcine epithelial intestinal IPEC-J2 cells were used in these studies. Phosphatidylcholine derivatives were dissolved in dimethyl sulfoxide (DMSO) prior to examination. Unfortunately, due to the poor solubility of compound 13 in DMSO, its cytotoxicity was not tested. The solvent did not show cytotoxicity against analyzed cell lines.
In the case of human leukemic HL-60 cells, the experiments revealed that ibuprofen (1) was non-toxic up to concentration of 100 µM. In contrast to ibuprofen (1), 2-lysophosphatidylcholine 7 was non-toxic even at concentration 200 µM (Fig. 6a, Table 1). However, phosphatidylcholines 9, 12 and 15 showed higher cytotoxic activity already at concentration 100 µM. The results of cytotoxicity studies of derivatives of ibuprofen against human colon carcinoma Caco-2 cells indicated that compounds 7, 9 and 12 at 200 µM concentration were less toxic than ibuprofen itself (Fig. 6b, Table 1). Phosphatidylcholines containing ibuprofen moieties at both sn-1 and sn-2 positions (15) showed higher toxicity in comparison to 1. Analysis of results of cytotoxic activity against normal porcine IPEC-J2 cells showed that compounds 7 and 9 were less toxic than ibuprofen (1) (Fig. 6c, Table 1). Phosphatidylcholines with ibuprofen in sn-2 position (12) and with two residues of ibuprofen (15) were more cytotoxic.

Viability of HL-60 cells (a) Caco-2 cells (b) and IPEC-J2 cells (c) grown in the presence of increased concentrations of ibuprofen (1) and phosphatidylcholines that contain ibuprofen in various sn positions (7, 9, 12 and 15). Percentage of viable cells was determined by the WST-1 assay, p < 0.01**, p < 0.001*** as described in Materials and Methods. Control – cells growing in appropriate complete media; DMSO – cells growing in the presence of DMSO.
Incubation of HL-60 cells revealed that naproxen (2) was non-toxic toward these cells up to concentration 100 µM. Among phosphatidylcholines containing naproxen, only compound 8 was significantly less cytotoxic (up to concentration of 200 µM) in comparison to free naproxen (Fig. 7a, Table 2). On the other hand, compound 16, and especially 10 were highly toxic for HL-60 cells. In the case of colon carcinoma Caco-2 cells naproxen (2) was less toxic in comparison to leukemic HL-60 cells. Interestingly, also in contrast to leukemic cells, compound 10 was non-toxic up to concentration of 200 µM against the colon carcinoma cells. These cells were only sensitive to compound 16 (Fig. 7b, Table 2). The activity of phosphatidylcholines 8, 10 and 16 have similar effect for both normal porcine IPEC-2 cells and colon carcinoma Caco-2 cells (Fig. 7b,c, Table 2). Phosphatidylcholines 8 and 10 were less toxic than naproxen (2) towards these cell lines at concentration 200 µM.

Viability of HL-60 cells (a) Caco-2 cells (b) and IPEC-J2 cells (c) grown in the presence of increased concentration of naproxen (2) and phosphatidylcholines that contain naproxen in various sn positions (8, 10 and 16). Percentage of viable cells was determined by the WST-1 assay, p < 0.01**, p < 0.001*** as described in Materials and Methods. Control – cells growing in appropriate complete media; DMSO – cells growing in the presence of DMSO.
The results of these studies showed that phosphatidylcholines containing NSAIDs at both sn-1 and sn-2 positions (15 and 16) were more toxic than ibuprofen or naproxen themselves. 2-Lysophosphatidylcholines (7 and 8) were less toxic against all tested cell lines. Phospholipids with NSAIDs in sn-1 and palmitic acid in sn-2 (9 and 10) were also less toxic against Caco-2 and normal cells (IPEC-J2).
Conclusions
In this study, we synthesized eight phosphatidylcholines with ibuprofen or naproxen in their sn-1, sn-2 or sn-1 and sn-2 positions, and the cytotoxicity of the resulting compounds towards two cancer cells (HL-60 and Caco-2) and normal IPEC-J2 cells was evaluated. The results from the biological studies indicated that 2-lysophosphatidylcholines with NSAIDs at sn-1 position (7 and 8) and phosphatidylcholines with NSAIDs at sn-1 and palmitic acid residue at sn-2 (9 and 10) were less toxic against Caco-2 and normal cells (IPEC-J2). Compounds 7 and 8 were also less toxic than ibuprofen or naproxen against HL-60 cells. These results confirmed our earlier observations that phospholipid derivatives of active compounds are less cytotoxic than the compounds themselves. These data indicate also that phospholipids could be used as carriers of NSAIDs. However, further biological studies are necessary in order to confirm this proposal.
Materials and Methods
Chemicals and Analysis Conditions
sn-Glycero-3-phosphocholine (GPC) was purchased from Bachem (Bubendorf, Switzerland). S-Ibuprofen (1), S-naproxen (2), N,N′-dicyclohexylcarbodiimide (DCC), 4-(N,N-dimethylamino)pyridine (DMAP), dibutyltin(IV) oxide (DBTO), triethylamine (TEA), palmitoyl chloride, oxalyl chloride, Dowex® 50WX8 H+ form (an ion-exchange resin), hexane, methanol, 2-propanol for liquid chromatography, and chloroform (CHROMASOLV®) were purchased from Sigma-Aldrich (Munich, Germany). Chloroform and methanol for column and thin-layer chromatography (TLC) were purchased from Stanlab (Lublin, Poland). TLC was performed on Merck Kieselgel 60 F254 plates (0.2 mm silica gel with fluorescent indicator UV254). As a visualization reagent, primuline spray (0.05% in acetone:water, 80:20, v/v) was used. Visualization was determined using UV light (λ = 365 nm). Column chromatography was performed on silica gel (Kieselgel 60, 0.040–0.063 mm, 230–400 mesh ASTM; Merck, Darmstadt, Germany) using an eluent mixture of CHCl3:CH3OH:H2O (65:25:4, v/v/v). The purity of all final phosphatidylcholines was determined by HPLC to be 96% or higher. HPLC was performed on an UltiMate 3000 apparatus (Dionex, Sunnyvale, CA, USA) with a charged aerosol detector (Corona CAD, ESA Biosciences, Chelmsford, USA). Nitrogen was used as a nebulizing gas at a pressure of 35 psi and at an acquisition range of 100 pA. A BETASIL Diol column (150 × 4.6 mm, 5 µm; Thermo Scientific, MA, USA) was used. The mobile phase was set at a flow rate of 1.5 mL/min and consisted of the following solvents: (A) water, (B) hexane, and (C) 2-propanol. The mobile phase was run as %A:%B:%C (v/v/v) using the following gradient timetable: 0 min - 0:43:57, 5 min -3:40:57, 8 min - 10:40:50, 13 min - 10:40:50, 13.1 min - 0:43:57, and 22 min - 0:43:57. All NMR spectra were recorded on an Avance II 600 MHz spectrometer (Brüker, Billerica, MA, USA) working at a frequency of 600 MHz for 1H, 150 MHz for 13C, and 243 MHz for 31P. Samples of all compounds were measured in a mixture of CDCl3:CD3OD (2:1, v/v). The chemical shifts were calibrated using: the methanol proton signals (δH = 3.31) in the 1H NMR spectra and the CDCl3 carbon atoms (δc = 77.0) in the 13C NMR spectra. H3PO4 (85%) was used as an external standard for the 31P NMR chemical shifts. HRMS spectra were recorded using the ESI technique on a spectrometer (ESI-Q-TOF Premier XE; Waters, Milford, MA, USA). Melting points (MPs, uncorrected) were determined on a Boetius apparatus.
The complexes of cadmium chloride and sn-glycero-3-phosphocholine (14, GPCxCdCl2), and 1-palmitoyl-2-hydroxy-sn-glycero-3-phosphocholine (11) were prepared according to previously reported procedures31,33.
Syntheses
General procedure for the preparation of 2-lysophosphatidylcholines 7 and 8. S-Ibuprofen (1, 0.3 g, 1.46 mmol) or S-naproxen (2, 0.34 g, 1.46 mmol) was dissolved in anhydrous methylene chloride (5 mL), and catalytic amount (2 drops) of anhydrous DMF was added to the mixture. Excess of oxalyl chloride (3 equiv, 376 μL, 4.38 mmol) was added dropwise to the stirring solution. The reaction was carried out at room temperature for 2 h and the solvent and excess of oxalyl chloride were subsequently evaporated. The resulting chloride 3 or 4 was immediately used for the synthesis of 2-lysophosphatidylcholine 7 or 8.
GPC (0.25 g, 0.97 mmol) and DBTO (0.242 g, 0.97 mmol) were suspended in 15 mL of 2-propanol and refluxed for 2 h. The mixture was cooled to room temperature and TEA (0.203 mL, 1.46 mmol) and corresponding chloride 3 or 4 were then added to it. The mixture was stirred for 30 min. After completion of the reaction (checked by TLC) the solvent was evaporated under reduced pressure and the residue was purified by silica gel column chromatography using chloroform:methanol:water (65:25:2 → 65:25:4) to give the corresponding 2-lysophosphatidylcholine (7 or 8).
1-[2′S-(4″-Isobutylphenyl)]propanoyl-2-hydroxy-sn-glycero-3-phosphocholine (7): Colorless, waxy product; yield 68% (0.292 g). 1H NMR (600 MHz, CDCl3:CD3OD, 2:1, v/v) δ: 0.86 (d, J = 6.7 Hz, 6 H, (CH3)2-CH-CH2-), 1.46 (d, J = 7.2 Hz, 3 H, CH3-3′), 1.81 (m, 1 H, (CH3)2-CH-CH2-), 2.41 (d, J = 7.2 Hz, 2 H, (CH3)2-CH-CH2-), 3.17 (s, 9 H, N(CH3)3), 3.58 (m, 2 H, CH2-β), 3.72 (q, J = 7.2 Hz, 1 H, H-2′), 3.81 (ddd, J = 11.0, 7.5, 5.7 Hz, 1 H, one of CH2-3), 3.88 (m, 1 H, one of CH2-3), 3.92 (m, 1 H, H-2), 4.02 (dd, J = 11.5, 6.1 Hz, 1 H, one of CH2-1), 4.18 (dd, J = 11.5, 5.0 Hz, 1 H, one of CH2-1), 4.23 (m, 2 H, CH2-α), 7.04–7.08 (m, 2 H, H-2″ and H-6″), 7.14–7.18 (m, 2 H, H-3″ and H-5″). 13C NMR (150 MHz, CDCl3:CD3OD, 2:1, v/v) δ: 17.85 (C-3′), 21.63 ((CH3)2-CH-CH2-), 29.68 ((CH3)2-CH-CH2-), 44.46 (C-2′), 44.53 ((CH3)2-CH-CH2-), 53.48, 53.51 and 53.53 (N(CH3)3), 58.67 (d, JC-P = 4.7 Hz, C-α), 64.76 (C-1), 65.87 (C-β), 66.34 (d, JC-P = 5.6 Hz, C-3), 68.11 (d, JC-P = 6.6 Hz, C-2), 126.62 (C-3″ and C-5″), 128.82 (C-2″ and C-6″), 137.06 (C-1″), 140.15 (C-4″), 174.80 (C-1′). 31P NMR (243 MHz, CDCl3:CD3OD, 2:1, v/v) δ: –0.34. ESI-MS: m/z calculated for C21H36NO7P [M + H]+: 446.2308. Found: 446.2319.
1-[2′S-(6″-Methoxynaphthalenyl)]propanoyl-2-hydroxy-sn-glycero-3-phosphocholine (8): Light pink powder; yield 70% (0.318 g); mp 162–164 °C. 1H NMR (600 MHz, CDCl3:CD3OD, 2:1, v/v) δ: 1.54 (d, J = 7.2 Hz, 3 H, CH3-3′), 3.08 (s, 9 H, N(CH3)3), 3.44 (m, 2 H, CH2-β), 3.79 (m, 1 H, one of CH2-3), 3.85 (m, 1 H, one of CH2-3), 3.88 (s, 3 H, O-CH3), 3.89–3.93 (m, 2 H, H-2′ and H-2), 4.07 (dd, J = 11.4, 6.1 Hz, 1 H, one of CH2-1′), 4.13 (m, 2 H, CH2-α), 4.18 (dd, J = 11.4, 5.0 Hz, 1 H, one of CH2-1), 7.09–7.13 (m, 2 H, H-5″ and H-7″), 7.37 (m, 1 H, H-10″), 7.64 (s, 1 H, H-2″), 7.67–7.70 (m, 2 H, H-4″ and H-9″). 13C NMR (150 MHz, CDCl3:CD3OD, 2:1, v/v) δ: 17.84 (C-3′), 44.85 (C-2′), 53.41, 53.44 and 53.46 (N(CH3)3), 54.68 (O-CH3), 58.55 (d, JC-P = 5.0 Hz, C-α), 64.75 (C-1), 65.80 (C-β), 66.24 (d, JC-P = 5.7 Hz, C-3), 68.06 (d, JC-P = 6.7 Hz, C-2), 105.14 (C-7″), 118.47 (C-5″), 125.41 (C-2″), 125.72 (C-10″), 126.72 and 128.77 (C-4″ and C-9″), 128.48 (C-3″), 133.30 (C-8″), 135.12 (C-1″), 157.24 (C-6″), 174.63 (C-1′). 31P NMR (243 MHz, CDCl3:CD3OD, 2:1, v/v) δ: –0.30. ESI-MS: m/z calculated for C22H32NO8P [M + H]+: 470.1944. Found: 470.1961.
General procedure for the preparation of phosphatidylcholines 9 and 10. Lysophosphatidylcholine 7 (0.089 g, 0.2 mmol) or 8 (0.094 g, 0.2 mmol) and DMAP (0.025 g, 0.2 mmol) were dissolved in dry methylene chloride (5 mL). The, a mixture of palmitic acid (2 equiv, 0.103 g, 0.4 mmol) and DCC (3 equiv, 0.124 g, 0.6 mmol) in the same solvent (5 mL) was added. The reaction was stirred for 72 h at room temperature. The suspension was filtered off and stirred with Dowex® resin (50WX8 H+) for 30 min. The resin was then filtered off and washed with a Folch mixture (15 mL). The solvent was evaporated and the crude product was purified by column chromatography on silica gel (CHCl3:CH3OH:H2O, 65:25:4) to give the corresponding phosphatidylcholines 9 or 10. The yields and the physical and spectroscopic data of the products are given below.
1-[2′-(4″-Isobutylphenyl)]propanoyl-2-palmitoyl-sn-glycero-3-phosphocholine (9): Colorless waxy product; yield 25% (0.034 g). 1H NMR (600 MHz, CDCl3:CD3OD, 2:1, v/v) δ: 0.84 (t, J = 7.1 Hz, 3 H, CH3-16″′), 0.86 (d, J = 6.6 Hz, 6 H, (CH3)2-CH-CH2-), 1.22–1.26 (m, 24 H, CH2-4″′-CH2-15″′), 1.43 and 1.44 (two d, J = 7.2 Hz, 3 H, CH3-3′), 1.47–1.55 (m, 2 H, CH2-3″′), 1.80 (m, 1 H, (CH3)2-CH-CH2-), 2.15 and 2.20 (two dt, J = 16.0, 7.6 Hz, 2 H, CH2-2″′), 2.40 (d, J = 7.2 Hz, 2 H, (CH3)2-CH-CH2-), 3.16 (s, 9 H, N(CH3)3), 3.55 (m, 2 H, CH2-β), 3.66 and 3.69 (two q, J = 7.2 Hz, 1 H, H-2′), 3.91–3.99 (two m, 2 H, CH2-3), 4.03 and 4.09 (two dd, J = 12.0, 7.3 Hz, 1 H, one of CH2-1), 4.20 (m, 2 H, CH2-α), 4.25 and 4.38 (two dd, J = 12.0, 3.1 Hz, 1 H, one of CH2-1), 5.19 (m, 1 H, H-2), 7.03–7.06 (m, 2 H, H-2″ and H-6″), 7.11–7.15 (m, 2 H, H-3″ and H-5″). 13C NMR (150 MHz, CDCl3:CD3OD, 2:1, v/v) δ: 13.44 (C-16″′), 17.76 (C-3′), 21.73 ((CH3)2-CH-CH2-), 24.36 (C-3″′), 28.68–31.46 (C-4″′-C-15″′), 29.76 ((CH3)2-CH-CH2-), 33.63 (C-2″′), 44.57 (C-2′), 44.59 ((CH3)2-CH-CH2-), 53.58, 53.60 and 53.63 (N(CH3)3), 58.67 (d, JC-P = 4.6 Hz, C-α), 62.65 (C-1), 63.28 (d, JC-P = 4.9 Hz, C-3), 65.99 (C-β), 69.79 (d, JC-P = 7.6 Hz, C-2), 126.68 (C-3″ and C-5″), 128.88 (C-2″ and C-6″), 136.94 (C-1″), 140.24 (C-4″), 173.10 (C-1″′), 174.43 (C-1′). 31P NMR (243 MHz, CDCl3:CD3OD, 2:1, v/v) δ: –0.76. ESI-MS: m/z calculated for C37H66NO8P [M + H]+: 684.4604. Found: 684.4619.
1-[2′-(6″-Methoxynaphthalenyl)]propanoyl-2-palmitoyl-sn-glycero-3-phosphocholine (10): Colorless waxy product; yield 57% (0.081 g). 1H NMR (600 MHz, CDCl3:CD3OD, 2:1, v/v) δ: 0.85 (t, J = 7.0 Hz, 3 H, CH3-16″′), 1.18–1.27 (m, 24 H, CH2-4″′-CH2-15″′), 1.35–1.41 (m, 2 H, CH2-3″′), 1.53 (d, J = 7.2 Hz, 3 H, CH3-3′), 1.98 (dt, J = 15.6, 7.6 Hz, 1 H, one of CH2-2″′), 2.07 (dt, J = 15.6, 7.6 Hz,1 H, one of CH2-2″′), 3.12 (s, 9 H, N(CH3)3), 3.47 (m, 2 H, CH2-β), 3.84 (q, J = 7.2 Hz, 1 H, H-2′), 3.88 (s, 3 H, O-CH3), 3.90–3.93 (m, 2 H, CH2-3), 4.14 (m, 2 H, CH2-α), 4.16 (dd, J = 12.0, 7.1 Hz, 1 H, one of CH2-1), 4.38 (dd, J = 12.0, 3.1 Hz, 1 H, one of CH2-1), 5.19 (m, 1 H, H-2), 7.09–7.12 (m, 2 H, H-5″ and H-7″), 7.34 (m, 1 H, H-10″), 7.61 (s, 1 H, H-2″), 7.66–7.69 (m, 2 H, H-4″ and H-9″). 13C NMR (150 MHz, CDCl3:CD3OD, 2:1, v/v) δ: 13.33 (C-16″′), 17.63 (C-3′), 24.17 (C-3″′), 22.12–31.39 (C-4″′-C-15″′), 33.43 (C-2″′), 44.83 (C-2′), 53.41, 53.43 and 53.46 (N(CH3)3), 54.61 (O-CH3), 58.54 (d, JC-P = 4.9 Hz, C-α), 62.60 (C-1), 63.09 (d, JC-P = 5.3 Hz, C-3), 65.82 (C-β), 69.64 (d, JC-P = 7.8 Hz, C-2), 105.08 (C-7″), 118.50 (C-5″), 125.40 (C-2″), 125.62 (C-10″), 126.69 and 128.72 (C-4″ and C-9″), 128.46 (C-3″), 133.30 (C-8″), 134.89 (C-1″), 157.25 (C-6″), 173.00 (C-1″′), 174.27 (C-1′). 31P NMR (243 MHz, CDCl3:CD3OD, 2:1, v/v) δ: –0.82. ESI-MS: m/z calculated for C38H62NO9P [M + H]+: 708.4240. Found: 708.4233.
General procedure for the preparation of phosphatidylcholines 12 and 13. To a solution of PLPC (11, 0.2 g, 0.4 mmol) and DMAP (0.049 g, 0.4 mmol) in anhydrous methylene chloride (5 mL), a mixture of S-ibuprofen (1, 0.163 g, 0.8 mmol) or S-naproxen (2, 0.184 g, 0.8 mmol) and DCC (0.165 g, 0.8 mmol) in the same solvent (3 mL) was added. The reaction was stirred for 72 h at room temperature. The resulting precipitate was filtered off and a Dowex® 50WX8 H+ form was added. After 30 min of stirring, the resin was filtered off on a Shott funnel and washed with a Folch mixture (15 mL). Products 12 and 13 were separated by column chromatography (CHCl3:CH3OH:H2O, 65:25:4).
1-Palmitoyl-2-[2′-[4″-isobutylphenyl)]propanoyl-sn-glycero-3-phosphocholine (12): Colorless waxy product; yield 41% (0.113 g). 1H NMR (600 MHz, CDCl3:CD3OD, 2:1, v/v) δ: 0.85 (t, J = 7.1 Hz, 3 H, CH3-16″′), 0.86 (d, J = 6.6 Hz, 6 H, (CH3)2-CH-CH2-), 1.23–1.26 (m, 24 H, CH2-4″′-CH2-15″′), 1.45 (m, 1 H, one of CH2-3″′), 1.44 (d, J = 7.1 Hz, CH3-3′), 1.53 (m, 1 H, one of CH2-3″′), 1.81 (m, 1 H, (CH3)2-CH-CH2-), 2.02–2.31 (two m, 2 H, CH2-2″′), 2.41 (d, J = 7.2 Hz, 2 H, (CH3)2-CH-CH2-), 3.19 (s, 9 H, N(CH3)3), 3.52 and 3.58 (m, 2 H, CH2-β), 3.67 and 3.71 (two q, J = 7.1 Hz, 1 H, H-2′), 3.91–3.99 (two m, 2 H, CH2-3), 4.05 and 4.11 (two dd, J = 11.9, 7.5 Hz, 1 H, one of CH2-1), 4.22 (m, 2 H, CH2-α), 4.26 and 4.39 (two dd, J = 11.9, 3.1 Hz, 1 H, one of CH2-1), 5.20 (m, 1 H, H-2), 7.03–7.08 (m, 2 H, H-2″ and H-6″), 7.12–7.17 (m, 2 H, H-3″ and H-5″). 13C NMR (150 MHz, CDCl3:CD3OD, 2:1, v/v) δ: 13.29 (C-16″′), 17.57 (C-3″), 21.59 ((CH3)2-CH-CH2-), 24.14 (C-3″′), 28.57–31.37 (C-4″′-C-15″′), 29.68 ((CH3)2-CH-CH2-), 33.25 (C-2″′), 44.44 and 44.48 (C-2′ and (CH3)2-CH-CH2-), 53.44, 53.46 and 53.49 (N(CH3)3), 58.60 (C-α), 62.03 (C-1), 63.19 (d, JC-P = 4.6 Hz, C-3), 65.86 (C-β), 70.21 (d, JC-P = 6.3 Hz, C-2), 126.56 (C-3″ and C-5″), 128.75 (C-2″ and C-6″), 136.97 (C-1″), 140.07 (C-4″), 173.35 (C-1″′), 174.10 (C-1′). 31P NMR (243 MHz, CDCl3:CD3OD, 2:1, v/v) δ: –0.78. ESI-MS: m/z calculated for C37H66NO8P [M + H]+: 684.4604. Found: 684.4617.
1-Palmitoyl-2-[2′S-(6″-methoxynaphthalenyl)]propanoyl-sn-glycero-3-phosphocholine (13): Colorless waxy product; yield 53% (0.152 g). 1H NMR (600 MHz, CDCl3:CD3OD, 2:1, v/v) δ: 0.85 (t, J = 6.8 Hz, 3 H, CH3-16″′), 0.98–1.24 (multiplets, 24 H, CH2-4″′-CH2-15″′), 1.25–1.27 (m, 2 H, CH2-3″′), 1.53 (d, J = 7.1 Hz, 3 H, CH3-3′), 1.79 and 1.84 (two dt, J = 15.6, 7.6 Hz, 2 H, CH2-2″′), 3.13 (s, 9 H, N(CH3)3), 3.46 (m, 2 H, CH2-β), 3.85 (m, 1 H, H-2′), 3.88 (s, 3 H, O-CH3), 3.95–3.99 (m, 2 H, CH2-3), 4.06 (dd, J = 11.9, 7.5 Hz, 1 H, one of CH2-1), 4.13 (m, 2 H, CH2-α), 4.24 (dd, J = 11.9, 3.1 Hz, 1 H, one of CH2-1), 5.21 (m, 1 H, H-2), 7.08–7.12 (m, 2 H, H-5″ and H-7″), 7.36 (m, 1 H, H-10″), 7.63 (s, 1 H, H-2″), 7.65–7.69 (m, 2 H, H-4″ and H-9″). 13C NMR (150 MHz, CDCl3:CD3OD, 2:1, v/v) δ: 13.42 (C-16″′), 17.56 (C-3′), 24.04 (C-3″′), 22.16–31.48 (C-4″′-C-15″′), 33.14 (C-2″′), 44.88 (C-2′), 53.50, 53.52 and 53.55 (N(CH3)3), 54.68 (O-CH3), 58.52 (d, JC-P = 4.9 Hz, C-α), 62.00 (C-1), 63.24 (d, JC-P = 5.2 Hz, C-3), 65.87 (C-β), 70.44 (d, JC-P = 7.8 Hz, C-2), 105.09 (C-7″), 118.54 (C-5″), 125.40 (C-2″), 125.72 (C-10″), 126.68 and 128.77 (C-4″ and C-9″), 128.48 (C-3″), 133.31 (C-8″), 134.99 (C-1″), 157.29 (C-6″), 173.34 and 173.99 (C-1′ and C-1″′). 31P NMR (243 MHz, CDCl3:CD3OD, 2:1, v/v) δ: –0.73. ESI-MS: m/z calculated for C38H62NO9P [M + H]+: 708.4240. Found: 708.4230.
General procedure for the synthesis of compounds 15 and 16. The GPC × CdCl2 complex (14, 0.2 g, 0.46 mmol) and DMAP (0.056 g, 0.46 mmol) were dissolved in anhydrous methylene chloride (5 mL). To this solution, a solution of DCC (0.380 g, 1.84 mmol) and S-ibuprofen (1, 0.380 g, 1.84 mmol) or S-naproxen (2, 0.424 g, 1.84 mmol) in anhydrous methylene chloride (5 mL) was added. The mixture was stirred for 48 h at 25 °C under nitrogen atmosphere and the progress of the reaction was monitored by TLC. The precipitated dicyclohexylurea was then filtered off and a Dowex® 50WX8 H+ form was added to the mixture. The solution was stirred for 30 min, after which the resin was filtered off on a Shott funnel. Then, the resin was washed with 15 mL of Folch mixture (CHCl3:CH3OH, 2:1) and the solvent was evaporated in vacuo. The residues were purified by column chromatography using a mixture of chloroform/methanol/water (65:25:4) as eluent to give a colorless waxy product 15 or 16.
1,2-Di-[2′-(4″-isobutylphenyl)]propanoyl-sn-glycero-3-phosphocholine (15): Colorless waxy product; yield 61% (0.177 g). 1H NMR (600 MHz, CDCl3:CD3OD, 2:1, v/v) δ: 0.84–0.88 (doublets, J = 6.6 Hz, 12 H, (CH3)2-CH-CH2-), 1.29–1.43 (doublets, J = 7.2 Hz, 6 H, CH3-3′a-b), 1.76–1.85 (two m, 2 H, 2 × (CH3)2-CH-CH2-), 2.39–2.43 (m, 4 H, (CH3)2-CH-CH2-), 3.11, 3.12, 3.15 and 3.16 (four s, 9 H, N(CH3)3), 3.43–3.69 (multiplets, 2 H, H-2′a-b), 3.49 and 3.55 (two m, 2 H, CH2-β), 3.84–3.96 (two m, 2 H, CH2-3), 3.98, 4.08–4.29, 4.35 and 4.42 (multiplets, 2 H, CH2-1), 4.11 and 4.19 (two m, 2 H, CH2-α), 5.15 and 5.20 (two m, 1 H, H-2), 7.02–7.16 (two m, 8 H, H-2″a-b, H-3″a-b, H-5″a-b and H-6″a-b). 13C NMR (150 MHz, CDCl3:CD3OD, 2:1, v/v) δ: 17.44, 17.51, 17.60, 17.66, 17.69, 17.78 and 17.87 (C-3′a-b), 21.58 ((CH3)2-CH-CH2-), 29.65 ((CH3)2-CH-CH2-), 44.24–44.58 (C-2′a-b and (CH3)2-CH-CH2-), 53.45 and 53.47 (N(CH3)3), 58.54 and 58.60 (dwa d, JC-P = 5.1 Hz,C-α), 62.29–63.11 (C-1 and C-3), 65.75–65.90 (C-β), 70.21 (d, JC-P = 8.0 Hz, C-2), 126.58, 126.60, 126.62 and 126.66 (C-3″a-b and C-5″a-b), 128.74, 128.76, 128.78 and 128.80 (C-2″a-b and C-6″a-b), 136.78, 136.80, 136.84, 136.85, 136.89, 136.92, 136.95 and 136.97 (C-1″a-b), 140.06, 140.10, 140.13 and 140.15 (C-4″a-b), 173.86, 173.94, 173.97, 174.05, 174.23, 174.25, 174.33 and 174.36 (C-1′a-b). 31P NMR (243 MHz, CDCl3:CD3OD, 2:1, v/v) δ: –1.02. ESI-MS: m/z calculated for C34H52NO8P [M + H]+: 634.3509. Found: 634.3510.
1,2-Di-[2′-(6″-methoxynaphthalenyl)]propanoyl-sn-glycero-3-phosphocholine (16): Colorless waxy product; yield 72% (0.225 g). 1H NMR (600 MHz, CDCl3:CD3OD, 2:1, v/v) δ: 1.23, 1.29, 1.33, 1.35, 1.42, 1.44, 1.48 and 1.49 (eight d, J = 7.2 Hz, 6 H, CH3-3′a-b), 2.90, 2.91, 3.01 and 3.03 (four s, 9 H, N(CH3)3), 3.08 and 3.36 (two m, 2 H, CH2-β), 3.39, 3.41, 3.54, 3.56 and 3.72–3.81 (quartets, J = 7.2 Hz, 2 H, H-2′a-b), 3.72–3.84 and 4.05 (multiplets, 2 H, CH2-α), 3.80–3.95 (two m, 2 H, CH2-3), 3.86 and 3.87 (two s, 6 H, O-CH3), 3.98, 4.11, 4.15 and 4.16 (four dd, J = 12.0, 7.0 Hz, 1 H, one of CH2-1), 4.23, 4.33, 4.41 and 4.44 (four dd, J = 12.0, 3.0 Hz, 1 H, one of CH2-1′a-b), 5.14 and 5.21 (two m, 1 H, H-2), 7.05–7.69 (multiplets, 12 H, H-2″a-b, H-4″a-b, H-5″a-b, H-7″a-b, H-9″a-b and H-10″a-b). 13C NMR (150 MHz, CDCl3:CD3OD, 2:1, v/v) δ: 17.22, 17.35, 17.41, 17.46, 17.57 and 17.66 (C-3′a-b), 44.42, 44.47, 44.70, 44.79 and 44.83 (C-2′a-b), 53.30 and 53.40 (N(CH3)3), 54.55, 54.57 and 54.61 (O-CH3a-b), 58.35 (d, JC-P = 5.0 Hz, C-α), 58.59 (d, JC-P = 4.9 Hz, C-α), 62.13, 62.40 and 62.42 (C-1), 62.96 and 63.03 (two d, JC-P = 5.2 Hz, C-3), 63.13 and 63.18 (two d, JC-P = 5.6 Hz, C-3), 65.41–65.77 (m, C-β), 70.24–70.47 (m, C-2), 105.04, 105.07 and 105.10 (C-7″a-b), 118.34, 118.43 and 118.45 (C-5″a-b), 125.27–125.74 (C-2″a-b and C-10″a-b), 126.56–128.78 (C-4″a-b and C-9″a-b), 128.32, 128.34, 128.43 and 128.45 (C-3″a-b), 133.20, 133.28 and 133.30 (C-8″a-b), 134.72, 134.87, 134.88, 134.92, 134.95 and 135.00 (C-1″a-b), 157.15, 157.21 and 157.23 (C-6″a-b), 173.73, 173.81, 173.82, 173.92, 174.09, 174.11, 174.21 and 174.25 (C-1′a-b). 31P NMR (243 MHz, CDCl3:CD3OD, 2:1, v/v) δ: –1.01 and –1.19. ESI-MS: m/z calculated for C36H44NO10P [M + H]+: 682.2781. Found: 682.2760.
Cells and cell culture
Human promyelocytic leukemia HL-60 cells and human colon cancer cell line Caco-2 were obtained from the American Type Culture Collection (Manassas, VA, USA). The HL-60 cells were cultured in an RPMI medium supplemented with 10% fetal bovine serum (FBS, Cytogen), 2 mM L-glutamine, and antibiotics (complete RPMI). The Caco-2 cells were cultured in an DMED medium supplemented with 20% fetal bovine serum (FBS, Cytogen), 2 mM glutamine and antibiotics (complete DMED). Intestinal epithelial IPEC-J2 cells from newborn porcine, a kind gift from Prof. P. Schierack (Institute of Biotechnology, Faculty of Environment and Natural Sciences, Brandenburg University of Technology Cottbus-Senftenberg, Senftenberg, Germany)34 were cultured in DMEM medium supplemented with 10% fetal bovine serum (FBS, Invitrogen, Carlsbad, USA), 2 mM glutamine and antibiotics (complete DMEM).
WST-1 cell proliferation assay
Cells (4 × 103/well) were grown for 24 h in 96-well plates (Sarstedt, Germany) in appropriate complete medium at 37 °C and atmosphere containing 5% CO2. Next day, increased concentrations of analyzed compounds (50, 100, 200 μM) dissolved in dimethyl sulfoxide (DMSO, Chempur, Poland) were added to cells growing in successive wells, and cell cultures were continued for 72 h. Due to the poor solubility of compound 13 in DMSO, its cytotoxicity was not tested. After indicated periods of time, WST-1 mixture was added to each well, and the cells were cultured for additional 6 h. The absorbance at 450 nm was measured on an EnSpire 2300 Multilabel Reader (Perkin-Elmer, USA), and data were analyzed using the program EnSpire 9.0 (Perkin-Elmer, USA). The experiments were performed in triplicates and repeated at least three times independently. Results were presented as mean IC50 (concentration of the tested compound, that inhibits cell proliferation by 50%) ± standard deviation. IC50 values were calculated using Cheburator 0.4 software35.
Statistical analysis
All statistical analyses were performed using Prism 5.0 (GraphPad, La Jolla, CA, USA). To statistically evaluate the significance of differences between cytotoxicity of reference drugs (ibuprofen and naproxen) and phosphatidylcholines containing ibuprofen or naproxen moieties non-parametric two-way Annova and multicomparsion Bonfferoni post-test was performed. The p-value significance threshold was set at 0.01 due to relatively low number of measured replicates. In all analyses, the results were considered statistically significant when p < 0.01.
References
Furst, D. E., Ulrich, R. W. & Prakash, S. In Basic and Clinical Pharmacology 636–642 (McGraw-Hill, 2012).
Shah, S. & Mehta, V. Controversies and Advances in Non-Steroidal Anti-Inflammatory Drug (NSAID) Analgesia in Chronic Pain Management. Postgrad. Med. J. 88, 73–78 (2012).
Brune, K. & Hinz, B. The Discovery and Development of Antiinflammatory Drugs. Arthritis Rheum. 50, 2391–2399 (2004).
Evans, A. M. Pharmacodynamics and Pharmacokinetics of the Profens: Enantioselectivity, Clinical Implications, and Special Reference to S(+)-ibuprofen. J. Clin. Pharmacol. 36, 7–15 (1996).
Reichel, C., Brugger, R., Bang, H., Geisslinger, G. & Brune, K. Molecular Cloning and Expression of a 2-Arylpropionyl-Coenzyme A Epimerase: A Key Enzyme in the Inversion Metabolism of Ibuprofen. Mol. Pharmacol. 51, 576–582 (1997).
Jin, S. G. et al. Mechanical Properties, Skin Permeation and In Vivo Evaluations of Dexibuprofen-Loaded Emulsion Gel for Topical Delivery. Arch. Pharm. Res. 38, 216–222 (2015).
Miranda, H. F., Sierralta, F., Aranda, N., Noriega, V. & Prieto, J. C. Pharmacological Profile of Dexketoprofen in Orofacial Pain. Pharmacol. Reports 68, 1111–1114 (2016).
Bjarnason, I. & Takeuchi, K. Intestinal Permeability in the Pathogenesis of NSAID-Induced Enteropathy. J. Gastroenterol. 44, 23–29 (2009).
Bjarnason, I., Hayllar, J., Macpherson, A. J. & Russell, S. Side Effects of Nonsteroidal Anti-Inflammatory Drugs on the Small and Large Intestine in Humans. Gastroenterology 104, 1832–1847 (1993).
Wallace, J. L. & Soldato, P. Del. The Therapeutic Potential of NO-NSAIDs. Fundam. Clin. Pharmacol. 17, 11–20 (2003).
Cheng, H. et al. Effects of Nitric Oxide-Releasing Nonsteroidal Anti-Inflammatory Drugs (NONO-NSAIDs) on Melanoma Cell Adhesion. Toxicol. Appl. Pharmacol. 264, 161–166 (2012).
Wong, C. C. et al. Phospho-NSAIDs Have Enhanced Efficacy in Mice Lacking Plasma Carboxylesterase: Implications for their Clinical Pharmacology. Pharm. Res. 32, 1663–1675 (2015).
Huang, L. et al. The Novel Phospho-Non-Steroidal Anti-Inflammatory Drugs, OXT-328, MDC-22 and MDC-917, Inhibit Adjuvant-Induced Arthritis in Rats. Br. J. Pharmacol. 162, 1521–1533 (2011).
MacKenzie, G. G. et al. Phospho-Sulindac (OXT-328), a Novel Sulindac Derivative, Is Safe and Effective in Colon Cancer Prevention in Mice. Gastroenterology 139, 1320–1332 (2010).
Sun, Y. et al. Phospho-Ibuprofen (MDC-917) Suppresses Breast Cancer Growth: An Effect Controlled by the Thioredoxin System. Breast Cancer Res. 14, R20 (2012).
Huang, L. et al. Chemotherapeutic Properties of Phospho-Nonsteroidal Anti-Inflammatory Drugs, a New Class of Anticancer Compounds. Cancer Res. 71, 7617–7627 (2011).
Lim, Y. J., Dial, E. J. & Lichtenberger, L. M. Advent of Novel Phosphatidylcholine-Associated Nonsteroidal Anti-Inflammatory Drugs with Improved Gastrointestinal Safety. Gut Liver 7, 7–15 (2013).
Lichtenberger, L. M. & Romero, J. J. & Dial, E. J. Gastrointestinal Safety and Therapeutic Efficacy of Parenterally Administered Phosphatidylcholine-Associated Indomethacin in Rodent Model Systems. Br. J. Pharmacol. 157, 252–257 (2009).
Kurinets, A. & Lichtenberger, L. M. Phosphatidylcholine-Associated Aspirin Accelerates Healing of Gastric Ulcers in Rats. Dig. Dis. Sci. 43, 786–790 (1998).
Lichtenberger, L. M. & Barron, M. & Marathi, U. Association of Phosphatidylcholine and NSAIDs as a Novel Strategy to Reduce Gastrointestinal Toxicity. Drugs of Today 45, 877–890 (2009).
Anand, B. S., Jim, J. J., Sanduja, S. K. & Lichtenberger, L. M. Phospholipid Association Reduces the Gastric Mucosal Toxicity of Aspirin in Human Subjects. Am. J. Gastroenterol. 94, 1818–1822 (1999).
Lichtenberger, L. M. et al. Nonsteroidal Anti-Inflammatory Drug and Phospholipid Prodrugs: Combination Therapy with Antisecretory Agents in Rats. Gastroenterology 111, 990–995 (1996).
Kohli, A. G., Kierstead, P. H., Venditto, V. J., Walsh, C. L. & Szoka, F. C. Designer Lipids for Drug Delivery: From Heads to Tails. J. Control. Release 190, 274–287 (2014).
Kłobucki, M. et al. Syntheses and Antiproliferative Activities of Novel Phosphatidylcholines Containing Dehydroepiandrosterone Moieties. Steroids 118, 109–118 (2017).
Niezgoda, N. et al. Phosphatidylcholine with cis-9, trans-11 and trans-10, cis-12 Conjugated Linoleic Acid Isomers: Synthesis and Cytotoxic Studies. Aust. J. Chem. 68, 1065–1075 (2015).
Kurz, M. & Scriba, G. K. E. Drug-Phospholipid Conjugates as Potential Prodrugs: Synthesis, Characterization, and Degradation by Pancreatic Phospholipase A2. Chem. Phys. Lipids 107, 143–157 (2000).
Gliszczyńska, A. et al. Synthesis and Biological Evaluation of Novel Phosphatidylcholine Analogues Containing Monoterpene Acids as Potent Antiproliferative Agents. PLoS One 11, 1–18 (2016).
Gliszczyńska, A., Niezgoda, N., Gładkowski, W., Świtalska, M. & Wietrzyk, J. Isoprenoid-phospholipid conjugates as potential therapeutic agents: Synthesis, characterization and antiproliferative studies. PLoS One 12, 1–27 (2017).
Ammazzalorso, A. et al. Asymmetric Synthesis of Arylpropionic Acids and Aryloxy Acids by Using Lactamides as Chiral Auxiliaries. European J. Org. Chem. 2, 4088–4091 (2006).
Yuchun, X., Huizhou, L. & Jiayong, C. Kinetics of base catalyzed racemization of ibuprofen enantiomers. Int. J. Pharm. 196, 21–26 (2000).
Fasoli, E. et al. Tin-Mediated Synthesis of Lyso-Phospholipids. Org. Biomol. Chem. 4, 2974–2978 (2006).
Niezgoda, N., Mituła, P., Kempińska, K., Wietrzyk, J. & Wawrzeńczyk, C. Synthesis of phosphatidylcholine with conjugated linoleic acid and studies on its cytotoxic activity. Aust. J. Chem. 66, 354–361 (2013).
Smuga, D. A., Smuga, M., Świzdor, A., Panek, A. & Wawrzeńczyk, C. Synthesis of Dehydroepiandrosterone Analogues Modified with Phosphatidic Acid Moiety. Steroids 75, 1146–1152 (2010).
Schierack, P. et al. Characterization of a porcine intestinal epithelial cell line for in vitro studies of microbial pathogenesis in swine. Histochem. Cell Biol. 125, 293–305 (2006).
Nevozhay, D. Cheburator software for automatically calculating drug inhibitory concentrations from in vitro screening assays. PLoS One 9, e106186 (2014).
Acknowledgements
Publication supported by Wroclaw Centre of Biotechnology, programme The Leading National Research Centre (KNOW) for years 2014–2018 (http://know.wroc.pl).
Author information
Authors and Affiliations
Contributions
M.K., A.U., B.K., M.U. and C.W. designed the experiments. M.K., A.U. and B.K. performed the experiments. M.K., G.M. and G.K. performed the HPLC and LC-MS analysis. M.K., A.U., A.G. and C.W. analyzed the data. M.K., A.G., M.U. and C.W. wrote the paper. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kłobucki, M., Urbaniak, A., Grudniewska, A. et al. Syntheses and cytotoxicity of phosphatidylcholines containing ibuprofen or naproxen moieties. Sci Rep 9, 220 (2019). https://doi.org/10.1038/s41598-018-36571-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-36571-1
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
