
Abstract
Most prostate cancer patients will progress to a castration-resistant state (CRPC) after androgen ablation therapy and despite the development of new potent anti-androgens, like enzalutamide (ENZ), which prolong survival in CRPC, ENZ-resistance (ENZR) rapidly occurs. Re-activation of the androgen receptor (AR) is a major mechanism of resistance. Interrogating our in vivo derived ENZR model, we discovered that transcription factor STAT3 not only displayed increased nuclear localization but also bound to and facilitated AR activity. We observed increased STAT3 S727 phosphorylation in ENZR cells, which has been previously reported to facilitate AR binding. Strikingly, ENZR cells were more sensitive to inhibition with STAT3 DNA-binding inhibitor galiellalactone (GPA500) compared to CRPC cells. Treatment with GPA500 suppressed AR activity and significantly reduced expression of Cyclin D1, thus reducing cell cycle progression into S phase and hindering cell proliferation. In vivo, GPA500 reduced tumor volume and serum PSA in ENZR xenografts. Lastly, the combination of ENZ and GPA500 was additive in the inhibition of AR activity and proliferation in LNCaP and CRPC cells, providing rationale for combination therapy. Overall, these results suggest that STAT3 inhibition is a rational therapeutic approach for ENZR prostate cancer, and could be valuable in CRPC in combination with ENZ.
Similar content being viewed by others

Introduction
With 1 in 8 Canadian males expected to be diagnosed with prostate cancer (PCa) in their lifetime and 23,600 new cases in 2014 in Canada alone, PCa is the most diagnosed cancer among Canadian males1. Despite early detection and localized surgery, cancer recurs in a significant number of patients and Androgen Deprivation Therapy (ADT) is used to block the growth-promoting effects of the Androgen Receptor (AR) and decrease tumor burden in this population. Though initially effective, the tumors eventually become castration resistant (CRPC) by replenishing AR activity2. Consequently, further use of AR Pathway Inhibitors (ARPI) is the cornerstone of CRPC treatment developed over the last decade. Enzalutamide (ENZ), a second generation anti-androgen, has significantly improved survival of CRPC patients3. However, as observed with ADT, the efficacy of ENZ treatment is short-lived and tumors become resistant4,5. As such, there is currently high demand for understanding the mechanisms driving ENZ resistance in these patients. Previous data from our laboratory demonstrates that, similar to the mechanisms of resistance in CRPC, ENZ resistance is often coupled with reactivation of AR. Under these conditions, AR activity can persist due to AR mutations, increase in steroidogenesis as well as altered expression and activation of co-regulators6,7,8. In this study, we attempt to investigate the role of Signal Transducer and Activator of Transcription 3 (STAT3) in ENZ resistance as a binding partner that can facilitate AR activity9.
STAT3 is a protein hub for several oncogenic signalling pathways and regulates the expression of key effectors in tumor cell survival (Bcl-xL, Bcl-2, Mcl-1)10, proliferation (Cyclin D1, D2, c-Myc)11,12, angiogenesis (bFGF, VEGF)13,14 and metastasis15. The canonical STAT3 pathway is identified by phosphorylation of STAT3 on Tyrosine 705 (Y705) (typically through Janus Associated Kinases (JAK’s) in response to cytokines from IL-6 and IL-10 family) and subsequent dimerization and nuclear localization of STAT3. Additionally, direct activation can also take place via phosphorylation by receptor tyrosine kinases (i.e. EGFR, VEGFR, IGFR) and non-receptor tyrosine kinases such as Src family kinases16. STAT3 activity is significant in PCa progression whether it’s treatment naïve, castrate-resistant or metastatic17,18,19,20. This can be attributed to STAT3’s role in regulating several driving forces of PCa progression including integration of signaling pathways involved in re-activation of AR (i.e. the PTEN/PI3k/AKT pathway)21,22. Furthermore, the activity of mTOR and MAPK pathways phosphorylate STAT3 at Serine 727 (S727) which results in direct interaction with the N-terminal domain of AR and enhances AR transcriptional activity23,24, while S727A mutation significantly reduces this interaction; hindering AR transcriptional activity and highlighting the significance of AR/STAT3 co-operation in PCa progression25.
Utilizing a unique model of ENZ resistance, our lab explores mechanisms of resistance that arise under the pressure of ENZ8,26. Studying STAT3 activity in these LNCaP-derived ENZ resistant cells, we discovered that inhibition of STAT3 by the DNA binding inhibitor galiellalactone (GPA500)27,28 disrupted the interaction between STAT3 and AR, reduced AR activity and reduced cell proliferation in PSA producing ENZ-resistant cells (PSAhi ENZR). Furthermore, combination of ENZ and GPA500 in LNCaP and CRPC cells in vitro had an additive effect on AR inhibition and cell proliferation. These effects translated well in vivo; GPA500 treatment reduced tumor volume of ENZR xenografts and reduced serum PSA levels. Taken together, our study provides proof-of-principle that STAT3 inhibition using galiellalactone is a viable treatment option as monotherapy in ENZR prostate cancer as well as a logical strategy for combination therapy with ENZ in CRPC.
Results
STAT3 is nuclear and is co-localized with AR in PSAhi ENZR cells
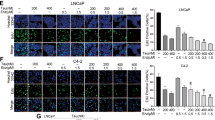
As with most transcription factors, STAT3’s primary activity occurs upon translocation into the nucleus. Interestingly, comparison of STAT3 localization in 16DCRPC and PSAhi ENZR cells (49 C and 49 F) showed that not only is STAT3 more localized to the nucleus in ENZR cells compared to CRPC, but also there is a clear co-localization between STAT3 and AR in these cells (Fig. 1A left, Supplementary Fig. S1). Nuclear localization of STAT3 in ENZR cells was further confirmed using a cytoplasmic/nuclear fractionation (Fig. 1A right). Conventionally, Y705 phosphorylation was considered as the prequel to S727 phosphorylation and STAT3 activation29; therefore, we explored the phosphorylation status of STAT3 in ENZR cells. Surprisingly, in comparison to 16DCRPC, 49 C and 49 F cells exhibit an increase in pSTAT3 S727, but not Y705 (Fig. 1B) (DU145 and IL6-treated LNCaPs were used as positive controls). Furthermore, long-term exposure of LNCaP and 16DCRPC cells to 10 µM ENZ clearly confirmed an increase in S727 phosphorylation in a time-dependent manner with no change in tyrosine phosphorylation (Fig. 1C). Strikingly, we observed a drastic increase in nuclear STAT3 and a clear co-localization with AR in LNCaP and 16DCRPC cells treated with ENZ (10 µM for 7 days) (Fig. 1D). This data supports previous studies documenting that S727 phosphorylation can activate STAT3 signaling independent of Y705 phosphorylation30,31.

STAT3 is nuclear and is co-localized with AR in PSAhi ENZR cells. (A) (Left) Immunofluorescence of STAT3 (red), AR (green) and the nucleus (DAPI, blue) in 16D CRPC and ENZR 49 F cells. Scale bar: 10 μm. (Middle) Graph visualization of nuclear and cytoplasmic levels of STAT3 and AR by calculating Spatial Signal Intensity. (Right) Cytoplasmic/Nuclear fractionation of STAT3, Vinculin and LaminB1 in 16D and 49 F cells. (B,C) Protein expression of p-STAT3S727, p-STAT3Y705, STAT3 and Vinculin in representative cell lines (B) DU145, LNCaP cells treated with/without IL6 (50 ng/mL), 16D CRPC, ENZR 49 F and 49 C and (C) LNCaP and 16D CRPC cells treated with 10 μM ENZ for indicated days. (D) Immunofluorescence of AR (green) and STAT3 (red) in LNCaP (Left) and 16D CRPC (Right) cells treated with 10 µM ENZ for 7 days. Scale bar: 10 μm.
Interestingly, while ENZR cells present with more nuclear STAT3 (Fig. 1A), analysis of RNA-seq in these cell lines revealed that only a subset of canonical STAT3 pathway genes32 were enriched (Supplementary Fig. S2, Table S1). Instead, several non-canonical STAT3 targets were upregulated (Supplementary Fig. S3, Table S2). These findings, support data showing that STAT3 not only binds DNA without Y705 phosphorylation33,34,35, but also results in activation of non-canonical STAT3 target genes. Taken together, these results indicate ENZ resistance increases STAT3 S727 phosphorylation and activates STAT3 signaling, suggesting that these cells might employ persistent STAT3/AR signaling activity as a mechanism of resistance.
PSAhi ENZR cells are sensitive to STAT3 inhibition by gallielalactone
Using our model of ENZR8,26, we examined the response of CRPC (16DCRPC) and PSAhi ENZR (49 C and 49 F) cells to the STAT3 inhibitor GPA500 and found that the PSAhi ENZR cells were more sensitive to STAT3 inhibition in comparison to 16DCRPC (Fig. 2A). Interrogating this effect, we discovered that GPA500 reduces the expression of canonical STAT3 target genes, Cyclin D1 and C-Myc, at the protein (Fig. 2B) and mRNA levels (Fig. 2C) more potently in ENZR cells. Moreover, we observed significant reduction in the mRNA levels of basic fibroblast growth factor (bFGF), a STAT3-regulated growth factor that promotes cell proliferation and angiogenesis (Fig. 2C).

PSAhi ENZR cells are sensitive to STAT3 inhibition by gallielalactone (GPA500). (A) Relative 72 hour cell proliferation assay using WST8 in 16D CRPC, 49 C and 49 F cells treated with indicated concentrations of GPA500 compared to untreated. (B) Protein expression of c-Myc, CyclinD1 and Vinculin and (C) Relative mRNA expression of c-Myc, CyclinD1 and bFGF in 16DCRPC and PSAhi ENZR cells (49 C and 49 F) treated with 0, 5 or 10 µM GPA500. (D) Cell cycle fraction of 16DCRPC and PSAhi ENZR cells 49 C and 49 F following GPA500 treatment (48 hour with indicated concentrations). All graphs represent pooled data from 3 independent experiments in triplicates.
Given the crucial role of Cyclin D1 in cell cycle progression, reduction of Cyclin D1 induces a G1 phase arrest11. In harmony with these findings, we discovered that GPA500 treatment in ENZR cells 49 C and 49 F triggered a more significant G1 phase arrest and a subsequent reduction of cells in S and G2/M phases in comparison to 16DCRPC cells (Fig. 2D). This reduction was also accompanied by a small increase in the sub-G0 fraction (Fig. 2D). Overall, these findings suggest that PSAhi ENZR cells, 49 C and 49 F, are dependent on STAT3 as an important proliferative transcription factor and are more sensitive to STAT3 inhibition when compared to CRPC cells.
Inhibition of STAT3 reduces AR activity in PSAhi ENZR cells in vitro
Re-activation of AR is one of the major factors in the emergence of ENZ resistant prostate cancer. In our model of ENZR8, we discovered that 75% of resistant tumors regained AR activity. To further explore the relationship between AR and STAT3 in these cell lines, we tested for changes in AR activity upon STAT3 inhibition. GPA500 treatment decreases the expression of AR target genes in PSAhi ENZR cells, 49 C and 49 F, in a dose dependent manner (Fig. 3A) and reduced AR activity (Supplementary Fig. S4A) without altering S727 phosphorylation (Fig. 3B). More interestingly, we found that there’s less interaction between STAT3 and AR in the presence of GPA500 (Supplementary Fig. S4B).

Inhibition of STAT3 reduces AR activity in PSAhi ENZR cells. (A) Relative mRNA expression of AR target genes PSA, NKX3-1 and FKBP5 and (B) Protein expression of p-STAT3S727, STAT3, PSA, AR and Vinculin in PSAhi ENZR cells 49 C and 49 F treated with 0, 5 or 10 µM GPA500 for 48 hours. (C) Protein expression of p-STAT3S727, STAT3, PSA, AR and Vinculin and (D) Relative AR activity assessed by luciferase assay and (E) Relative mRNA expression of AR target genes PSA, FKBP5, NKX3-1 and TMPRSS2 in LNCaP (Left) and 16D CRPC cells (Right) treated with 10 µM ENZ or GPA500 or combination of both for 48 hours. (F) Relative 72 hour cell proliferation assay using WST8 in LNCaP (Left) and 16D CRPC cells (Right) treated with indicated ENZ and GPA500 concentration. All graphs represent pooled data from 3 independent experiments and relative luciferase activity is compared to non-treated samples (Control = 1).
To investigate the effects of simultaneous AR/STAT3 inhibition, LNCaP and 16DCRPC cells were treated with GPA500 or ENZ alone and in combination. Consistent with our previous results (Fig. 2C), we found that ENZ treatment induced Ser727 phosphorylation of STAT3 and decreased PSA in LNCaP and 16DCRPC cells without affecting AR expression (Fig. 3C). Also, combination of both GPA500 and ENZ further reduced PSA (Fig. 3C). This data was supplemented by an AR transactivation assay (Fig. 3D) and qRT-PCR of AR target genes PSA, FKBP51, NKX3-1, TMPRSS2 (Fig. 3E). Combination of both drugs reduced AR activity more than either monotherapy in LNCaP (Fig. 3E Left) and 16DCRPC cells (Fig. 3E Right). Finally, in a cell growth assay for these cell lines we discovered GPA500 and ENZ reduced cell proliferation in an additive manner (Fig. 3F). These results suggest that STAT3 may play an important role for growth of PCa cells under pressure of anti-androgen treatment and co-targeting both pathways may provide additional effect.
Inhibition of STAT3 reduces AR activity in PSAhi ENZR cells in vivo
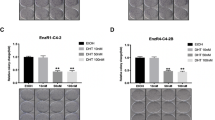
The efficacy of GPA500 on PSAhi ENZR 49 F cells that were subcutaneously injected into castrated male nude mice was tested. Once the tumor volume reached 200 mm3, mice were randomized and treated with vehicle control or 5 mg/kg/day of GPA500. Consistent with our in vitro data (Fig. 2A), treatment with GPA500 reduced tumor growth compared to control (Fig. 4A). Consequently, the serum PSA in the treated mice was also lower (Fig. 4B). The reduction in serum PSA could be attributed to the lower tumor volume; as such we tested the expression of PSA in the tumors. PSA expression in the tumors of mice treated with GPA500 was lower than control (Fig. 4C). Additionally, the expression of Cyclin D1 was also reduced in the tumors treated with GPA500 (Fig. 4C) which recapitulated our previous findings in vitro (Fig. 2C Right). In summary, our findings suggest that targeting STAT3 in PSA producing ENZ-resistant tumors is a rational approach to reduce tumor burden.

Inhibition of STAT3 reduces AR activity in PSAhi ENZR cells in vivo. (A) Weekly mean tumour volume (Left) and serum prostate-specific antigen (PSA) levels (Right) in mice treated with GPA500 5 mg/kg/day versus vehicle. Treatment was started when tumours reached 200 mm3 and was given intraperitoneally for 5 days on and 2 days off. (B) Protein expression of PSA, CyclinD1 and Vinculin in collected tumors treated with vehicle (control M1, M2 and M3) or GPA500 5 mg/kg (M4, M5 and M6).
Discussion
Prostate cancer is the 2nd most diagnosed cancer in men around the world and unfortunately, development of treatment resistance is the inevitable fate of prostate cancer patients put on 2nd generation ARPI’s like ENZ. Exploring and understanding the various mechanisms of resistance that contribute to AR re-activation after receiving ENZ may guide us in designing rational treatment regimens in the future. To this end, this study shows that in our model of ENZ resistance the STAT3/AR interaction facilitates not only sustained AR activity but also STAT3 signaling. Moreover, inhibition with the STAT3 DNA-binding inhibitor galiellalactone (GPA500)28 suppresses both AR and STAT3 target genes. We also demonstrate that despite the conventional dogma that STAT3 dimerization and nuclear translocation requires Y705 phosphorylation, the STAT3/AR complex in ENZ-resistant cell lines can occur independent of phospho-Y705. Ultimately, we demonstrate that GPA500 shows efficacy in ENZ-resistant tumors as monotherapy in vivo and it displays potential for combination with Enzalutamide at earlier stages of the disease in vitro.
Over the past 2 decades, STAT3 activity has been repeatedly implicated in PCa initiation and progression. Loss of the tumor suppressor PTEN is the most frequent deletion in PCa and combining PTEN loss with STAT3 activation accelerates development of adenocarcinoma36. STAT3 is intricately linked to key transcription factors associated with PCa development such as HIF1α37 and NF-κB38 resulting in regulation of multiple oncogenic proteins including but not limited to HER2, BCL-2 and BCL-3, VEGF and TWIST139. Moreover, there is a considerable body of work evaluating the interplay between STAT3 and AR, the fundamental driver of PCa. For example, IL-6 treatment and subsequent signaling through the canonical JAK/STAT3 pathway can induce AR expression/activity21. STAT3 was also shown to enhance AR signaling through direct interaction with the N-terminus domain of the AR23. Moreover, the crucial mediator in this direct interaction was found to be S727 phosphorylation on STAT325 which was shown to be a driver of tumorigenesis in prostate cancer independent of Y705 phosphorylation30. Interestingly, S727 phosphorylation was found to be expressed more in higher Gleason Score tissues17,25,30 further establishing its clinical importance in localized PCa.
STAT3 signaling also plays a role in treatment-resistant prostate cancer phenotypes such as CRPC40, regulation of cancer stem cell phenotype41 as well as development of resistance to 2nd generation anti-androgen, ENZ42. An investigation by Liu et al. showed that constitutive activation of STAT3 can overcome the anti-proliferative effects of ENZ and STAT3 inhibition in LNCaP cells reverses ENZ-resistance43. While insightful, this data doesn’t reflect the natural progression to ENZ resistance. The effect on STAT3 was not evaluated as a response to ENZ, rather, the authors studied the reduced efficacy of ENZ in response to artificially activated STAT343.
Studying the cell lines from our naturally derived ENZ-resistance tumors8 we found that similar to castration resistance16, the STAT3 pathway still plays a considerable role in ENZ resistance. Similar to LNCaP cells which they are originated from, there is little to no canonical IL6/STAT3 activity in these PSAhi ENZR cells; however, pSTAT3 S727 and not Y705 is upregulated compared to 16DCRPC cells. Previous research from our lab demonstrated that two of the kinase pathways upstream of Ser727 phosphorylation, ERK (MAPK1/2) and AKT, are activated in response to ENZ treatment6, possibly explaining the upregulation of this phosphorylation in response to ENZ. Interestingly, another group showed that treatment of LNCaP cells with ENZ does not affect the tyrosine phosphorylation of STAT344, although they didn’t examine the S727 phosphorylation.
Resistance acquired against ENZ is a collection of complex events comprised of both AR dependent (direct or indirect) and AR independent mechanisms45. Supplementing the previously identified AR/STAT3 interaction, our study reveals that in PSAhi ENZR cells, STAT3 binds AR and leads to continued expression of both AR and STAT3 target genes. However, it is important to note that this association does not upregulate all canonical STAT3 genes. Evidence for this is apparent in the RNA-seq data where only a subset of classic STAT3 genes are upregulated in the ENZR cells and the canonical pathway is not enriched overall. The increased sensitivity of the ENZR cells to STAT3 inhibition may be due to the concomitant reduction of both STAT3 and AR target genes. Indeed, combination of ENZ and galiellalactone on LNCaP and CRPC derived cells had an additive effect on AR inhibition and reduced cell proliferation more than ENZ monotherapy. In summary, our study further establishes STAT3’s pivotal role in development of ENZ resistance as it can promote oncogenic signaling not only through its canonical pathway, but also by re-activation of AR and shows that STAT3 inhibitor, galiellalactone, decreases ENZ-resistant cell proliferation in vitro and in vivo.
Materials and Methods
Generation of ENZR Xenografts and Cell Lines
The detailed procedure for generation of CRPC and ENZR tumors and cell lines can be found in our previously published report8,26.
Cell Line Culture and Reagents
LNCaP cells were kindly provided by Dr. Leland W.K. Chung (Emory University) and authenticated in January 2013. CRPC and ENZR cell lines were generated from LNCaP cells, tested, and authenticated by whole-genome and whole-transcriptome sequencing (Illumina Genome Analyzer IIx, 2012). Cells were maintained in RPMI-1640 (+10 μmol/L ENZ (Haoyuan Chemexpress) for ENZR or DMSO (Sigma-Aldrich) for LNCaP and 16DCRPC. Cells were seeded at a density of 1 × 106 cells/10 mL media, treated the following day with galiellalactone (GPA500, Glactone Pharma) and harvested after 48 hours unless otherwise noted.
Cell Growth Assay
16DCRPC (without ENZ in media), 49C ENZR and 49F ENZR (with ENZ in media) cells were plated in 96 well plates (Corning) at 4000 cells per well. Treatments were done next day, proliferation was quantified using WST-8 assay (Dojindo) as per manufacturer’s protocol.
RNA Sequencing
RNA sequencing data was extracted from microarray gene expression previously performed8.
Quantitative reverse transcription PCR (qRT-PCR)
Total RNA was extracted from cells using TRIzol reagent (Life Technology) and 2 μg was reversed-transcribed using MMLV reverse transcriptase and random hexamers (Invitrogen). Real-time PCR was performed using SyberGreen ROX Master Mix (Roche Applied Science). Target gene expression was normalized to GAPDH levels in three experimental replicates per sample. For primer sequences, please see Supplementary Table (Table S3).
Luciferase Assay
Luciferase reporter assay was performed as described previously46. In short, 50,000 cells per well are plated in 12-well plates are transfected with ARR3-luc reporter using Mirus TransIT 20/20. 24 hours later, cells are treated and lysed for bioluminescence analysis 48 hours later.
Cell cycle analysis using Flow Cytometry
49CENZR and 49FENZR (with ENZ) cells were plated in 10 cm dishes and treated with ENZ and/or GPA500 at indicated concentration. After incubation for 48 hr, cell-cycle fraction was analyzed using Propidium Iodide (PI). Briefly, cells were trypsinized, washed with cold PBS and fixed with 70% ethanol while light vortexing to prevent clumping. Cells were then stained with PI staining solution containing RNase. Stained cells were run through FACS CANTO II machine and analyzed using FlowJo.
Immunoprecipitation and Western Blotting
Whole-cell extracts were obtained upon lysis of cells in cold RIPA buffer (Thermo) with 1x concentration of PhoSTOP and Protease Inhibitor (Roche). Once protein concentration was determined using BCA assay (Thermo), 40 μg samples were boiled for 5 mins in SDS sample buffer and ran on SDS-PAGE gel. Immunoprecipitation was performed using ImmunoCruz™ IP/WB Optima B System (Santa Cruz) based on the manufacturer’s guideline. 2 µg of primary antibody, or immunoglobulin G (IgG) was used for immunoprecipitation and control respectively. Transfer was done onto PVDF membranes, blocked with Odyssey Blocking Buffer (Licor), probed with primary antibodies at 1:1000 dilution. Antibodies for STAT3, pSTAT3 S727 and Y705, PSA, c-MYC, Cyclin D1 and PARP were all obtained from Cell Signaling. Vinculin antibody was purchased from Sigma while AR and LaminB1 were purchased from Santa Cruz. Image acquisition and intensity quantifications were carried out using Licor Odyssey Scanner with Application Software V3.0 and normalized to Vinculin intensity. Full length blots are provided as Supplementary Figs S5–S9.
Immunofluorescence
Cells were plated at 25,000 per in 12 well plates on circular glass slides in a 12 well plate. Cells were fixed and stained as previously described47 using antibodies against STAT3 (1:100) and AR (1:100). DAPI was used to visualize nuclei and then the pictures were taken using Zeiss LSM confocal microscope. Results are representative of random pictures taken from three independent experiments.
Animal treatment
All experimental protocols used in this study were approved by the Canadian Council on Animal Care and the University of British Columbia (UBC) Animal Care Committee (certificate number A106-0246). All methods carried out in this study are in accordance with guidelines and regulations of both organizations. Castrated male athymic mice (Sprague Dawley; Harlan, Inc., Indianapolis, IN) were injected subcutaneously with 1 × 106 49FENZR cells (suspended in 0.1 mL Matrigel; BD Biosciences). Once tumors were 200 mm3, mice were randomly assigned to (i) vehicle, (ii) GPA500 (5 mg/kg) and treated Intraperitoneally 5 times per week. Tumor volume and PSA measurements were performed once weekly. Serum PSA measurements were performed by cobas e 411 analyzer (Roche). After sacrifice, tumors were harvested.
Statistical Analysis and Data Representation
In bar graphs, unpaired, two-tailed, Student t tests were performed to analyze statistical significance between groups using GraphPad Prism (GraphPad Software). Significance is indicated as follows: *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001. Graphs show pooled data with error bars representing SEM obtained from at least three independent experiments.
References
CCSsACoC, S. Canadian cancer statistics 2014. Toronto, ON: Canadian Cancer Society 2014 (2014).
Hotte, S. J. & Saad, F. Current management of castrate-resistant prostate cancer. Curr Oncol 17(Suppl 2), S72–79 (2010).
Scher, H. I. et al. Increased survival with enzalutamide in prostate cancer after chemotherapy. The New England journal of medicine 367, 1187–1197, https://doi.org/10.1056/NEJMoa1207506 (2012).
Watson, P. A., Arora, V. K. & Sawyers, C. L. Emerging mechanisms of resistance to androgen receptor inhibitors in prostate cancer. Nat Rev Cancer 15, 701–711, https://doi.org/10.1038/nrc4016 (2015).
Antonarakis, E. S. et al. AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer. N Engl J Med 371, 1028–1038, https://doi.org/10.1056/NEJMoa1315815 (2014).
Toren, P., Kim, S., Johnson, F. & Zoubeidi, A. Combined AKT and MEK Pathway Blockade in Pre-Clinical Models of Enzalutamide-Resistant Prostate Cancer. PLoS One 11, e0152861, https://doi.org/10.1371/journal.pone.0152861 (2016).
Toren, P. et al. Combination AZD5363 with Enzalutamide Significantly Delays Enzalutamide-resistant Prostate Cancer in Preclinical Models. Eur Urol 67, 986–990, https://doi.org/10.1016/j.eururo.2014.08.006 (2015).
Bishop, J. L. et al. The Master Neural Transcription Factor BRN2 Is an Androgen Receptor-Suppressed Driver of Neuroendocrine Differentiation in Prostate Cancer. Cancer Discov 7, 54–71, https://doi.org/10.1158/2159-8290.CD-15-1263 (2017).
Yamamoto, T. et al. Molecular interactions between STAT3 and protein inhibitor of activated STAT3, and androgen receptor. Biochemical and biophysical research communications 306, 610–615 (2003).
Fukada, T. et al. Two signals are necessary for cell proliferation induced by a cytokine receptorgp130: involvement of STAT3 in anti-apoptosis. Immunity 5, 449–460 (1996).
Leslie, K. et al. Cyclin D1 is transcriptionally regulated by and required for transformation by activated signal transducer and activator of transcription 3. Cancer Res 66, 2544–2552, https://doi.org/10.1158/0008-5472.CAN-05-2203 (2006).
Bromberg, J. F. et al. Stat3 as an oncogene. Cell 98, 295–303 (1999).
Niu, G. et al. Constitutive Stat3 activity up-regulates VEGF expression and tumor angiogenesis. Oncogene 21, 2000–2008, https://doi.org/10.1038/sj.onc.1205260 (2002).
Huang, Y. H. et al. STAT1 activation by venous malformations mutant Tie2-R849W antagonizes VEGF-A-mediated angiogenic response partly via reduced bFGF production. Angiogenesis 16, 207–222, https://doi.org/10.1007/s10456-012-9313-x (2013).
Huang, S. Regulation of metastases by signal transducer and activator of transcription 3 signaling pathway: clinical implications. Clin Cancer Res 13, 1362–1366, https://doi.org/10.1158/1078-0432.CCR-06-2313 (2007).
Bishop, J. L., Thaper, D. & Zoubeidi, A. The Multifaceted Roles of STAT3 Signaling in the Progression of Prostate Cancer. Cancers 6, 829–859, https://doi.org/10.3390/cancers6020829 (2014).
Cocchiola, R. et al. Analysis of STAT3 post-translational modifications (PTMs) in human prostate cancer with different Gleason Score. Oncotarget 8, 42560–42570, https://doi.org/10.18632/oncotarget.17245 (2017).
Don-Doncow, N. et al. Expression of STAT3 in Prostate Cancer Metastases. Eur Urol 71, 313–316, https://doi.org/10.1016/j.eururo.2016.06.018 (2017).
Tam, L. et al. Expression levels of the JAK/STAT pathway in the transition from hormone-sensitive to hormone-refractory prostate cancer. Br J Cancer 97, 378–383, https://doi.org/10.1038/sj.bjc.6603871 (2007).
Mohanty, S. K. et al. STAT3 and STAT5A are potential therapeutic targets in castration-resistant prostate cancer. Oncotarget 8, 85997–86010, https://doi.org/10.18632/oncotarget.20844 (2017).
Yang, L. et al. Interleukin-6 differentially regulates androgen receptor transactivation via PI3K-Akt, STAT3, and MAPK, three distinct signal pathways in prostate cancer cells. Biochemical and biophysical research communications 305, 462–469 (2003).
Lin, D. L., Whitney, M. C., Yao, Z. & Keller, E. T. Interleukin-6 induces androgen responsiveness in prostate cancer cells through up-regulation of androgen receptor expression. Clinical cancer research: an official journal of the American Association for Cancer Research 7, 1773–1781 (2001).
Ueda, T., Bruchovsky, N. & Sadar, M. D. Activation of the androgen receptor N-terminal domain by interleukin-6 via MAPK and STAT3 signal transduction pathways. The Journal of biological chemistry 277, 7076–7085, https://doi.org/10.1074/jbc.M108255200 (2002).
Chen, T., Wang, L. H. & Farrar, W. L. Interleukin 6 activates androgen receptor-mediated gene expression through a signal transducer and activator of transcription 3-dependent pathway in LNCaP prostate cancer cells. Cancer Res 60, 2132–2135 (2000).
Hsu, F. N. et al. Cyclin-dependent kinase 5 modulates STAT3 and androgen receptor activation through phosphorylation of Ser(7)(2)(7) on STAT3 in prostate cancer cells. Am J Physiol Endocrinol Metab 305, E975–986, https://doi.org/10.1152/ajpendo.00615.2012 (2013).
Kuruma, H. et al. A novel antiandrogen, Compound 30, suppresses castration-resistant and MDV3100-resistant prostate cancer growth in vitro and in vivo. Mol Cancer Ther 12, 567–576, https://doi.org/10.1158/1535-7163.MCT-12-0798 (2013).
Hellsten, R. et al. Galiellalactone is a novel therapeutic candidate against hormone-refractory prostate cancer expressing activated Stat3. Prostate 68, 269–280, https://doi.org/10.1002/pros.20699 (2008).
Don-Doncow, N. et al. Galiellalactone is a direct inhibitor of the transcription factor STAT3 in prostate cancer cells. J Biol Chem 289, 15969–15978, https://doi.org/10.1074/jbc.M114.564252 (2014).
Yu, H., Lee, H., Herrmann, A., Buettner, R. & Jove, R. Revisiting STAT3 signalling in cancer: new and unexpected biological functions. Nat Rev Cancer 14, 736–746, https://doi.org/10.1038/nrc3818 (2014).
Qin, H. R. et al. Activation of signal transducer and activator of transcription 3 through a phosphomimetic serine 727 promotes prostate tumorigenesis independent of tyrosine 705 phosphorylation. Cancer Res 68, 7736–7741, https://doi.org/10.1158/0008-5472.CAN-08-1125 (2008).
Androutsellis-Theotokis, A. et al. Notch signalling regulates stem cell numbers in vitro and in vivo. Nature 442, 823–826, https://doi.org/10.1038/nature04940 (2006).
Azare, J. et al. Constitutively activated Stat3 induces tumorigenesis and enhances cell motility of prostate epithelial cells through integrin beta 6. Molecular and cellular biology 27, 4444–4453, https://doi.org/10.1128/MCB.02404-06 (2007).
Yang, J. et al. Novel roles of unphosphorylated STAT3 in oncogenesis and transcriptional regulation. Cancer Res 65, 939–947 (2005).
Yang, J. & Stark, G. R. Roles of unphosphorylated STATs in signaling. Cell research 18, 443–451, https://doi.org/10.1038/cr.2008.41 (2008).
Cimica, V., Chen, H. C., Iyer, J. K. & Reich, N. C. Dynamics of the STAT3 transcription factor: nuclear import dependent on Ran and importin-beta1. PloS one 6, e20188, https://doi.org/10.1371/journal.pone.0020188 (2011).
Smith, D. A., Kiba, A., Zong, Y. & Witte, O. N. Interleukin-6 and oncostatin-M synergize with the PI3K/AKT pathway to promote aggressive prostate malignancy in mouse and human tissues. Molecular cancer research: MCR 11, 1159–1165, https://doi.org/10.1158/1541-7786.MCR-13-0238 (2013).
Pawlus, M. R., Wang, L. & Hu, C. J. STAT3 and HIF1alpha cooperatively activate HIF1 target genes in MDA-MB-231 and RCC4 cells. Oncogene 33, 1670–1679, https://doi.org/10.1038/onc.2013.115 (2014).
Yu, Z., Zhang, W. & Kone, B. C. Signal transducers and activators of transcription 3 (STAT3) inhibits transcription of the inducible nitric oxide synthase gene by interacting with nuclear factor kappaB. The Biochemical journal 367, 97–105, https://doi.org/10.1042/BJ20020588 (2002).
Carpenter, R. L. & Lo, H. W. STAT3 Target Genes Relevant to Human Cancers. Cancers (Basel) 6, 897–925, https://doi.org/10.3390/cancers6020897 (2014).
Shiota, M. et al. Hsp27 regulates epithelial mesenchymal transition, metastasis, and circulating tumor cells in prostate cancer. Cancer Res 73, 3109–3119, https://doi.org/10.1158/0008-5472.CAN-12-3979 (2013).
Schroeder, A. et al. Loss of androgen receptor expression promotes a stem-like cell phenotype in prostate cancer through STAT3 signaling. Cancer Res 74, 1227–1237, https://doi.org/10.1158/0008-5472.CAN-13-0594 (2014).
Wang, C. et al. Blocking the Feedback Loop between Neuroendocrine Differentiation and Macrophages Improves the Therapeutic Effects of Enzalutamide (MDV3100) on Prostate Cancer. Clin Cancer Res 24, 708–723, https://doi.org/10.1158/1078-0432.CCR-17-2446 (2018).
Liu, C. et al. Inhibition of constitutively active Stat3 reverses enzalutamide resistance in LNCaP derivative prostate cancer cells. Prostate 74, 201–209, https://doi.org/10.1002/pros.22741 (2014).
Handle, F. et al. SOCS3 Modulates the Response to Enzalutamide and Is Regulated by Androgen Receptor Signaling and CpG Methylation in Prostate Cancer Cells. Mol Cancer Res 14, 574–585, https://doi.org/10.1158/1541-7786.MCR-15-0495 (2016).
Claessens, F. et al. Emerging mechanisms of enzalutamide resistance in prostate cancer. Nature reviews. Urology 11, 712–716, https://doi.org/10.1038/nrurol.2014.243 (2014).
Vahid, S., Thaper, D., Gibson, K. F., Bishop, J. L. & Zoubeidi, A. Molecular chaperone Hsp27 regulates the Hippo tumor suppressor pathway in cancer. Scientific reports 6, 31842, https://doi.org/10.1038/srep31842 (2016).
Thaper, D. et al. Targeting Lyn regulates Snail family shuttling and inhibits metastasis. Oncogene, https://doi.org/10.1038/onc.2017.5 (2017).
Acknowledgements
This work was supported by the Canadian Institutes of Health Research (Grant Number 201410GSD-352466-230421); and Glactone Pharma (which provided GPA500).
Author information
Authors and Affiliations
Contributions
Conceptualization, design and interpretation of data: D.T., S.V., J.L.B. and A.Z. Writing, review and editing: S.V., D.T., J.L.B., M.J. and A.Z. In vitro data acquisition: S.V., D.T., S.K., R.K., S.N. In vivo data acquisition: D.T., J.L.B.; Supervision: A.Z.
Corresponding author
Ethics declarations
Competing Interests
Martin Johansson is the CEO of Glactone Pharma Development AB which provided galiellalactone. Other authors declare no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Thaper, D., Vahid, S., Kaur, R. et al. Galiellalactone inhibits the STAT3/AR signaling axis and suppresses Enzalutamide-resistant Prostate Cancer. Sci Rep 8, 17307 (2018). https://doi.org/10.1038/s41598-018-35612-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-35612-z
Keywords
This article is cited by
-
Molecular panorama of therapy resistance in prostate cancer: a pre-clinical and bioinformatics analysis for clinical translation
Cancer and Metastasis Reviews (2024)
-
MicroRNA-375 is a therapeutic target for castration-resistant prostate cancer through the PTPN4/STAT3 axis
Experimental & Molecular Medicine (2022)
-
PIM kinase inhibition: co-targeted therapeutic approaches in prostate cancer
Signal Transduction and Targeted Therapy (2020)
-
STAT3 inhibition with galiellalactone effectively targets the prostate cancer stem-like cell population
Scientific Reports (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
