
Abstract
As multidrug resistant pathogenic microorganisms are a serious health menace, it is crucial to continuously develop novel medicines in order to overcome the emerging resistance. The methylerythritol phosphate pathway (MEP) is an ideal target for antimicrobial development as it is absent in humans but present in most bacteria and in the parasite Plasmodium falciparum. Here, we report the synthesis and the steady-state kinetics of a novel potent inhibitor (MEPN3) of Escherichia coli YgbP/IspD, the third enzyme of the MEP pathway. MEPN3 inhibits E. coli YgbP/IspD in mixed type mode regarding both substrates. Interestingly, MEPN3 shows the highest inhibitory activity when compared to known inhibitors of E. coli YgbP/IspD. The mechanism of this enzyme was also studied by steady-state kinetic analysis and it was found that the substrates add to the enzyme in sequential manner.
Similar content being viewed by others


Introduction
Drug resistance is an ever-growing concern that poses a major challenge for new drug development. In the field of antibiotic discovery, the situation is alarming as some infections are already impossible to treat due to resistance. Enterobacteriaceae, in particular Klebsiella pneumonaiae and Escherichia coli, are highly pervasive in community-acquired and nosocomial infections. The worldwide emergence of carbapenemase-producing Enterobacteriaceae represents a serious public health threat as carbapenems are often the last option for treatment of patients infected by these bacteria1. WHO already estimated in 2014 that 44% of its member states reported E. coli strains resistant to third-generation cephalosporins and fluoroquinolones and highlighted the high resistance rates of E. coli strains to the last-generation drugs2. In September 2017, WHO classified carbapenem-resistant and third-generation cephalosporin resistant Enterobacteriaceae among the most critical priority for Research and Development of new antibiotics as strains that cannot be fought by any antibiotic on the market are emerging worldwide3. Given the severe threat from organisms resistant to conventional antibacterial agents, targeting the MEP pathway responsible for the biosynthesis of the universal isoprenoid precursors in most bacteria and in the parasite responsible for malaria was proposed as an attractive strategy in the search for new antimicrobial agents4,5,6,7.
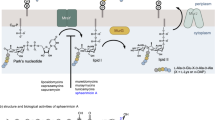
Isoprenoids are the most diverse family of natural products that comprises over 55000 known compounds. They are found in all living organisms and are involved in numerous essential biological processes such as electron transport, cell-wall biosynthesis, and protein prenylation8,9. Isoprenoids are synthesised through multiple condensation of two main building blocks: dimethylallyl diphosphate (DMADP, 1) and isopentenyl diphosphate (IDP, 2, Fig. 1)8. The MEP pathway, absent in humans, is an alternative to the well-known mevalonate pathway existing in animals10 for the formation of IDP and DMADP. The MEP pathway (Fig. 1) starts with condensation of pyruvate (3) and glyceraldehyde 3-phosphate (4) to form 1-deoxy-D-xylulose 5-phosphate (5), which is further converted to 2-C-methyl-D-erythritol 4-phosphate (MEP, 6). MEP reacts with cytidine triphosphate (CTP) to generate 4-diphosphocytidyl-2-C-methyl-D-erythritol (CDP-ME, 7), which is further phosphorylated to yield 4-diphosphocytidyl-2-C-methyl-D-erythritol 2-phosphate (CDP-MEP, 8). After cyclisation and cytidine monophosphate (CMP) release, 2-C-methyl-D-erythritol 2,4-cyclodiphosphate (ME-cPP, 9) forms. ME-cPP then generates (E)-4-hydroxy-3-methylbut-2-en-1-yl diphosphate (HMBDP, 10) that further produces DMADP and IDP.

MEP pathway.
To date only one compound targeting the MEP pathway, namely fosmidomycin, an inhibitor of the second enzyme 1-deoxyxylulose 5-phosphate reductoisomerase (DXR), is under clinical trial as an antimalarial agent in combination with clindamycin and piperaquine11, highlighting the potential of the MEP pathway for drug development12. Exploring new inhibitors of the MEP pathway could be a source of new therapeutic agents that are urgently needed to fight life-threatening infections. Here we report the synthesis and inhibition studies of MEPN3, (11, Fig. 2a) as a potential inhibitor of E. coli YgbP (also called IspD), the third enzyme of the MEP pathway.

Synthesis of MEPN3 (11) and BCN-TMBPP (23). (a) Synthetic route to MEPN3. (a) (CH3)3SOI, NaH, DMSO, 10 min, r.t; (b) NaN3, NH4Cl, DMF, 2 h, 60 °C; (c) TBAF, THF, 0 °C to r.t; (d) (CH3O)2POCl, DMAP, DCM, 2 h, 0 °C; (e) DOWEX (H+), MeOH, 48 h, r.t; (f) (CH3)3SiBr, H2O, DCM, 0 °C to r.t. (b) Synthetic scheme for BCN-TMPP. (a) Ethylenediamine, TEA, DMF, 2 h, r.t; (b) Bromoacetic acid N-hydroxysuccinimide ester, toluene, 0.5 h, r.t; (c) NEt3, DCM, 16 h, r.t.
Results and Discussion
E. coli YgbP/IspD
E. coli YgbP/IspD (EC 2.7.7.60) is encoded by the ygbP gene and catalyses the transformation of MEP and CTP into 4-diphosphocytidyl-2-C-methyl-D-erythritol (CDP-ME; 7) and inorganic diphosphate (PPi) (Fig. 1) in the presence of a divalent cation such as Mn2+, Mg2+ or Co2+13. YgbP was checked for activity using a similar method as described previously and based on the transformation of inorganic diphosphate to phosphate by inorganic pyrophosphatase followed by the quantification of the resulting phosphate by complexation with malachite green ammonium molybdate14. This method is robust, simple, fast, reliable and inexpensive for checking the activity of YgbP. The activity of YgbP limiting the concentration of CTP to 200 µM (as substrate inhibition was reported at high concentrations15) was 3.47 µmol.min−1.mg−1 and is in the same range as published14. The kinetic parameters of YgbP/IspD were determined using either varied MEP concentrations and a fixed CTP concentration (200 µM) or varied CTP concentrations and a fixed MEP concentration (250 µM). The reaction rates could be fitted according to the Michaelis–Menten equation and apparent Km values of 40 ± 7 µM for MEP (kcat = 1.77 s−1) and 84 ± 9 µM for CTP (kcat = 6.87 s−1) were estimated, which are in agreement with the reported values using the same method for the quantification of the activity14. A similar Km for MEP (32 ± 3 µM) was reported by Cane et al.16 using a radiolabeled assay. Rohdich et al.13 published a smaller Km for MEP (3.14 µM) but a similar Km for CTP (131 µM) when the detection of inorganic diphosphate was achieved indirectly after its consumption through a cascade of reactions leading to the reduction of NADP+. Richard et al.15 reported 370 ± 60 µM (10-fold higher than our result) Km value for MEP but used different conditions with high CTP concentrations (above 7 mM).
Our aim was to design an E. coli YgbP/IspD inhibitor that would be suitable for fragment-based drug discovery approaches. We searched a position for the insertion of an azide functionality while introducing minimal structural perturbation on the MEP substrate and keeping chemical stability. Therefore, we tested MEPN3 (11, Fig. 2), a MEP analogue harbouring an azido on the methyl group.
Synthesis of MEPN3
The route to MEPN3, (11) is outlined in Fig. 2 and starts with ketone 12 which was previously described by Coates and coworkers17. Submitted to a Corey–Chaykovsky reaction, ketone 12 was diastereoselectively converted to epoxide 1318,19; this represents a valuable improvement compared to Coates’ synthesis of 13, which featured a two-step “olefin formation-epoxidation” procedure that led to a diastereomeric mixture17. Epoxide 13 was further transformed into azido alcohol 14 with sodium azide, followed by deprotection of the primary alcohol with tetra-n-butylammonium fluoride. The resulting azido diol 15 was phosphorylated with dimethyl chlorophosphate to yield compound 16. Hydrolysis of benzylidene acetal with an acidic resin followed by deprotection of the phosphate group using a McKenna reaction20 afforded MEPN3 (11).
MEPN3 is a poor substrate of YgbP/IspD
As MEPN3 (11) structurally resembles MEP, YgbP was further assayed using MEPN3 as a substrate. Poulter et al.21 have previously reported that 2-C-ethyl-D-erythritol phosphate was a substrate for Agrobacterium tumefaciens YgpP/IspD showing replacement of the methyl at C-2 of MEP by an alternative substituent could still allow catalysis. Initial studies showed that MEPN3 was a substrate of YgbP but the enzymatic reaction velocity declined at higher MEPN3 concentrations (>500 µM), revealing substrate inhibition (Fig. S1). From the obtained data, the activity of YgbP (at 300 µM of MEPN3) was 100-fold less than the activity of YgbP with MEP (at 250 µM) showing that MEPN3 is a poor substrate.
The previous assay was based on the detection of the released diphosphate but not on the detection of CDP-MEN3 (18) (Fig. 3a) that should be produced if MEPN3 behaved like MEP in the active site of YgbP. In this context, strain-promoted alkyne-azide cycloaddition (SPAAC) using BCN ((1R,8S,9S)-bicyclo[6.1.0]non-4-yn-9-yl)methanol) derivative 23 encompassing a TMPP (tris(2,4,6-trimethoxyphenyl phosphonium, 21, Fig. 2) tag was employed to detect CDP-MEN3 (18) in the YgbP assay with MEPN3 as substrate. TMPP had been previously applied as a charge derivatisation agent for small biological molecules to enhance their detectability using positive ion ESI-MS analysis, as it carries a permanent positive charge22,23,24. BCN-TMPP (23) was prepared as described in Fig. 2. BCN was activated with p-nitrochloroformate to generate molecule 1925 which was further converted into compound 20 using ethylenediamine26. In parallel, TMPP and bromoacetic acid N-hydroxysuccinimide ester were used to produce TMPP derivative 22. Reaction of BCN derivative 20 with the activated TMPP (22) afforded the target molecule 23 in good yield.

LC-MS analysis of MEPN3 or its product in the YgbP catalysed reaction after derivatisation with TMPP-BCN (23). (a) Structure of CDP-MEN3 (b) Structure of MEPN3 or of its product after derivatisation with TMBPP-BCN. (c) Mass-to-charge ratio of MEPN3 clicked with TMPP-BCN. (d) Mass-to-charge ratio of the product of 11 in the YgbP catalysed reaction clicked with 23.
In a preliminary experiment, BCN-TMPP was incubated with MEPN3 and after LC-MS analysis (C18 column, UV detection for LC, positive mode detection for MS) the major product was detected at m/z 534 Da (calculated mass = 1067 Da) consisting of BCN-TMPP-MEPN3 (24) (Fig. 3b) carrying a second positive charge in addition to TMPP’s permanent charge (Fig. 3c). In order to identify the product of YgbP using MEPN3 as substrate, the completed enzymatic reaction was incubated with BCN-TMPP, and after LC-MS analysis, a product displaying m/z 686 Da corresponding to 25 (calculated mass = 1372 Da) harboring two charges was detected confirming that MEPN3 was turned over by YgbP to CDP-MEN3 (Fig. 3d). The fact that MEPN3 was a poor substrate compared to MEP was surprising as the only difference between both molecules was the azide function. To better understand the slow turnover of MEPN3 by YgbP, we further evaluated the inhibition potential of 11 on YgbP.
Inhibition of E. coli YgbP/IspD by MEPN3
Preliminary experiments revealed the YgbP reaction rate using its natural substrates (MEP and CTP) was decreasing when MEPN3 was present. In order to further determine the inhibition parameters and the inhibition mode of MEPN3, steady-state inhibition kinetic studies were performed (Fig. 4, a,d). The data were fitted according to the double reciprocal analysis (Fig. 4b,e) and highlighted that MEPN3 inhibited YgbP in a mixed type inhibition mode with respect to both substrates. From the replots (Fig. 4c,f), we have found Ki = 21 ± 3 µM and αKi = 54 ± 4 µM (α = 2.5) when MEP was the varied substrate while Ki = 47 ± 6 µM and αKi = 105 ± 24 µM (α = 2.2) when CTP was the varied substrate. As αKi value was higher than Ki value in both cases, it suggests that MEPN3 has a higher affinity for the free enzyme than for the enzyme–substrate complex.

Inhibition of E. coli YgbP by MEPN3. (a) Steady state kinetics at variable MEP concentrations and fixed MEPN3 concentrations (0 µM, r2 = 0.99; 20 µM, r2 = 0.98; 40 µM, r2 = 0.99; 60 µM, r2 = 0.99). (b) Double reciprocal plot of (a) (0 µM, r2 = 0.99; 20 µM, r2 = 0.99; 40 µM, r2 = 0.98; 60 µM, r2 = 0.99) (c) Replot of slope and intercept of (b) (slope, r2 = 0.98; intercept, r2 = 0.95). (d) Steady state kinetics at variable CTP concentrations and fixed MEPN3 concentrations (0 µM, r2 = 0.98; 30 µM, r2 = 0.99; 60 µM, r2 = 0.99; 90 µM, r2 = 0.98). (e) Double reciprocal plot of (d) (0 µM, r2 = 0.99; 30 µM, r2 = 0.98; 60 µM, r2 = 0.98; 90 µM, r2 = 0.99). (f) Replot of slope and intercept of (e) (slope, r2 = 0.99; intercept, r2 = 0.95). Mean and SEM values are displayed, n ≥ 3.
Although there are number of reports describing potent inhibitors of YgbP from malaria parasites27,28 and Mycobacteria29,30 as well as from the plant Arabidopsis thaliana31,32,33, there are hardly any inhibitors reported for E. coli YgbP (Fig. 5) and they all display very high IC50 reflecting their poor inhibition potential. The first inhibitor described for E. coli YgbP was D-erythrol-4-phosphate (26) with IC50 value of 1.36 mM which was also a substrate and reduced the turnover rate compared to natural substrate MEP34. Two years later, L-erythrol-4-phosphate (27) was reported to be a weak competitive inhibitor of MEP in E. coli YgbP, displaying a Ki value of 240 mM35. Interestingly fosmidomycin (28), the only inhibitor of MEP pathway currently in clinical trial and targeting DXR, was also reported to be an inhibitor of YgbP. Odom and co-workers reported an increase of MEP level and decrease of CDP-ME level when Plasmodium falciparum was treated with fosmidomycin and further showed that 28 inhibited E. coli YgbP activity with an IC50 value of 20.4 mM36. Interestingly, in the course of this work, Freel Meyers and co-workers37 described (5S)-D-methylerythritol monofluoromethyl phosphonate (29) as a new inhibitor of E. coli YgbP displaying an IC50 value of 0.7 mM. Even though no detailed kinetics were reported, these results encouraged us in our approach. MEPN3 is a mixed type inhibitor of E. coli YgbP regarding both substrates with Ki values in the micromolar range (21 µM and 47 µM). IC50 for MEPN3 was estimated to be around 41.5 µM (see Fig. S4), making MEPN3 the best inhibitor of E. coli YgbP known to date.

Structures of E. coli YgbP described (26–28) and new (11) inhibitors. Ki values are displayed when available, otherwise they are replaced by the reported IC50 values.
As MEPN3 is a substrate analogue of MEP, we expected the inhibition to be competitive with MEP and uncompetitive with CTP. In order to further understand the mixed type inhibition observed for MEPN3 on YgbP regarding both substrates, set of docking studies were performed to investigate the binding mode of 11.
Docking experiments of MEPN3 with E. coli YgbP
Three different X-ray structures of E. coli YgbP have been reported38: the apo form of the protein (1INJ) and its complexes with CTP (1I52) or CDP-ME (1INI). However, no structure of E. coli YgbP in complex with MEP has been obtained to date. To identify the mode of binding of 11, we attempted to solve the crystal structure of E. coli YgbP in complex with 11 but we were unsuccessful. The lack of crystal structure of YgbP in complex with MEP or with its structural analogue MEPN3 may be due to an ordered sequential mechanism, in which CTP binds first to the enzyme followed by MEP, as proposed by Cane and co-workers15. To further investigate the binding mode of MEPN3, docking experiments were performed using the E. coli YgbP homodimeric CTP form (1I52). Indeed, this form appears to be the most suitable since it has the highest resolution and is the only one that displays a well-defined P-loop (residues 17–25)38. Two sets of docking were done: the first one using a protein target containing a bound CTP and a second one using an empty protein target. To avoid bias linked to the use of a too small docking area, we defined, for both cases, a search area covering the entire YgbP CTP/CDP-ME binding site. To validate our docking procedure, CTP for which the crystallographic structure in complex with YgbP is available, was submitted to our docking protocol. The docked CTP superimposed very well onto the CTP observed in the crystal structure. The closest docking pose displays a docking score of −11.27 and a RMSD value of 0.11 Å (Fig. 6).

In silico docking results. Docking experiments were performed with the X-ray structure of E. coli YgbP: CTP complex (PDB ID: 1I52). The best docking poses and their corresponding docking scores (D.S) are reported. (A,B) CTP and respectively MEPN3 docked using a target with an empty binding pocket. (C) to (F) docking poses revealing the two possible binding sites observed for MEP (C,E) or MEPN3 (D,F) when the docking experiments were performed with a target already containing bound CTP. The compounds were docked using Glide in extra precision (XP) mode and the Glide docking score was used to rank the docking poses.
When using a target already containing CTP, we were able to identify two binding sites for MEP and MEPN3 (Fig. 6): one deeply buried at the bottom of the CDP-ME pocket with docking scores of −7.86 and −7.96 for MEPN3 and MEP respectively, and a second site located closest to the surface of the protein which appears to be more favorable for the binding of the two molecules given the slightly better docking scores obtained in that case (−9.28 and −8.64 for MEPN3 and MEP respectively). When the empty protein (no CTP bound) was used as a target, we observed that MEPN3 preferentially docked into the CTP binding pocket with a docking score for the best pose of −8.85.
The docking results confirmed that MEPN3 can bind either to the CTP binding pocket or to the hypothetical MEP binding pocket explaining the mixed type inhibition of MEPN3 for E. coli YgbP revealed by the kinetic data. These results show that the very high inhibition potential of MEPN3 compared to the other E. coli YgbP described inhibitors is most probably due to the fact that MEPN3 can bind to both substrate pockets. The design of such inhibitors has never been achieved previously. Interestingly, fosmidomycin (28), has also been reported to bind to the CTP binding site of E. coli YgbP according to docking experiments36. The high potential of fosmidomycin as a drug compared to the other known DXR inhibitors might be linked to this additional binding property. The druggability of the CTP-binding pocket in the homologue protein of M. tuberculosis has actually been reported by Hirsch and co-workers39. In this context, the binding of MEPN3 into the CTP-binding site should be considered as a starting point for new antibacterial development.
As MEPN3 is an analogue of MEP, we further checked whether MEP could also bind to the CTP pocket of E. coli YgbP. Using the target without bound CTP, our experiments revealed that MEP also preferentially docks to this pocket with a docking score of −9.40 (Fig. S5). No other evidence suggesting the binding of MEP to the CTP-binding pocket has been reported but if this hypothesis based on docking were to be true, the multiple binding sites revealed here for MEP would be puzzling from the catalytic point of view. This has prompted us to further investigate the mechanism of E. coli YgbP using a more detailed kinetic analysis.
Investigation of E. coli YgbP mechanism by using a bi-substrate kinetic analysis
Cane and co-workers15 highlighted, using pulse-chase experiments, that the formation of CDP-ME was accomplished by an ordered sequential mechanism, in which CTP binds first to the enzyme followed by MEP binding. Nucleophilic attack on α-phosphate group of CTP by the phosphate moiety of MEP will then afford a pentacoordinate intermediate that will subsequently collapse to produce CDP-ME and diphosphate15,38. However, the ordered sequential catalytic mechanism of YgbP has never been characterised using a complete bisubstrate kinetic analysis. Such a kinetic analysis was performed here by measuring the velocity of different assays in which one of the substrate concentration was varied at different but fixed concentrations of the second substrate. Double reciprocal plots of initial velocity for both substrates resulted in lines intersecting left to the vertical axis and above to the horizontal axis (Fig. 7a,c) confirming that YgbP mechanism is sequential where ternary complex forms before any product release40,41.

Bi-substrate steady-state kinetic analysis of E. coli YgbP. (a) Double reciprocal plot of initial velocities at variable MEP concentrations and fixed CTP concentrations (30 µM, r2 = 0.99; 50 µM, r2 = 0.99; 80 µM, r2 = 0.96; 240 µM, r2 = 0.99). (b) Slope and intercept replot of (a) (slope, r2 = 0.97; intercept, r2 = 0.90). (c) Double reciprocal plot of initial velocities at variable CTP concentrations and fixed MEP concentrations (10 µM, r2 = 0.99; 20 µM, r2 = 0.96; 40 µM, r2 = 0.95; 120 µM, r2 = 0.98). (d) Slope and intercept replot of (c) (slope, r2 = 0.97; intercept, r2 = 0.92). Mean and SEM values are displayed, n ≥ 3.
After fitting the data to the rate equation for sequential bi-substrate mechanism (See supporting information for more details), the complete kinetic scheme for E. coli YgbP was obtained for the first time (Fig. 8). Km values of 149 μM (KiA) for CTP and 46 μM (KiB) for MEP were retrieved and were found to be in agreement with the Km values obtained using the classical Michaelis–Menten equations (Km = 84 μM for CTP and Km = 40 μM for MEP). The dissociation constant of YgbP-CTP complex for MEP (KB) was revealed to be very low (20 μM) while the dissociation constant of YgbP-MEP complex for CTP (KA) was thirteen times higher (265 μM). These values show that if CTP binds to the free enzyme first, the affinity of MEP for YgbP-CTP complex is increased (low dissociation constant) leading to the production of CDP-ME. If MEP binds to the free enzyme first, the affinity of CTP for YgbP-MEP complex will be very low (high dissociation constant) and the YgbP-MEP complex might not be productive and would dissociate to regenerate the free enzyme. This achievement is compatible with the hypothesis that MEP could bind to the CTP pocket and if this happens, the resulting complex would be expected to dissociate to allow the binding of CTP in the CTP pocket. Then, once MEP is present in the active site, the reaction would proceed.

E. coli YgbP bi-substrate steady state kinetic analysis. For reasons of clarity, the scheme was simplified as in the YgbP-MEP complex, MEP could also bind in the CTP binding site. See Fig. S3 for detailed reaction mechanism.
Selectivity of MEPN3
Fosmydomycin is the only inhibitor of the MEP pathway currently under clinical trial. It targets DXR but also to a weaker extend YgbP. This double targeting of fosmidomycin for the MEP pathway is very interesting and may be at the origin of the high potential of fosmidomycin as an antibiotic compared to the other MEP pathway inhibitors. This unique property of fosmidomycin encouraged us to investigated whether MEPN3 is selective for YgbP or if it targets another enzyme of the MEP pathway. In this context, DXR was investigated as a potential target for our inhibitor, as it lays just before YgbP in the MEP pathway and produces MEP. Docking experiments were carried out based on the structure of E. coli YgbP in complex with 1-deoxy-D-xylulose 5-phosphate (5) and NADPH (1Q0Q). Two sets of docking were performed: one with only NADPH bound and another with the empty target. Interestingly, we observed that MEPN3 binds in the binding site of the substrate 5 with a docking score of −9.09 for the best pose (Fig. S6). These results need to be further confirmed using kinetic investigations on DXR but they already highlight that MEPN3 is a good starting point in the search for new drugs as it might target several enzymes of the MEP pathway.
Conclusion
We have successfully synthesised MEPN3, the best inhibitor of E. coli YgbP/IspD known to date and the first inhibitor shown to bind to either or both substrate binding sites. This special binding feature appears to be at the origin of the potency of MEPN3. In addition, our in-depth kinetic studies of YgbP, using a bi-substrate model for the first time also highlighted that the binding of MEP to the free enzyme disfavored the formation of the product. Building on the knowledge gained from our study, new inhibitors derived from MEPN3 might be further elaborated either by developing new analogs bearing this dual binding profile or via structure-based fragment selection and in situ chemistry since MEPN3 is already a good starting point for such strategy. With this aim, fragments could be among other MEP analogues or other molecules binding to the CTP pocket. In this context, preliminary docking experiments were performed using the empty target and a ligand obtained by replacing the azido function of 11 by a methyltriazole moiety. Docking scores and the pose obtained when 11 is in the CTP pocket show that fragment growing using click chemistry is feasible.
Therefore, the discovery of MEPN3 as a new YgbP inhibitor as well as its unusual mode of action paves the way for original approaches toward the discovery of drug candidates that are urgently needed for the treatment of antimicrobial-resistant Enterobacteriacea infections.
Materials and Methods
General conditions for enzyme kinetics
Colorimetric assay reported by Bernal et al.14 was used with some modifications. The standard reaction mixture contained 50 mM Tris-HCl pH = 8, 1 mM MgCl2, 1 mM DTT, 133 mU/mL of inorganic pyrophosphatase, 200 µM CTP (when MEP was the variable substrate), 250 µM MEP (when CTP was the variable substrate) unless otherwise stated and 0.065 µg YgbP enzyme in the final volume of 400 µL. Assays were initiated by addition of YgbP and incubated eight min at 30 °C before being quenched with 100 µL dye reagent (for preparation see SI). The assays were further incubated for ten min before measuring OD at 630 nm. A blank reaction that contained every component except YgbP was carried out at the same time for each assay and the corresponding OD630 value of the blank was subtracted from the OD630 value measured for the assay. The phosphate concentration of the assays was determined from standard curves obtained by measuring OD630 values of different phosphate standards with concentrations varying from 2 to 30 µM. Data were fitted with the least-squares method to the corresponding equations using GraphPad Prism 7.
YgbP kinetic parameter determination
MEP concentrations were 15, 30, 45, 60, 100, 150 and 250 µM when MEP was the variable substrate and CTP concentration was fixed at 200 µM. CTP concentrations were 30, 60, 90, 120, 180, 270 and 450 µM when CTP was the variable substrate and MEP concentration was fixed at 250 µM.
YgbP inhibition kinetic studies
Steady-state kinetic constants were determined from different assays at several fixed inhibitor concentrations and varying the concentration of one substrate and keeping the concentration of the other substrate constant. The initial velocities and concentrations were fitted according to the appropriate model of inhibition42.
YgbP mechanism determination
YgbP bi-substrate kinetic assays were performed, first by varying MEP concentrations at several fixed CTP concentrations and second by varying CTP concentrations at several fixed MEP concentrations. The data were fitted to the corresponding equations to determine kinetic values as described in SI.
YgbP kinetic studies with MEPN3 as a substrate
MEPN3 concentrations were 40, 150, 300, 400, 600, 1000, 1500, 2000 and 3000 µM and CTP concentration was fixed at 200 µM.
MEPN3 as substrate of YgbP
MEPN3 (0.2 mM) was added to a mixture of CTP (1 mM), MgCl2 (5 mM), DTT (1 mM) in a final volume of 200 μL buffer (Tris HCl, 50 mM pH = 8). E. coli YgbP (34 µg) was added to initiate the reaction. The reaction mixture was incubated at 30 °C for one h then MeCN (200 µL) was added, and the mixture was left at 0 °C for 20 min to precipitate proteins. The precipitate was removed by centrifugation (13000 rpm, 10 min). BCN-TMPP (0.8 mM) was added to the supernatant that was further incubated at 37 °C overnight. The mixture was analysed by LC-MS (Waters Alliance 2690 LC system with C18 column coupled with Waters ACQUITY QDa mass detector) using 10 µL of injection volume.
Docking experiments
In silico docking experiments were carried out with the Schrödinger suite (Schrödinger LLC, New York, NY, USA). The X-ray structure of the CDP-ME synthase with CTP (PDB ID 1I52) was used for the studies38. The protein structure was processed with the protein preparation wizard tool. Ligand 3D structures, tautomers and ionisation states were produced with LigPrep. The CDP-ME binding pocket was used to generate the docking grid. We defined a docking area of 32 Å × 32 Å × 32 Å centered on the reaction product (CDP-ME). No constraints (such as hydrogen bond or atom position) were applied to guide the binding. All the compounds were docked using Glide43 in extra precision (XP) mode44. The Glide docking score was used to rank the docking poses.
References
Grundmann, H. et al. Occurrence of carbapenemase-producing Klebsiella pneumoniae and Escherichia coli in the European survey of carbapenemase-producing Enterobacteriaceae (EuSCAPE): a prospective, multinational study. Lancet. Infect. Dis 17, 153–163, https://doi.org/10.1016/S1473-3099(16)30257-2 (2017).
ANTIMICROBIAL RESISTANCE Global Report on Surveillance. (World Health Organization, 2014).
Prioritization of pathogens to guide discovery, research and development of new antibiotics for drug resistant bacterial infections, including tuberculosis. Report No. WHO/EMP/IAU/2017.12, (World Health Organization, 2017).
Rohmer, M., Grosdemange-Billiard, C., Seemann, M. & Tritsch, D. Isoprenoid biosynthesis as a novel target for antibacterial and antiparasitic drugs. Curr Opin Investig Drugs 5, 154–162 (2004).
Gräwert, T., Groll, M., Rohdich, F., Bacher, A. & Eisenreich, W. Biochemistry of the non-mevalonate isoprenoid pathway. Cell Mol Life Sci 68, 3797–3814 (2011).
Zhao, L. S., Chang, W. C., Xiao, Y. L., Liu, H. W. & Liu, P. H. Methylerythritol Phosphate Pathway of Isoprenoid Biosynthesis. Annu. Rev. Biochem 82, 497–530, https://doi.org/10.1146/annurev-biochem-052010-100934 (2013).
Masini, T. & Hirsch, A. K. H. Development of Inhibitors of the 2C-Methyl-D-erythritol 4-Phosphate (MEP) Pathway Enzymes as Potential Anti-Infective Agents. J. Med. Chem. 57, 9740–9763, https://doi.org/10.1021/jm5010978 (2014).
Rohmer, M. The discovery of a mevalonate-independent pathway for isoprenoid biosynthesis in bacteria, algae and higher plants. Nat. Prod. Rep. 16, 565–574 (1999).
Heuston, S., Begley, M., Gahan, C. G. M. & Hill, C. Isoprenoid biosynthesis in bacterial pathogens. Microbiology 158, 1389–1401, https://doi.org/10.1099/mic.0.051599-0 (2012).
Bloch, K. Sterol Molecule - Structure, Biosynthesis, and Function. Steroids 57, 378–383 (1992).
Evaluation of Fosmidomycin and Piperaquine in the Treatment of Acute Falciparum Malaria, https://ClinicalTrials.gov/show/NCT02198807 (2015).
Wells, T. N. C., van Huijsduijnen, R. H. & Van Voorhis, W. C. Malaria medicines: a glass half full? Nat. Rev. Drug. Discov. 14, 424–442, https://doi.org/10.1038/nrd4573 (2015).
Rohdich, F. et al. Cytidine 5′-triphosphate-dependent biosynthesis of isoprenoids: YgbP protein of Escherichia coli catalyzes the formation of 4-diphosphocytidyl-2-C-methylerythritol. Proc. Natl. Acad. Sci. USA 96, 11758–11763, https://doi.org/10.1073/pnas.96.21.11758 (1999).
Bernal, C., Palacin, C. & Boronat, A. & Imperial, S. A colorimetric assay for the determination of 4-diphosphocytidyl-2-C-methyl-D-erythritol 4-phosphate synthase activity. Anal. Biochem. 337, 55–61, https://doi.org/10.1016/j.ab.2004.10.011 (2005).
Richard, S. B. et al. Kinetic Analysis of Escherichia coli 2-C-Methyl-D-erythritol-4-phosphate Cytidyltransferase, Wild Type and Mutants, Reveals Roles of Active Site Amino Acids. Biochemistry. 43, 12189–12197, https://doi.org/10.1021/bi0487241 (2004).
Cane, D. E., Chow, C., Lillo, A. & Kang, I. Molecular cloning, expression and characterization of the first three genes in the mevalonate-independent isoprenoid pathway in Streptomyces coelicolor. Biorg. Med. Chem. 9, 1467–1477, https://doi.org/10.1016/S0968-0896(01)00050-5 (2001).
Lagisetti, C., Urbansky, M. & Coates, R. M. The Dioxanone Approach to (2S,3R)-2-C-Methylerythritol 4-Phosphate and 2,4-Cyclodiphosphate, and Various MEP Analogues. J. Org. Chem. 72, 9886–9895, https://doi.org/10.1021/jo0711900 (2007).
Corey, E. J. & Chaykovsky, M. Dimethyloxosulfonium Methylide ((CH3)2SOCH2) and Dimethylsulfonium Methylide ((CH3)2SCH2). Formation and Application to Organic Synthesis. J. Am. Chem. Soc. 87, 1353–1364, https://doi.org/10.1021/ja01084a034 (1965).
Mazitschek, R., Huwe, A. & Giannis, A. Synthesis and biological evaluation of novel fumagillin and ovalicin analogues. Org Biomol Chem 3, 2150–2154 (2005).
Błażewska, K. M. McKenna Reaction—Which Oxygen Attacks Bromotrimethylsilane? J. Org. Chem. 79, 408–412, https://doi.org/10.1021/jo4021612 (2014).
Krasutsky, S. G. et al. Synthesis of Methylerythritol Phosphate Analogues and Their Evaluation as Alternate Substrates for IspDF and IspE from Agrobacterium tumefaciens. J. Org. Chem. 79, 9170–9178, https://doi.org/10.1021/jo501529k (2014).
Cartwright, A. J., Jones, P., Wolff, J.-C. & Evans, E. H. Detection of phosphorus tagged carboxylic acids using HPLC-SF-ICP-MS. J. Anal. At. Spectrom. 20, 75–80, https://doi.org/10.1039/B415962D (2005).
Woo, H.-K. et al. Phosphonium labeling for increasing metabolomic coverage of neutral lipids using electrospray ionization mass spectrometry. Rapid Commun. Mass Spectrom. 23, 1849–1855, https://doi.org/10.1002/rcm.4076 (2009).
Huang, Z.-H. et al. A Picomole-Scale Method for Charge Derivatization of Peptides for Sequence Analysis by Mass Spectrometry. Anal. Chem. 69, 137–144, https://doi.org/10.1021/ac9608578 (1997).
Ursuegui, S., Recher, M., Krężel, W. & Wagner, A. An in vivo strategy to counteract post-administration anticoagulant activity of azido-Warfarin. Nature Communications 8, 15242, https://doi.org/10.1038/ncomms15242 https://www.nature.com/articles/ncomms15242#supplementary-information (2017).
D’Alessandro, P. L. et al. Bioorthogonal Probes for the Study of MDM2-p53 Inhibitors in Cells and Development of High-Content Screening Assays for Drug Discovery. Angew. Chem. Int. Ed. 55, 16026–16030 (2016).
Imlay, L. S. et al. Plasmodium IspD (2-C-Methyl-D-erythritol 4-Phosphate Cytidyltransferase), an Essential and Druggable Antimalarial Target. ACS Infect Dis 1, 157–167 (2015).
Price, K. E. et al. Molecular Mechanism of Action of Antimalarial Benzoisothiazolones: Species-Selective Inhibitors of the Plasmodium spp. MEP Pathway enzyme, IspD. Sci Rep 6, 36777, https://doi.org/10.1038/srep36777 (2016).
Gao, P. et al. Identification and validation of a novel lead compound targeting 4-diphosphocytidyl-2-C-methylerythritol synthetase (IspD) of mycobacteria. Eur. J. Pharmacol. 694, 45–52, https://doi.org/10.1016/j.ejphar.2012.08.012 (2012).
Varikoti, R. A., Gangwal, R. P., Dhoke, G. V., Ramaswamy, V. K. & Sangamwar, A. T. Structure based de novo design of IspD inhibitors as anti-tubercular agents. Nat. Preced (2012).
Witschel, M. C. et al. Inhibitors of the Herbicidal Target IspD: Allosteric Site Binding. Angew. Chem. Int. Ed. 50, 7931–7935, https://doi.org/10.1002/anie.201102281 (2011).
Kunfermann, A. et al. Pseudilins: Halogenated, Allosteric Inhibitors of the Non-Mevalonate Pathway Enzyme IspD. Angew. Chem. Int. Ed. 53, 2235–2239, https://doi.org/10.1002/anie.201309557 (2014).
Witschel, M., Rohl, F., Niggeweg, R. & Newton, T. In search of new herbicidal inhibitors of the non-mevalonate pathway. Pest Manag. Sci. 69, 559–563, https://doi.org/10.1002/ps.3479 (2013).
Wungsintaweekul, J. PhD thesis thesis, Technical University of Munich (2001).
Lillo, A. M., Tetzlaff, C. N., Sangari, F. J. & Cane, D. E. Functional expression and characterization of EryA, the erythritol kinase of Brucella abortus, and enzymatic synthesis of L-erythritol-4-phosphate. Bioorg. Med. Chem. Lett. 13, 737–739 (2003).
Zhang, B. et al. A Second Target of the Antimalarial and Antibacterial Agent Fosmidomycin Revealed by Cellular Metabolic Profiling. Biochemistry. 50, 3570–3577, https://doi.org/10.1021/bi200113y (2011).
Bartee, D., Wheadon, M. J. & Freel Meyers, C. L. Synthesis and Evaluation of Fluoroalkyl Phosphonyl Analogs of 2-C-Methylerythritol Phosphate as Substrates and Inhibitors of IspD from Human Pathogens. J. Org. Chem., https://doi.org/10.1021/acs.joc.8b00686 (2018).
Richard, S. B. et al. Structure of 4-diphosphocytidyl-2-C-methylerythritol synthetase involved in mevalonate-independent isoprenoid biosynthesis. Nat. Struct. Biol. 8, 641–648, https://doi.org/10.1038/89691 (2001).
Masini, T., Kroezen, B. S. & Hirsch, A. K. H. Druggability of the enzymes of the non-mevalonate-pathway. Drug Discovery Today 18, 1256–1262 (2013).
Copeland, R. A. Evaluation of enzyme inhibitors in drug discovery. A guide for medicinal chemists and pharmacologists. Methods Biochem. Anal. 46, 1–265 (2005).
Borra, M. T., Langer, M. R., Slama, J. T. & Denu, J. M. Substrate specificity and kinetic mechanism of the Sir2 family of NAD(+)-dependent histone/protein deacetylases. Biochemistry. 43, 9877–9887 (2004).
Segel, I. H. Enzyme kinetics: Behavior and Analysis of Rapid Equilibrium and Steady State Enzyme Systems. (Wiley, 1975).
Friesner, R. A. et al. Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem. 49, 6177–6196 (2006).
Halgren, T. A. et al. Glide: A new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J. Med. Chem. 47, 1750–1759 (2004).
Acknowledgements
This work was supported by a grant from “Centre International de Recherche aux Frontieres de la Chimie”, by the Centre National de la Recherche Scientifique (CNRS) and University of Strasbourg. We are thankful to Magali Parisse for technical assistance. We appreciate the help from the staff of the computing facility provided by the Commissariat à l’Energie Atomique (CEA/DSV/GIPSI), Saclay, France, and the Centre de Calcul Recherche et Technologie (CEA/CCRT), Bruyères-le-Châtel, France.
Author information
Authors and Affiliations
Contributions
Z.B., A.W. and M.S. conceived the study, designed the experiments and prepared the manuscript. Z.B. synthesised all compounds and performed in vitro enzymatic assays under the guidance of A.W. and M.S. P.C. provided MEP which was used in the enzymatic assays and participated in the data interpretations. F.B. and J.L.F. performed the docking experiments. All the authors reviewed the final version of the manuscript.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Baatarkhuu, Z., Chaignon, P., Borel, F. et al. Synthesis and Kinetic evaluation of an azido analogue of methylerythritol phosphate: a Novel Inhibitor of E. coli YgbP/IspD. Sci Rep 8, 17892 (2018). https://doi.org/10.1038/s41598-018-35586-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-35586-y
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
