
Abstract
Seeds exhibit wide variation in the fatty acid composition of their storage oil. However, the genetic basis of this variation is only partially understood. Here we have used a multi-parent advanced generation inter-cross (MAGIC) population to study the genetic control of fatty acid chain length in Arabidopsis thaliana seed oil. We mapped four quantitative trait loci (QTL) for the quantity of the major very long chain fatty acid species 11-eicosenoic acid (20:1), using multiple QTL modelling. Surprisingly, the main-effect QTL does not coincide with FATTY ACID ELONGASE 1 and a parallel genome wide association study suggested that LYSOPHOSPHATIDYLCHOLINE ACYLTRANSFERASE 2 (LPCAT2) is a candidate for this QTL. Regression analysis also suggested that LPCAT2 expression and 20:1 content in seeds of the 19 MAGIC founder accessions are related. LPCAT is a key component of the Lands cycle; an acyl editing pathway that enables acyl-exchange between the acyl-Coenzyme A and phosphatidylcholine precursor pools used for microsomal fatty acid elongation and desaturation, respectively. We Mendelianised the main-effect QTL using biparental chromosome segment substitution lines and carried out complementation tests to show that a single cis-acting polymorphism in the LPCAT2 promoter causes the variation in seed 20:1 content, by altering the LPCAT2 expression level and total LPCAT activity in developing siliques. Our work establishes that oilseed species exhibit natural variation in the enzymic capacity for acyl editing and this contributes to the genetic control of storage oil composition.
Similar content being viewed by others

Introduction
Seed maturation is associated with the deposition of storage reserves, such as oil (triacylglycerol), carbohydrates and proteins1. The most common reserve is oil, which can account for up to ~70% of seed weight in some species2. The physiological role of storage oil is to provide a source of carbon to support post-germinative growth, thereby allowing seedling establishment and the completion of the plant’s life cycle3. However, seed storage oils also serve as a primary source of nutrition for humans and livestock, and provide renewable chemical feedstock for a variety of industrial applications4. The fatty acid composition of seed storage oils varies greatly5 and is believed to have adaptive significance6. For example, biogeographic studies indicate that the degree of fatty acid unsaturation has played a role in local adaptation to temperature on a micro- and a macroevolutionary scale7,8. Understanding how plants control seed storage oil content and fatty acid composition is of both strategic and fundamental interest1,9.
The model plant Arabidopsis thaliana has served as a powerful research tool to study many aspects of seed biology. Much of our molecular understanding of storage oil deposition in dicotyledonous seeds is founded on Arabidopsis mutant studies1. Forward and reverse genetic screens have identified the network of transcriptional master regulators that orchestrate the seed maturation program10,11, as well as many downstream components of the metabolic machinery that partitions imported sucrose into triacylglycerol (TAG)1,9. Much of the underpinning knowledge obtained from the study of Arabidopsis has also proved useful in understanding crop species, such as oilseed rape (Brassica napus), which is also a member of the Brassicaceae.
Arabidopsis has a wide geographical distribution and exhibits significant natural variation in seed TAG content and composition12,13. Several studies have used recombinant inbred populations derived from bi-parent crosses to map quantitative trait loci (QTL) controlling oil composition6,14,15,16. The power to detect QTL with this method is high because the minor allele frequency (p) is ~0.5, but mapping resolution is often relatively poor (~5 to 20 cM)17. Branham et al., (2016) have also performed a genome-wide association study (GWAS) of oil composition using ~200,000 sequence variants in a panel of ~400 accessions18. The mapping resolution of this approach is superior because linkage disequilibrium decays rapidly in natural accessions19. However, QTL discovery rate can be lower because p is <0.5 for many alleles20 and marker coverage is usually incomplete21. Complex population structure20 and allelic heterogeneity at causal loci22 can also lead to spurious and ghost associations. Although previous studies have identified many genomic regions that are associated with seed TAG composition6,14,16,18, only one QTL has ever been fine-mapped and the causal sequence variant determined15.
The development of multi-parent advanced generation inter-cross (MAGIC) populations allows the complementary use of both linkage and association methodologies, without any confounding caused by population structure23. Kover et al., (2009) created a large Arabidopsis MAGIC population that encompasses the genetic variation within 19 founder accessions and consists of >500 recombinant inbred lines (RILs)24. Both the founder accessions and the RILs, have been sequenced providing comprehensive marker coverage, consisting of ~3 million individual sequence variants25,26. Arabidopsis seed TAG is primarily composed of polyunsaturated fatty acids (PUFAs) and very long chain fatty acids (VLCFAs)12,13. We have recently used the MAGIC population to study the genetic control of fatty acid desaturation in seeds27,28. The aim of this study was to investigate the control of fatty acid elongation and to identify both QTL and their underlying causal sequence variants.
Results
FAE1 is not the major determinant of seed 20:1 content in the MAGIC population
Arabidopsis thaliana seed TAG contains a high proportion of VLCFAs, of which 11-eicosenoic acid (20:1) is the predominant form12,13. Kover et al., (2009) previously selected 19 Arabidopsis founder accessions, representing a wide range of genotypic and phenotypic diversity, to construct a MAGIC population24. To investigate whether this population would contain significant variation in 20:1 content, we first analysed the fatty acid composition of seeds from the 19 founder accessions28. The 20:1 content ranged between 18.2 ± 0.2 and 22.1 ± 0.3 mol% (n = 5, s.e.m.). We therefore analysed the fatty acid composition of 427 RILs from the MAGIC population, that were grown as three biological replicates in a random block design experiment28. The broad-sense heritability (H2) for 20:1 was high (0.85) and line averages for the RILs ranged between 16.7 ± 0.3 and 22.9 ± 0.1 mol% (n = 3, s.e.m.).
We used the seed 20:1 content line averages to carry out both QTL analysis and GWAS, exploiting genomic resources and software tools developed for the MAGIC population by Richard Mott and colleagues24,25,26. Using multiple QTL modelling, we identified four 20:1 QTL, with a genome-wide P value < 0.01 (Supplementary Table 1). 20:1q2 accounted for most of the phenotypic variation in the trait and is situated on Chromosome 1 at ~23.3 Mb. The 90% confidence interval (CI) for this QTL is ~0.8 Mb24. The other three more minor QTL (20:1q1, 3 and 4) are situated on Chromosomes 1, 4 and 5 at ~0.6, ~17.4 and ~11.8 Mb, respectively (Supplementary Table 1). The 90% CI for 20:1q3 corresponds approximately to the location of FATTY ACID ELONGASE 1 (FAE 1), which is situated at ~16.5 Mb on Chromosome 4. FAE 1 encodes the β-ketoacyl-Coenzyme A synthase activity of the fatty acid elongase complex in developing seeds29 and has previously been shown to be a major-effect QTL that underlies variation in seed VLCFA / 20:1 content among several Arabidopsis accessions6,15,16.
LPCAT2 is a candidate for the main-effect QTL for 20:1 content in the MAGIC population
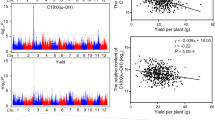
In parallel to the QTL analysis, we also performed GWAS to identify individual polymorphisms associated with seed 20:1 content (Fig. 1). This analysis used all ~3 million sequence variants within the imputed genomes of the MAGIC RILs25. Polymorphisms with a –log10(p) score above the genome-wide significance threshold were detected only within the 90% CIs for 20:1q1 and 20:1q2 (Fig. 1). The polymorphisms were ranked by –log10(p) score and genes that map within 1 kb up or downstream were identified and listed, together with their gene names/descriptions from The Institute for Genome Research (Supplementary Dataset 1). The highest-ranking polymorphisms within the 20:1q2 CI are all confined to a ~70 kb region and lie within 1 kb of 14 genes (Supplementary Table 2). The function of these genes was investigated by searching relevant databases such as ARALIP9, the Arabidopsis Information Resource (https://www.Arabidopsis.org/) and KnetMiner30. The strongest candidate amongst the 14 genes is LYSOPHOSPHATIDYLCHOLINE ACYLTRANSFERASE 2 (LPCAT2), based on prior knowledge of gene function9,31,32.

Manhattan plot showing association of ~3 million individual sequence variants with seed 20:1 content in the MAGIC population. Individual plants for 427 RILs were grown in three randomised blocks (n = 3) in the glasshouse and the seed for each plant were harvested separately and analysed. GWAS was performed using the ‘magic_src_v4.0.tar.gz’ package. The 90% CIs for corresponding 20:1 QTL are bracketed and the grey line marks the genome-wide significance threshold.
LPCAT2 is the predominant LPCAT isoform in developing Arabidopsis seeds and catalyses the esterification of 1-lysophospatidylcholine (1-LPC) with acyl-Coenzyme A (acyl-CoA) to form phosphatidylcholine (PC)31,32,33. This reversible reaction34,35 is a key component of the Lands cycle, or acyl editing pathway, that allows newly synthesised fatty acids in the acyl-CoA pool to enter the sn-2 position of PC for desaturation and subsequent assembly into TAG31,32. An alternative fate for fatty acids in the acyl-CoA pool is to undergo elongation to form VLCFAs such as 20:19. Loss of LPCAT2 function has been shown to increase seed 20:1 content, mainly at the expense of PUFAs32,36, while gain of function leads to a decrease in 20:132. It is therefore conceivable that allelic variation in LPCAT2 could underlie 20:1q2 and account for much of the phenotypic variation in seed 20:1 content within the MAGIC population.
Seed 20:1 content in the MAGIC founder accessions is related to LPCAT2 expression
Gan et al.25 have previously shown that transcript abundance data for the 19 founder accessions of the MAGIC population can be used to identify potential cis-acting variants associated with expression (i.e. cis-eQTL)25. The polymorphisms in LPCAT2, which are most strongly associated with seed 20:1 content, are located within the promoter (P1), first intron (P2) and 3′ intergenic region (P3 and P4) (Supplementary Table 3). If one or more of these polymorphisms cause the phenotypic variation in seed 20:1 content, they would most likely act by modifying the level of LPCAT2 expression. We therefore used quantitative RT-PCR to measure LPCAT2 transcript abundance in seeds from each of the 19 founder accessions. Linear regression analysis suggests that there is a significant negative relationship between LPCAT2 transcript abundance and seed 20:1 content (R2 = 0.652; P < 0.05) (Fig. 2). Furthermore, LPCAT2 expression level was also related to variation in the four polymorphisms at this locus (P < 0.05), which constitute a haplotype that distinguishes Colombia-0 (Col-0) and Rschew (Rsch-4) from the remaining 17 founder accessions (Fig. 2), including Landsberg erecta (Ler-0)25,37. LPCAT2 transcript abundance was ~5-fold higher in Col-0 seeds than in Ler-0 (Fig. 2).

Relationship between LPCAT2 expression and 20:1 content in seeds of the 19 founder accessions of the MAGIC population. Quantitative RT-PCR was performed on RNA from mature seeds of five separate plants of each genotype (n = 5) and values are normalised to Col-0 and expressed as the mean ± s.e.m. Seed 20:1 content is also the mean of measurements on five separate plants (n = 5) and all s.e.m. are < 0.3. Closed symbols are accessions that have the Col-0 LPCAT2 haplotype (Supplementary Table 3) and square symbols highlight Col-0 and Ler-0. Regression analysis supports a negative linear relationship (R2 = 0.652; P < 0.05). The dotted line marks the 95% confidence interval.
The main-effect QTL 20:1q2 can be Mendelianised using biparental substitution lines
To investigate whether allelic variation between Col-0 and Ler-0 can explain the main-effect QTL 20:1q2, we obtained three chromosome segment substitution lines with single Ler-0 introgressions on chromosome 1 in a Col-0 background38. SRL1 0–3, SRL1 0–24 and SRL1 0–84 contain introgressions between 0 and 3–24, 24–42 and 84–115 cM, respectively38. PCR-based genotyping using polymorphic markers showed that the Ler-0 introgression in SRL1 0–84 extends beyond ~90 cM (renamed SRL1 0–90) and therefore encompasses the 90% CIs of both 20:1q1 and 20:1q2 (Fig. 3a). Analysis of fatty acid composition showed that the 20:1 content of SRL1 0–90 seed is significantly higher than that of Col-0 (P > 0.05), whereas 20:1 content of SRL1 0–3 and SRL1 0–24 seed is not significantly different from Col-0 (Fig. 3b). These data are consistent with the presence of a 20:1q2 allele in Ler-0 that is hypomorphic to Col-0. We backcrossed SRL1 0–90 to Col-0 and used PCR-based polymorphic markers to obtain a new near isogenic line (NIL) from the F2 progeny. SRL1 84–90 contains a single Ler-0 introgression between 61–84 and 90–115 cM that encompasses 20:1q2, and not 20:1q1 (Fig. 3a). Fatty acid analysis of SRL1 84–90 seed confirmed that the 20:1 content is significantly higher than Col-0 (P > 0.05; Fig. 3b).

Mendelianisation of 20:1q2 using biparental substitution lines. (a) The positions of single Ler-0 introgressions on Chromosome 138 are shown. Crossovers lie within the grey regions between markers. (b) The fatty acid composition of seeds from five separate plants of each genotype (n = 5) was determined and 20:1 content is the mean ± s.e.m. The asterisk denotes values that are significantly different (P > 0.05) from Col-0 (LSD-test).
A 20:1q2 NIL has a lpcat2 seed fatty acid profile and reduced LPCAT expression and activity
20:1q2 maps to the location of LPCAT2 (Fig. 1; Supplementary Table 2) and lpcat2 null mutants in the same genetic background as SRL1 84–90 (Col-0) also have elevated 20:1 content32,36. Seed of lpcat2 exhibit additional characteristic changes in fatty acid profile, including a reduction in PUFA content32, that results from reduced acyl-entry into the PC substrate pool for desaturation31,32. We therefore analysed the total fatty acid composition of SRL1 84–90 seed and found that oleic acid (18:1), linoleic acid (18:2) and linolenic acid (18:3) content are also significantly reduced (P > 0.05) relative to Col-0 (Fig. 4) and that the total fatty acid profile mirrors that of lpcat2-232. To determine whether SRL1 84–90 is defective in LPCAT2 function we preformed quantitative RT-PCR analysis and microsomal enzyme activity on siliques containing developing seeds at curled to green cotyledons stage (i.e. stage 8 to 9)32. LPCAT2 transcript abundance was reduced by ~80% in SRL1 84–90 siliques (Fig. 5a) and total LPCAT activity was reduced by ~60% (Fig. 5b). For comparison, LPCAT2 transcripts were absent in lpcat2-2 siliques and LPCAT activity was reduced by ~70% (Fig. 5b).

Seed fatty acid profile of Col-0, lpcat2-2 and SRL1 84–90, carrying a single Ler-0 introgression in the region of 20:1q2. The fatty acid composition of seeds from five separate plants of each genotype (n = 5) was determined and values are the mean ± s.e.m. The asterisk denotes values that are significantly different (P > 0.05) from Col-0 (LSD-test).

LPCAT expression and activity and Col-0, lpcat2-2 and SRL1 84–90, carrying a single Ler-0 introgression in the region of 20:1q2. (a) Quantitative RT-PCR and (b) microsomal enzyme assays were performed on siliques containing seeds at stages 8 to 9 of development60. Values are the mean ± s.e.m. of measurements on three separate extracts (n = 3) and expression is normalised to that of Col-0. The asterisk denotes values that are significantly different (P > 0.05) from Col-0 (LSD-test).
A complementation test suggests 20:1q2 and LPCAT2 are synonymous
To test whether variation in LPCAT2 allele function between Col-0 and Ler-0 is sufficient to explain 20:1q2, we performed a complementation test15. We carried out reciprocal crosses between wild-type Col-0, the homozygous lpcat2-2 mutant32 and SRL1 84-90, which carries a single Ler-0 introgression at 20:1q2. We then analysed the fatty acid composition of heterozygous F1 seed and self-pollinated homozygous F1 seed (Fig. 6). The 20:1 content of Col-0/SRL1 84–90 and Col-0/lpcat2-2 seed was not significantly different from Col-0 (P < 0.05), suggesting that lpcat2-2 and Ler-0 20:1q2 are both recessive hypomorphic mutant alleles (Fig. 6). When we measured the 20:1 content of lpcat2-2/SRL1 84–90 seed it was significantly higher than Col-0 (P < 0.05) (Fig. 6). This lack of complementation by recessive alleles suggests that LPCAT2 and 20:1q2 are synonymous.

Complementation test between 20:1q2 near isogenic line SRL1 84–90 and lpcat2-2. Reciprocal crosses were performed between Col-0, lpcat2-2 and SRL1 84–90, carrying a single Ler-0 introgression in the region of 20:1q2. The fatty acid composition of heterozygous (and homozygous) F1 seed from five plants of each genotype (n = 5) was measured and 20:1 content is the mean ± s.e.m. The asterisk denotes values that are significantly different (P > 0.05) from Col-0 (LSD-test). For heterozygous F1 seed the maternal parent is listed first.
An INDEL in the LPCAT2 promoter is responsible for the variation in seed 20:1 content
Four polymorphisms in LPCAT2 are strongly associated with both seed 20:1 content and LPCAT2 expression level in the MAGIC population (Fig. 2, Supplementary Table 3). These polymorphisms form a haplotype that distinguishes Col-0 from Ler-0 LPCAT2. P1 is an insertion-deletion (INDEL) that lies early in the promoter of LPCAT2, P2 is a single nucleotide polymorphism (SNP) in the first intron and P3 and P4 are SNPs that lie downstream in the 3′ intergenic region (Supplementary Table 3). To test whether one or more of these polymorphisms cause variation in LPCAT2 function between Col-0 and Ler-0, we transformed lpcat2-2 with three different T-DNA constructs (Fig. 7a). The first construct (CL) contained a ~3.3 kb genomic region of Col-0 LPCAT2 encompassing P1 and P2. The second construct (LL) contained the corresponding genomic region of Ler-0 LPCAT2. Finally, the third construct (LP) was the same as LL, but contained the Col-0 variant of P1. Fatty acid analysis performed on seeds from three independent homozygous transgenic lines containing each construct showed that CL and LP could both complement the seed 20:1 content phenotype of lpcat2-2, whereas LL could not (Fig. 7b). Although we cannot rule out a contribution from other polymorphisms in the region of LPCAT2, our data suggest that P1 (a 3 bp INDEL at −27 bp in the promoter) is sufficient to explain allelic variation in LPCAT2 function between Col-0 and Ler-0, and by extension 20:1q2 in the MAGIC population.

Identification of the polymorphism that causes variation in LPCAT2 function between Col-0 and Ler-0 alleles. (a) T-DNA constructs containing a ~3.3 kb genomic region of Col-0 LPCAT2 (CL), Ler-0 LPCAT2 (LL) or LL with a Col-0 variant of P1 (LP) were transformed into lpcat2-2. (b) Fatty acid analysis was performed on seeds from three homozygous transformants (1–3) for each construct and 20:1 content is the mean ± s.e.m. of measurements on five plants (n = 5) of each genotype. The asterisk denotes values that are not significantly different (P < 0.05) from Col-0 (LSD-test).
Discussion
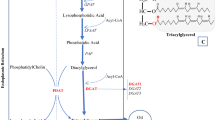
In this study, we show that natural variation in LPCAT231,32,33 is a determinant of seed storage oil composition in Arabidopsis. LPCAT catalyses the reversible acylation of 1-LPC34,35, and in doing so enables acyl-exchange (or ‘acyl editing’) between the acyl-CoA and PC pools that are the respective sites of microsomal fatty acid elongation and desaturation31,32 (Fig. 8). Previous work has established that acyl-editing makes a major contribution to acyl flux into TAG in several oilseed species31,32,39,40,41,42 and disruption of LPCAT in Arabidopsis decreases fatty acid unsaturation and increases chain length31,32. There has long been speculation that LPCAT activity contributes to the regulation of seed TAG composition, particularly in PUFA-rich species32,42. However, acyl editing also takes place without any acyl modification31,32,41,42. Our study establishes that natural variation in acyl editing exists in oilseeds and is a factor that contributes to the genetic regulation of TAG composition.

A simplified diagram of fatty acid synthesis, modification and assembly into triacylglycerol in developing Arabidopsis seeds8. The diagram illustrates the role of LPCAT in acyl partitioning between the cytosolic acyl-Coenzyme A (CoA) and phosphatidylcholine (PC) pools used for microsomal elongation and desaturation. Major acyl groups made (or present) in the plastidial acyl-acyl carrier protein (ACP) and cytosolic acyl-CoA and PC pools are shown. 2:0 is acetic acid; 16:0 is palmitic acid, 18:0 is stearic acid, 18:1 is oleic acid, 20:0 is eicosanoic acid, 20:1 is eicosenoic acid, 18:2 is linoleic acid and 18:3 is linolenic acid. DAG is diacylglycerol, TAG is triacylglycerol, LPCAT is acyl-CoA:lysophosphatidylcholine acyltransferase, FAE is fatty acid elongase, FAD is fatty acid desaturase, PDCT is PC:DAG cholinephosphotransferase, PDAT is phospholipid:DAG acyltransferase and DGAT is acyl-CoA:DAG acyltransferase. The dotted arrow represents the first three catalytic steps of the Kennedy pathway responsible for acyl-CoA dependent formation of DAG from glycerol-3-phosphate.
Within the MAGIC population24, we found that a causal sequence variant in LPCAT2 (P1) is a small INDEL situated in the promoter. Arabidopsis contains two LPCAT genes and Wang et al., (2012) showed that LPCAT2 is the predominant form in developing seeds32. It is not known precisely how LPCAT2 expression is regulated. However, initiation of transcription by RNA polymerase II requires assembly of a basal transcription apparatus at the core promoter, a region of ~70 bp flanking the transcription start site (TSS)43. P1 is situated just 27 bp upstream of the LPCAT2 TSS44 (Supplementary Fig. 1) and it may therefore affect transcriptional initiation, either by modifying a recognition element42 or by changing promoter context45. Extensive cis regulation of gene expression is thought to exist in Arabidopsis46 and Gan et al.25 previously reported that potential cis-acting sequence variants, associated with differential gene expression in seedlings of the MAGIC founder accessions, are concentrated in the ~100 bp promoter region25. Bioinformatic analysis of 1,135 Arabidopsis genomes sequences from the 1001 Genomes Consortium47 suggests that P1 is not a rare allele. The hypermorphic Col-0 variant is present in ~55% of accessions. Furthermore, there is a significant relationship between the genotype at P1 and latitude at the accession collection site47 (Col-0 variant = 48.96° ± 0.29, n = 622 and Ler-0 variant = 47.43° ± 0.33, n = 510; P = 0.0003, two-tailed t-test). An increased capacity for acyl editing at higher latitude is consistent with a need for more substrate to support microsomal fatty acid desaturation al lower temperatures28,48. Hence, allelic variation at P1 may be significant in local adaptation to temperature in natural populations of Arabidopsis48.
Natural variation is seed TAG composition has been studied quite extensively in Arabidopsis and many QTL have previously been identified6,14,15,16,18,27,28. In several studies, the main-effect QTL for VLCFA (or 20:1) content has mapped to the location of FAE16,15,16. The identification of LPCAT2 as a main-effect QTL for 20:1 content in the MAGIC population was therefore surprising, but this just reflects a lack of causal allelic variation in FAE1 within the 19 MAGIC founder accessions. These founder accessions are not polymorphic for the nonsynonymous causal sequence variant in FAE1 that was identified by Jasinski et al., (2012) and no polymorphisms exist between Col-0 and Ler-0 within a region ~300 bp up and downstream of FAE16,25,37. A minor QTL (20:1q3) in the MAGIC population did map to the approximate location of FAE1. However, we could not identify individual sequence variants that are significantly associated with 20:1 content within the 90% CI for 20:1q3.
We could identify individual sequence variants that are significantly associated with 20:1 content within the 90% CI of one other minor QTL (20:1q1). 20:1q1 corresponds approximately to the location of a 20:1/18:1 (oleic acid) ratio QTL previously identified by O’Neill et al. (2012) in a Wietze (Wt-5) × Catania (Ct-1) biparental mapping population16. Ct-1 is one of the 19 MAGIC founder accessions24. We found associated sequence variants within 1 kb up or downstream of several genes in the 20:1q1 90% CI, including three U-box containing proteins that are potential E3 ubiquitin ligases49, three transcription factors, a ATP-binding cassette transporter and a glycerol-3-phosphate acyltransferase (GPAT4) (Supplementary Data 1). Among these genes, only GPAT4 has previously been ascribed a function in lipid metabolism. However, GPAT4 is unlikely to be 20:1q1 because it has been shown to have a specialised role in producing oxygenated sn-2 acyl glycerol monomers for the extracellular polymer cutin50,51. It is noteworthy that 20:1q1 is located relatively close to LPCAT1 and lpcat1 seeds exhibit a slight increase in 20:132. However, LPCAT1 lies outside the 20:1q1 90% CI and no significantly associated sequence variants were detected near this gene (Fig. 1). Further work will be required to identify the causal polymorphism(s) for 20:1q1.
In conclusion, we have found that natural variation in a gene encoding the acyl editing enzyme LPCAT2 influences Arabidopsis seed TAG composition. LPCATs partition newly synthesised fatty acids between the acyl-CoA and PC substrate pools used for microsomal fatty acid elongation and desaturation, respectively31,32. Previous studies have identified natural variation in the enzymes that are directly responsible for fatty acid modification, such as FAE115 and FATTY ACID DESATURASE 228. In vivo pulse radiolabelling studies have shown that, in addition to acyl editing, PC-diacylglycerol (DAG) interconversion also makes a major contribution to acyl flux into TAG in several PUFA-rich oilseed species42 (Fig. 8). The main mechanism for PC-DAG interconversion in Arabidopsis is head group exchange, catalysed by phosphatidylcholine:diacylglycerol cholinephosphotransferase (PDCT)31,52. Phospholipid:diacylglycerol acyltransferase (PDAT) also transfers acyl groups to TAG directly from the sn-2 position in PC53 (Fig. 8) and LPCAT2 is required to re-esterify the 1-LPC co-product36. It will therefore be interesting to explore whether natural variation also exists in PDCT and PDAT, which like LPCAT are not directly involved in acyl modification.
Materials and Methods
Plant material and growth conditions
The Arabidopsis thaliana MAGIC population founder accessions and recombinant inbred lines (RILs) (N782242) and the STAIRS single recombinant lines (SRLs) (N721831) were obtained from the European Arabidopsis Stock Centre (University of Nottingham, UK). The lpcat2-2 mutant32 has been described previously. Seeds were sown on moist Levington F2 compost in P40 trays and vernalized for 6 weeks where necessary before being transferred to a controlled environment chamber or an air-conditioned glasshouse set to a 16-h light (22 °C)/8-h dark (16 °C) cycle. After one week seedlings were individually transplanted to 7 cm2 pots. For the initial analysis of the MAGIC RILs, the pots were arranged into a random block design in the glasshouse28. The plants were bagged individually at the onset of flowering54 and the seeds were then harvested at maturity.
Analysis of seed fatty acid composition
The total fatty acid composition of seeds was measured by gas chromatography55, using the combined digestion and fatty acid methyl ester formation method56.
Genetic analysis
Quantitative Trait Loci (QTL) mapping with the MAGIC population was carried out as described by Kover et al. (2009), using the ‘HAPPY R’ package from, http://archive.is/mus.well.ox.ac.uk 24. Genome-wide association studies (GWAS) were performed with the ‘magic_src_v4.0.tar.gz’ package, which can be obtained from the same site and includes detailed instructions. In brief, the ‘reconstruction’ program generates imputed genomes for the RILs with a mosaic breakpoint accuracy of >2 kb, using polymorphism calls derived from low coverage sequence and 1.2 M biallelic variants from the complete genomes of the 19 MAGIC founder accessions26. The ‘genome_scan’ program then performs association mapping, using all ~3 M individual sequence variants imputed from the 19 MAGIC founders. Specific sequence variants in the genomes of the 19 MAGIC founders were visualised with the Rätsch lab GBrowse tool (http://gbrowse.inf.ethz.ch/gb/gbrowse/thaliana-19magic). Sequence variants in 1,135 Arabidopsis genomes were visualised using Polymorph 1001 (http://tools.1001genomes.org/polymorph/).
Gene expression analysis
RNA was purified from mature seeds and developing siliques and DNase-treated using the RNeasy kit from Qiagen with modifications described previously57. Single-stranded cDNA synthesis was performed using SuperScript II RNase H- reverse transcriptase from Invitrogen. A MyiQ Single-Color real-time PCR detection system (Bio-Rad) was used to carry out real-time PCR with the qPCR Mastermix Plus from Eurogentec. The data were analyzed using Bio-Rad iQ5, Optical System Software, version 2.0. The real-time PCR primer pairs were LPCAT2_Q (5′-tgcggttcagattccgcttttct-3′ and 5′-gttgccaccggtaaatagctttcg-3′) and 18S-Q (5′-tcctagtaagcgcgagtcatc-3′ and 5′- cgaacacttcaccggatcat-3′).
Genotyping substitution lines
STAIRS single recombinant lines (SRLs) and the F2 progeny of a backcross to Col-0 were genotyped using PCR-based simple sequence length polymorphic markers38, as described by Koumproglou et al., (2002). In addition to using the existing markers nga 59, F20D23, nga 392, T27K12Sp6, nga 208, F5I14-49495 and nga 11138, we also created two new markers, using flanking INDELs situated ~1 kb 5′ and 3′ of the LPCAT2 transcribed region. The primer pairs for PCR genotyping with these markers were LPCAT2_5′ (5′-aaaataacatgattttgagttgttgt3′ and 5′-ttgcaaataaatcataatatctaccaa-3′) and LPCAT2_3′ (5′-cgataaggcgctagatgctc-3′ and 5′-cacggcctctcttttcttctt-3′).
Enzyme assay
Microsomal fractions were prepared from homogenates of ~1 g of developing siliques of each genotype and the acylation of lysophosphatidylcholine was then assayed following the methods described previously32.
Cloning and plant transformation
A ~3.3 kb region of Col-0 and Ler-0 genomic DNA containing LPCAT2 was amplified by PCR using primer pair (5′-ccacaggagggcgtcgaattttggtg-3′ and 5′-tggtccactcatcgtctcgctaatgt-3′) and cloned into the pENTR/D-TOPO vector. The Quikchange Lightning Site-Directed Mutagenesis Kit from Agilent Technologies (http://www.agilent.com) and primer pair (5′-aacttcacacaaacctcgtcaagatcgaaaccaaacccac-3′ and 5′-gtgggtttggtttcgatcttgacgaggtttgtgtgaagtt-3′) were then used to introduce the Col-0 variants of polymorphisms P1 (Supplementary Table 3) into the Ler-0 allele, following the manufacturer’s instructions. The gene cassettes were then cloned into the destination vector pEarleyGate30158 using the Gateway LR clonase enzyme mix from Invitrogen Ltd (http://www.thermofisher.com). Heat shock was used to transform the plasmids into Agrobacterium tumefaciens strain GV3101 and Arabidopsis transformation was then carried out using the floral-dip method59. Glufosinate resistance was used to select T0 primary transgenic lines and homozygous T3 lines were subsequently recovered and analysed.
Statistical Analyses
The number of biological replicates (n) and the standard error of the mean (s.e.m.) are shown. ANOVA (one-way analysis of variance) was used to assess differences between genotypes for seed fatty acid measurements. Following significant (P < 0.05) F-test results, means were compared using the appropriate LSD (least significant difference) value at the 5% (P = 0.05) level of significance, on the corresponding df (degrees of freedom). These analyses were performed using GenStat (18th edition, VSN International Ltd, Hemel Hempstead, UK). Linear regression analysis was also performed using the function in SigmaPlot v14.0 (Systat Software Inc.).
Data Availability
The datasets generated during and/or analysed during the current study are available from the corresponding author on reasonable request.
References
Baud, S. & Lepiniec, L. Physiological and developmental regulation of seed oil production. Prog. Lipid Res. 49, 235–249 (2010).
Levin, D. A. The oil content of seeds: An ecological perspective. Am. Nat. 108, 193–206 (1974).
Graham, I. A. Seed storage oil mobilization. Annu. Rev. Plant Biol. 59, 115–142 (2008).
Carlsson, A. S., Yilmaz, J. L., Green, A. G., Stymne, S. & Hofvander, P. Replacing fossil oil with fresh oil - with what and for what? Eur. J Lipid Sci. Technol. 113, 812–831 (2011).
Ohlrogge, J. et al. PlantFAdb: A Resource for Exploring Hundreds of Plant Fatty Acid Structures Synthesized by Thousands of Plants and their Phylogenetic Relationships. Plant J. https://doi.org/10.1111/tpj.14102 (2018).
Sanyal, A. & Linder, R. C. Quantitative trait loci involved in regulating seed oil composition in Arabidopsis thaliana and their evolutionary implications. Theor Appl Genet. 124, 723–738 (2012).
Linder, C. Adaptive evolution of seed oils in plants: accounting for the biogeographic distribution of saturated and unsaturated fatty acids in seed oils. Am. Nat. 156, 442–458 (2000).
Sanyal, A. & Linder, C. R. Plasticity and constraints on fatty acid composition in the phospholipids and triacylglycerols of Arabidopsis accessions grown at different temperatures. BMC Plant Biol. 13, 63 (2013).
Li-Beisson, Y. et al. Acyl-Lipid Metabolism in The Arabidopsis Book, Rockville, MD: American Society of Plant Biologists (2013).
Vicente-Carbajosa, J. & Carbonero, P. Seed maturation: developing an intrusive phase to accomplish a quiescent state. Int. J Dev. Biol. 49, 645–651 (2005).
Santos-Mendoza, M. et al. Deciphering gene regulatory networks that control seed development and maturation in Arabidopsis. Plant J. 54, 608–620 (2008).
Millar, A. A. & Kunst, L. The natural genetic variation of the fatty-acyl composition of seed oils in different ecotypes of Arabidopsis thaliana. Phytochemistry 52, 1029–1033 (1999).
O’Neill, C. M., Gill, S., Hobbs, D., Morgan, C. & Bancroft, I. Natural variation for seed oil composition in Arabidopsis thaliana. Phytochemistry 64, 1077–1090 (2003).
Hobbs, D. H., Flintham, J. E. & Hills, M. J. Genetic control of storage oil synthesis in seeds of Arabidopsis. Plant Physiol. 136, 3341–3349 (2004).
Jasinski, S. et al. Natural variation in seed very long chain fatty acid content is controlled by a new isoform of KCS18 in Arabidopsis thaliana. PLoS One 7, e49261 (2012).
O’Neill, C. M. et al. Towards the genetic architecture of seed lipid biosynthesis and accumulation in Arabidopsis thaliana. Heredity 108, 115–123 (2012).
Kearsey, M. J. & Farquhar, A. G. QTL analysis in plants; where are we now? Heredity 80, 137–142 (1998).
Branham, S. E., Wright, S. J., Reba, A. & Linder, C. R. Genome-Wide Association Study of Arabidopsis thaliana Identifies Determinants of Natural Variation in Seed Oil Composition. J Heredity 107, 248–256 (2016).
Nordborg, M. & Weigel, D. Next-generation genetics in plants. Nature 456, 720–723 (2008).
Clark, R. M. et al. Common sequence polymorphisms shaping genetic diversity in Arabidopsis thaliana. Science 317, 338–342 (2007).
Kim, S. et al. Recombination and linkage disequilibrium in Arabidopsis thaliana. Nat. Genet. 39, 1151–1155 (2007).
Atwell, S. et al. Genome-wide association study of 107 phenotypes in Arabidopsis thaliana inbred lines. Nature 465, 627–631 (2010).
Cavanagh, C., Morell, M., Mackay, I. & Powell, W. From mutations to MAGIC: resources for gene discovery, validation and delivery in crop plants. Curr. Opin. Plant Biol. 11, 215–221 (2008).
Kover, P. X. et al. A Multiparent Advanced Generation Inter-Cross to fine-map quantitative traits in Arabidopsis thaliana. PLoS Genet. 5, e1000551 (2009).
Gan, X. et al. Multiple reference genomes and transcriptomes for Arabidopsis thaliana. Nature 477, 419–423 (2011).
Imprialou, M. et al. Genomic Rearrangements Considered as Quantitative Traits. Genetics 205, 1425–1441 (2017).
Bryant, F. M. et al. ACYL-ACYL CARRIER PROTEIN DESATURASE2 and 3 Are Responsible for Making Omega-7 Fatty Acids in the Arabidopsis Aleurone. Plant Physiol. 172, 154–162 (2016).
Menard, G. N. et al. Genome Wide Analysis of Fatty Acid Desaturation and Its Response to Temperature. Plant Physiol. 173, 1594–1605 (2017).
James, D. W. et al. Directed tagging of the Arabidopsis FATTY ACID ELONGATION1 (FAE1) gene with the maize transposon activator. Plant Cell 7, 309–319 (1995).
Hassani-Pak, K. et al. Developing integrated crop knowledge networks to advance candidate gene discovery. Applied & Translational Genomics 11, 18–26 (2016).
Bates, P. D. et al. Acyl editing and headgroup exchange are the major mechanisms that direct polyunsaturated fatty acid flux into triacylglycerols. Plant Physiol. 160, 1530–1539 (2012).
Wang, L. et al. Metabolic interactions between the Lands cycle and the Kennedy pathway of glycerolipid synthesis in Arabidopsis developing seeds. Plant Cell 24, 4652–4669 (2012).
Ståhl, U., Stålberg, K., Stymne, S. & Ronne, H. A family of eukaryotic lysophospholipid acyltransferases with broad specificity. FEBS Lett. 582, 305–309 (2008).
Stymne, S. & Stobart, A. K. Evidence for the reversibility of the acyl-CoA:lysophosphatidylcholine acyltransferase in microsomal preparations from developing safflower (Carthamus tinctorius L.) cotyledons and rat liver. Biochem. J. 223, 305–314 (1984).
Lager, I. et al. Plant acyl-CoA:lysophosphatidylcholine acyltransferases (LPCATs) have different specificities in their forward and reverse reactions. J. Biol. Chem. 288, 36902–36914 (2013).
Xu, J. et al. Triacylglycerol synthesis by PDAT1 in the absence of DGAT1 activity is dependent on re-acylation of LPC by LPCAT2. BMC Plant Biol. 12, 4 (2012).
Zapata, L. et al. Chromosome-level assembly of Arabidopsis thaliana Ler reveals the extent of translocation and inversion polymorphisms. Proc. Natl. Acad. Sci. USA 113, E4052–4060 (2016).
Koumproglou, R. et al. STAIRS: a new genetic resource for functional genomic studies of Arabidopsis. Plant J. 31, 355–364 (2002).
Slack, C. R., Roughan, P. G. & Balasingham, N. Labelling of glycerolipids in the cotyledons of developing oilseeds by [1–14C] acetate and [2-3H] glycerol. Biochem. J. 170, 421–433 (1978).
Griffiths, G., Stymne, S. & Stobart, A. K. The utilisation of fatty-acid substrates in triacylglycerol biosynthesis by tissue-slices of developing safflower (Carthamus tinctorius L.) and sunflower (Helianthus annuus L.) cotyledons. Planta 173, 309–316 (1988).
Bates, P. D., Durrett, T. P., Ohlrogge, J. B. & Pollard, M. Analysis of acyl fluxes through multiple pathways of triacylglycerol synthesis in developing soybean embryos. Plant Physiol. 150, 55–72 (2009).
Allen, D. K., Bates, P. D. & Tjellström, H. Tracking the metabolic pulse of plant lipid production with isotopic labelling and flux analyses: past, present and future. Prog. Lipid Res. 58, 97–120 (2015).
Molina, C. & Grotewold, E. Genome wide analysis of Arabidopsis core promoters. BMC Genomics 6, 25 (2005).
Morton, T. et al. Paired-end analysis of transcription start sites in Arabidopsis reveals plant-specific promoter signatures. Plant Cell 26, 2746–2760 (2014).
Liu, L. et al. Induced and natural variation of promoter length modulates the photoperiodic response of FLOWERING LOCUS T. Nature Comm. 5, 4558 (2014).
Keurentjes, J. J. et al. Regulatory network construction in Arabidopsis by using genome-wide gene expression quantitative trait loci. Proc. Natl. Acad. Sci. USA 104, 1708–1713 (2007).
1001 Genomes Consortium. 1,135 Genomes Reveal the Global Pattern of Polymorphism in Arabidopsis thaliana. Cell 166, 481–491 (2016).
Branham, S. E., Wright, S. J., Reba, A., Morrison, G. D. & Linder, C. R. Genome-wide association study in Arabidopsis thaliana of natural variation in seed oil melting point: a widespread adaptive trait in plants. J. Hered. 107, 257–265 (2016).
Yang, C. W. et al. The E3 ubiquitin ligase activity of Arabidopsis PLANT U-BOX 17 and its functional tobacco homolog ACRE276 are required for cell death and defense. Plant Cell 18, 1084–1098 (2006).
Li, Y. et al. Identification of acyltransferases required for cutin biosynthesis and production of cutin with suberin-like monomers. Proc. Natl. Acad. Sci. USA 104, 18339–18344 (2007).
Yang, W. et al. A distinct type of glycerol-3-phosphate acyltransferase with sn-2 preference and phosphatase activity producing 2-monoacylglycerol. Proc. Natl. Acad. Sci. USA 107, 12040–12045 (2010).
Lu, C., Xin, Z., Ren, Z., Miquel, M. & Browse, J. An enzyme regulating triacylglycerol composition is encoded by the ROD1 gene of Arabidopsis. Proc. Natl. Acad. Sci. USA 106, 18837–18842 (2009).
Dahlqvist, A. et al. Phospholipid:diacylglycerol acyltransferase: an enzyme that catalyzes the acyl-CoA-independent formation of triacylglycerol in yeast and plants. Proc. Natl. Acad. Sci. USA 97, 6487–6492 (2000).
van Erp, H., Kelly, A. A., Menard, G. & Eastmond, P. J. Multigene engineering of triacylglycerol metabolism boosts seed oil content in Arabidopsis. Plant Physiol. 165, 30–36 (2014).
van Erp, H., Menard, G. & Eastmond, P. J. Seed Storage Reserve Analysis. Bio Protoc. 4, pii: e1263 (2014).
Browse, J., McCourt, P. J. & Somerville, C. R. Fatty acid composition of leaf lipids determined after combined digestion and fatty acid methyl ester formation from fresh tissue. Anal. Biochem. 152, 141–145 (1986).
Mendes, A. et al. bZIP67 regulates the omega-3 fatty acid content of Arabidopsis seed oil by activating FATTY ACID DESATURASE3. Plant Cell 25, 3104–3116 (2013).
Earley, K. W. et al. Gateway‐compatible vectors for plant functional genomics and proteomics. Plant J. 45, 616–626 (2006).
Clough, S. J. & Bent, A. F. Floral dip: a simplified method for Agrobacterium-mediated transformation of Arabidopsis thaliana. Plant J. 16, 735–743 (1998).
Winter, D. et al. An “Electronic Fluorescent Pictograph” browser for exploring and analyzing large-scale biological data sets. PLoS ONE 2, e718 (2007).
Acknowledgements
We thank Dr Sue Welham for her assistance with statistical analysis and Dr Nicki Adams, Dr Ana Mendes, Eve Shaw, Osama Butt, Jerome Dussard-McFarlane and Daniel Tomkins for their assistance in harvesting, cleaning seed, and performing fatty acid analysis. We also thank the horticultural staff at the University of Warwick and Rothamsted Research for their assistance with plant growth. The work was supported by the UK Biotechnology and Biological Sciences Research Council through grants BB/P012663/1, BB/K002147/1 and BB/E022197/1.
Author information
Authors and Affiliations
Contributions
P.J.E. conceived the original research plans and P.J.E., G.N.M., A.A.K. and S.K. supervised the experiments; G.N.M., F.M.B., A.A.K., C.P.C., I.L. and P.J.E. performed the experiments; P.J.E. designed the experiments and G.N.M., F.M.B., A.A.K., S.K., K.H-P. and P.J.E. analysed the data; P.J.E. conceived the project and wrote the article with contributions of the authors.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Menard, G.N., Bryant, F.M., Kelly, A.A. et al. Natural variation in acyl editing is a determinant of seed storage oil composition. Sci Rep 8, 17346 (2018). https://doi.org/10.1038/s41598-018-35136-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-35136-6
Keywords
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
