
Abstract
Analysis of pathogen genome variation is essential for informing disease management and control measures in farmed animals. For farmed fish, the standard approach is to use PCR and Sanger sequencing to study partial regions of pathogen genomes, with second and third-generation sequencing tools yet to be widely applied. Here we demonstrate rapid and accurate sequencing of two disease-causing viruses affecting global salmonid aquaculture, salmonid alphavirus (SAV) and infectious salmon anaemia virus (ISAV), using third-generation nanopore sequencing on the MinION platform (Oxford Nanopore Technologies). Our approach complements PCR from infected material with MinION sequencing to recover genomic information that matches near perfectly to Sanger-verified references. We use this method to present the first SAV subtype-6 genome, which branches as the sister to all other SAV lineages in a genome-wide phylogenetic reconstruction. MinION sequencing offers an effective strategy for fast, genome-wide analysis of fish viruses, with major potential applications for diagnostics and robust investigations into the origins and spread of disease outbreaks.
Similar content being viewed by others

Introduction
Pathogen genome sequencing greatly enhances the study of viral disease evolution, phylogeography and epidemiology1, including human epidemics such as Ebola2, HIV3, and influenza4,5. Second-generation sequencing platforms (e.g. Illumina) are now used routinely for genome-wide monitoring and investigations of viral disease, and generate accurate short-read data at massive throughput6,7,8, typically requiring computationally-intensive analysis pipelines. Third-generation platforms, including single-molecule real time (SMRT)9 and Oxford Nanopore10 show high promise for genome-wide analysis of viruses11,12, and bring the additional benefit of longer sequencing reads offset by higher error rates. The MinION nanopore sequencer is a particularly promising technology for viral research and diagnostics, owing to several unique features (i.e. portability, low start-up costs, real-time data generation and straightforward application) that have, for example, allowed human pathogens to be rapidly characterized in the field without high-power computing or major laboratory infrastructure13,14.
Aquaculture is the fastest growing food production sector15, yet its sustainability and expansion is threatened by infectious diseases. Among a list of concerning pathogens, several known viral disease agents cause major animal health and welfare issues, accompanied by massive financial losses through mortalities, slow growth, poor flesh quality, treatment interventions and control protocols (e.g. culling)16,17. Accurate diagnosis of viral diseases is an essential part of strategic planning to manage existing and limit future outbreaks, and is especially important considering the lack of fully-effective treatments and vaccines for most fish viral pathogens (e.g.18,19,20). Recommended diagnostic procedures of viral disease include demonstration of clinical pathology coupled to the presence of pathogen DNA/RNA, followed by culturing to establish the presence of viable pathogen21. Diagnostic sequencing of aquatic viruses is typically done by PCR and Sanger sequencing, which benefits from high accuracy and established protocols. However, such approaches are limited to relatively short sequences (i.e. up to 1500 bp when sequencing both directions) and cannot gain a genome-wide representation of viruses and their variants without non-routine effort. Second and third generation sequencing tools hold promise for the characterization of aquatic viruses (reviewed in22,23), including pathogens affecting global fish aquaculture, yet they are being up-taken relatively slowly. The utility of such approaches have been demonstrated by the characterisation of novel pathogens such as Tilapia Lake Virus (TiLV) using Ion Torrent sequencing24, the discovery of Piscine Reovirus (PRV)25 and Piscine myocarditis virus (PMCV)26 with pyrosequencing, and the analysis of Cyprinid herpesvirus 3 genomes using a target enrichment and Illumina sequencing approach to identify mixed genotype infections27. However, as far as we are aware, to date no published studies have successfully used MinION sequencing to study viral diseases impacting farmed fish.
In this study, we demonstrate rapid genome-wide sequencing of fish viral pathogens using nanopore sequencing on the MinION platform. We focussed on two disease agents affecting farmed Atlantic salmon (Salmo salar L.), salmonid alphavirus (SAV) and infectious salmon anaemia virus (ISAV). SAV is a single-strand positive-strand RNA virus (Family Togaviridae) and the causative agent of pancreas disease, prevalent across European salmon aquaculture, with six SAV subtypes (SAV1-6) established28. All SAV sequences published to date have been generated using the Sanger method, including full genomes for SAV1-329,30,31,32,33, and partial genomic regions primarily encoding a glycoprotein (E2) or a non-structural protein (NsP3) (neither representing known virulence markers), for samples representing all six subtypes (e.g.28,34). ISAV is a highly pathogenic, segmented, negative-strand RNA virus (Family Orthomyxoviridae) often resulting in high mortality rates35,36, with containment and culling being the only effective mitigation strategy37. ISAV genomes have been Sanger-sequenced from several ‘genogroups’38,39,40,41,42,43, while segments 5 and 6, which contain known virulence markers and respectively encode the fusion and hemagglutinin surface proteins, are routinely used for Sanger genotyping, but have also been characterized using Illumina sequencing44. Overall, in common with other fish viruses, there is a lack of genome-wide data for SAV and ISAV, limiting power to define virulence markers and understand the evolution of different viral lineages. This study linked MinION sequencing to standard PCR enrichment to accurately sequence and genotype both SAV and ISAV. In addition to reporting the first full genome sequence for SAV6, we discuss the potentially transformative applications of MinION sequencing in diagnostics and molecular epidemiology of viruses impacting aquaculture.
Results and Discussion
SAV genome-wide sequencing
Using primers matching conserved regions of the SAV genome (Table 1), three overlapping PCR amplicons (approx. 4 kb each; Supplementary Fig. 1) were obtained from two samples known to represent SAV1 (SCO/4640/08) and SAV6 (F1045-96) (Table 2) and sequenced on separate MinION flow cells (R9.4) for 2–3 hours (sequencing details provided in Supplementary Table 1). Over 98% of each SAV genome was recovered with 90 bp missing at the 5′ and 30 bp at the 3′ region of the genome due to the location of the highly conserved primer binding sites. The average read length from both sequencing runs was ~3800 bp per amplicon, indicating limited DNA shearing during the library preparation. The sequencing of sample SCO/4640/08 was stopped after 3-hours producing over 400 Mb of ‘pass’ reads (Q-score ≥ 7), resulting in almost 40,000x coverage throughout the genome (Table 3). By mapping against the Sanger sequenced SAV1 reference sequence for SCO/4640/0832
As the above approach led to an accurate representation of a verified SAV genome sequence, we can be confident in its application to discovering entirely novel variation. For this reason, we decided to sequence SAV6 (sample F1045-96), which has only been identified once, as partial E2 and NsP3 sequences, from a single Irish sample28, and is highly distinct from all other subtypes. After two hours of sequencing, a genome-wide average of 21,000x coverage was achieved. The SAV6 genome consensus showed 100% similarity to Sanger-sequenced NsP3 (EF675499) and E2 (EF675547) gene sequences. Table 4 shows consistent genome-wide pairwise similarities contrasting the genome of SAV6 to the other SAV sub-types at both nucleotide and amino acid level (88.6–89.2% and 93.8–94.6% respectively). Variability among SAV subtypes differed based on the gene of interest and the greatest variability was seen in the NsP3 gene (82.0–83.8% and 87.7–89.8% nucleotide/amino acid similarity). In conclusion, these data gained by MinION sequencing confirm for the first time using genome-wide evidence that SAV6 represents a highly-divergent SAV subtype.
Genome-wide SAV phylogeny
Previous studies have failed to establish the position of SAV6 within the SAV phylogeny based on E2 and NsP3 sequences (e.g.28,34). We performed genome-wide phylogenetic reconstructions incorporating the new SAV6 genome gained by MinION sequencing, along with 17 SAV genomes available in NCBI, and 5 new (i.e. previously unpublished) Sanger-sequenced genomes for SAV2, 4 and 5 (isolate details in Supplementary Table 2). We used two probabilistic methods, the first a Bayesian approach incorporating a relaxed clock model45 allowing estimation of the tree root46 and the second an unrooted maximum-likelihood (ML) approach (Fig. 1). The root of the SAV phylogeny was estimated with high confidence (posterior probability: 0.97), and split SAV6 from all other SAV sub-types. Branching of other subtypes was maximally supported (posterior probability: 1.0; ML bootstrap values > 95%), with SAV3 and 2 forming a monophyletic group separate from a clade containing SAV1, 4 and 5 (Fig. 1). The basal phylogenetic position of SAV6 highlights particular importance for the new MinION genome sequence in future investigations of the evolution and phylogeography of the major SAV lineages.

Genome-wide Bayesian phylogeny for SAV lineages including the SAV6 sequence generated by MinION sequencing (shown in red). The data represents an 11,638 bp nucleotide alignment and the analysis was done using the best-fitting nucleotide substitution model (GTR) and a relaxed molecular clock model. Branch posterior probability support is shown with comparable ML bootstrap support given in parentheses. Branch lengths are scaled in relative time as the relaxed clock model was uncalibrated. Root posterior probability (RPP) estimated by RootAnnotator46 is shown in red font.
ISAV segment 5 and 6 sequencing
To test MinION sequencing on a distinct fish virus, we focused our efforts on ISAV, which exists in eight genomic segments (length: 740–2169 bp). This inherent aspect of the virus limits one of the main benefits of MinION sequencing: its capacity to generate genome-wide representation of a virus with a small number of overlapping PCR amplicons, as done successfully for SAV. We instead focused on ISAV segments 5 and 6, which are widely studied and known to contain ISAV virulence markers, this time testing a barcoding approach to sequence multiple samples on a single MinION flow cell. PCR amplicons (primers in Table 1) amplifying 97% of segment 5 and 93% of segment 6 including both virulence markers, were obtained from seven ISAV isolates (Table 2) and pooled in equimolar amounts for sequencing after barcoding. After 3 hours, approx. 9,000x mean coverage was achieved per sample. Only one of the isolates used in this study has a reference Sanger sequence (NO/Glessvær/2/90); basecalling accuracy was estimated for segments 5 and 6 of this isolate and 100% similarity was observed.
ISAV segment 6 contains a highly polymorphic region (HPR) at the 3′ end of the gene which is a known virulence marker. The putatively non-pathogenic ISAV, called HPR0, is characterized by a full length of the HPR comprising 35 amino acids and all pathogenic ISAV strains to date (called HPR-deleted) contain a deletion in the HPR region of varying length47. While none of the isolates used in this study were HPR0, the HPR of all the ISAV isolates used in this study were successfully classified with several different deletions being identified including three samples CA/NB04-85-1/04, CA/NB7178/08, CA/F679/99 which have a deletion previously found only once before and not yet fully characterised48 (Table 3). In addition, the consensus sequences for each segment 5 captured another proposed virulence marker, the substitution Q266L39,49,50, with all but one isolate (CA/NB04-85-1/04) possessing the L variant. CA/NB04-85-1/04 instead encodes for a proline at this position which while unusual, is also present in a Canadian isolate from the EU/NA genogroup (EF432567)51. These data thus demonstrate that MinION sequencing effectively recaptures sequence-level virulence markers.
Optimal sequence coverage
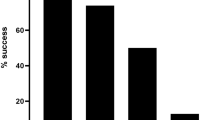
Future studies would benefit from establishing the necessary coverage required to determine confident consensus sequences using MinION. Thus, we randomly sampled MinION reads mapping to segments 5 and 6 of one ISAV sample (NO/Glessvær/2/90) and the SAV1 genome (sample: SCO/4640/08) at different coverages to establish the impact on consensus sequence accuracy (Fig. 2A–C). 50x and 500x coverage of either ISAV segment achieved a consensus sequence >99% and 100% identical to the Sanger reference, respectively (Fig. 2A,B). For SAV1, just 20x coverage led to 99% similarity with the Sanger reference, while 1,000x coverage led to 99.97% similarity (Fig. 2C). Thus, despite its high error rate (e.g.10), a highly-accurate consensus sequence can be generated with very modest MinION sequencing time.

Impact of MinION read coverage on accuracy of consensus sequence generation. ‘% identity’ is shown between reference Sanger sequences and consensus sequences generated from randomly sampling MinION reads at multiple sequence coverages for: (A) Segment 5 of ISAV NO/Glessvær/2/90; (B) Segment 6 of ISAV NO/Glessvær/2/90; (C) SAV1 genome (sample SCO/4640/08).
Broader perspectives and comparisons with other platforms
Rapid sequencing of two structurally-distinct fish RNA viruses was achieved with high accuracy using MinION sequencing coupled with PCR. While the samples used were from cultured viruses, we have had equal success using the same protocols and infected tissues with much lower virus titres (data not shown). The methods described were achieved within 24 hours lab-time, exploiting PCR primers matching conserved genomic regions, which allowed a highly divergent viral genome (SAV6) to be sequenced with little prior knowledge of sequence variation. Combining such turn-around and ease of application with the accuracy gained from moderate sequencing coverage opens the doorway to routine high-confidence viral genotyping at shallow phylogenetic scales, sufficient for robust diagnostics supporting disease management and regulatory decisions. Elsewhere, it has also been shown that MinION sequencing can be used to recover viral RNA genomes from infected samples without prior PCR enrichment, which has advantages in the field52 and can also potentially identify viruses beyond the target pathogen. The ease of generating genome-wide sequencing data for non-segmented viruses such as SAV has revolutionary potential for diversifying the relatively restricted current repertoire of publicly-available fish virus genomes, bringing benefits for fundamental research and disease management. However, it is important to acknowledge that our approach is best-suited to generating consensus viral genome sequences, and less useful for identifying population variation within samples, which is well-established for RNA viruses53,54,55,56,57,58, as the PCR enrichment may introduce biases toward particular variants, and the high sequencing error rate of MinION reduces power to call low frequency variants de novo.
Future efforts should also aim to reduce the cost of genome-wide sequencing using multiplexing to exploit the high coverage possible on a single MinION flow cell. We estimate that the single SAV genomes (~12 kb) generated in our study cost approx. £850 each, including all consumables and an entire flow cell; however, multiplexing using 96 samples and the same approach would reduce this cost to approx. £50–60 per sample. By comparison, it would not be possible to perform a direct-equivalent Sanger sequencing approach, as the amplicon length exceeds the possible length of sequenced reads. Assuming an SAV genome was tiled across 7 PCR amplicons (e.g.32) and sequenced directly using Sanger with no cloning step (which would add further costs), we estimate a cost of approx. £100 per SAV consensus genome, including all reagents and bi-directional sequencing. In addition to a per-genome saving, the MinION approach is more convenient and time-efficient when a large number of genomes need to be sequenced, being done in-house in a single sequencing run with fewer amplicons, avoiding the need for cloning and the use of an external Sanger provider. It is more challenging to directly compare costs of our MinION strategy with alternative high-throughput approaches, as there are many platforms and variations in library preparation strategy, and this would also be affected by the extent of sub-contracting to an external provider. However, we estimate that the costs of generating complete SAV genomes using Illumina at the same scale (i.e. 96 samples), assuming the same amplicon strategy followed by in-house library preparation/indexing (Nextera XT DNA kit) and sequencing on the MiSeq platform by an external provider to be approx. £50–65 (i.e. very comparable). While Illumina brings advantages in terms of data accuracy, e.g. giving more scope for detecting viral population variation, the MinION avoids use of an external provider, which typically leads to a lag of weeks to months for delivery. Overall, our MinION approach has some cost and/or time advantages when compared to Sanger and Illumina approaches if the aim is to recover a consensus SAV genome with high accuracy, and future work is needed to develop this approach for robust analysis of viral population variation.
In conclusion, once low cost MinION sequencing of fish viral genomes is achieved, considering the unique portability of the sequencer alongside the modest computational power needed to analyse the resultant data, it seems reasonable to anticipate in-field diagnostic applications in the near future, including the monitoring of viral genotypes and subtypes directly on fish farms and in the field.
Materials and Methods
Sample preparation and PCR
Total RNA was extracted from SAV and ISAV samples (Table 2) using a phenol-chloroform extraction method, except for the SAV6 sample, which was extracted using a Viral RNA Isolation kit (Qiagen). cDNA was synthesised using Protoscript II (New England Biolabs) reverse transcriptase and a mix of random hexamer and oligo dT (dT23VN) primers (New England Biolabs) as per the manufacturers’ instructions. First-strand cDNA was used as template for long-range PCR reactions.
To amplify the SAV1/6 genomes, degenerate PCR primers targeting three ~4 kb overlapping amplicons were designed in regions of the genome conserved in the five subtypes where sequence data is available (Table 1). PCR was conducted using LongAmp polymerase (New England Biolabs) with cycling conditions as follows: 30 s at 94 °C, followed by 35 cycles of 15 s at 94 °C, 1 min at 56 °C and 3 min 50 s at 65 °C, with a final extension for 10 min at 65 °C. ISAV segments 5 and 6 were amplified using the same approach and primers designed to conserved 5′ and 3′ regions of segment 5/6 (Table 2) under the same conditions, except that the PCR extension time was 2 min 30 s. PCR products were visualised on a 1% agarose gel, purified using QIAquick Gel Extraction Kit (Qiagen) and stored at −80 °C until sequencing.
Sanger sequencing of novel SAV genomes
Seven overlapping PCRs were performed in triplicates for five SAV isolates (Supplementary Table 2) according to the methods published by Matejusova et al.32. The complete SAV genomes were generated by Sanger sequencing, assembled using Sequencher v5.4.6 and used in the phylogenetic analysis presented in Fig. 1.
Preparation of SAV Library and sequencing
1000 ng of equimolar pooled amplicon from each SAV isolate was the input to a library generated with the Ligation Sequencing Kit 1D SQK-LSK108 (Oxford Nanopore Technologies). Before ligating sequencing adaptors, DNA was end-repaired using the NEBNext Ultra II End Repair/dA Tailing kit (New England Biolabs), purified using AMPure XP beads (Beckman Coulter) in a ratio of 1:1 volume of beads per sample and eluted in 30 µl of nuclease-free water (Sigma). Sequencing adapters (AMX1D) (ONT) were ligated to the DNA using Blunt/TA Ligation Master Mix (New England Biolabs) by incubation at room temperature for 10 min. The adapter-ligated DNA library was purified with AMPure XP beads in a ratio of 1:2.5 volume of beads per sample, followed by a wash with Adapter Bead Binding buffer (ABB) (ONT) and elution in 15 µl nuclease-free water. DNA concentrations were determined between each step using a Qubit fluorimeter (Fisher Thermo). Each cleaned library was loaded onto a separate MinION Flow Cell Mk1 R9.4 (ONT) and run via MinKNOW software (without real-time basecalling) for 2 and 3 hours for SAV6 (F1045-96) and SAV1 (SCO/4640/08) respectively.
Preparation of ISAV Library and Sequencing
The ISAV library was prepared using the Ligation Sequencing Kit 1D SQK-LSK108 and a Native Barcoding Kit EXP-NBD103 (Oxford Nanopore Technologies). Segments 5 and 6 from the same virus isolate were pooled in equimolar amounts and 300 ng of each isolate end-repaired using the NEBNext Ultra II End Repair/dA Tailing kit. DNA was purified using AMPure XP beads in a ratio of 1:1 volume of beads per sample and eluted in 30 µl nuclease-free water. Native barcodes were ligated to 200 ng of end-repaired DNA using Blunt/TA Ligation Master Mix. The barcoded DNA was purified using AMPure XP beads in a ratio of 1:1 volume of beads to sample to remove excess barcodes and eluted in 26 µl nuclease-free water. The barcoded samples were pooled in equimolar amounts to a total of 200 ng library DNA (~0.2 pmol as per Oxford Nanopore Technologies instructions). Barcode adapter mix (BAM) (ONT) was ligated to the library DNA using NEBNext Quick Ligation Reaction Buffer and Quick T4 DNA Ligase (New England Biolabs), and incubated at room temperature for 10 min. Library DNA was purified using AMPure XP beads in a ratio of 1:2.5 volume of beads per sample and subsequently washed with Adapter Bead Binding buffer (ABB) before elution in 15 µl nuclease-free water. DNA concentrations were determined between each step as above. Libraries were loaded according to the native barcoding kit protocol (ONT) onto a MinION Flow Cell Mk1 R9.5., using a 3-hour sequencing run via MinKNOW without real-time basecalling.
Basecalling and consensus assembly
MinION data basecalling and demultiplexing for barcoded ISAV samples was performed using Albacore v.2.1.7 on Windows command line. Base-called FASTQ files were loaded into Geneious v.1059 for mapping and analysis. SAV1 (SCO/4640/08) sample reads were mapped to the SAV1 Sanger-reference sequence32. SAV6 (F1045-96) sample reads were individually mapped to the partial gene E2 and NsP3 sequences of SAV628 and reference genomes for SAV132, SAV229, SAV333, SAV4 (generated in this study; isolate SAV 04-44) and SAV5 (generated in this study; isolate SCO10-684). In order to reconstruct the whole SAV6 genome, mapping was set at 5 iterations and a 65% consensus threshold. The 5 generated SAV consensus sequences were then manually inspected and any single base ambiguities resolved by parsimony, giving a final F1045-96 (SAV6) consensus sequence. For example, at position 2235, 4 out of 5 consensus sequences were the base G, whereas one consensus sequence was A: in this case, G was adopted for the final consensus. The ISAV samples were individually mapped to the previously sequenced segment 5 and 6 of the Scot157/08 isolate60 using the same parameters.
Reads for ISAV NO/Glessvær/2/90 segments 5 and 6, and SAV1 (SCO/4640/08) were subjected to random subsampling to determine the depth of coverage necessary to generate an accurate consensus (i.e. Fig. 2). Subsampling was performed in Geneious v.10 using the ‘Randomly Sample Sequences’ workflow. Subsampled reads were realigned to the reference sequences using the same mapping methods as above and consensus sequences were generated from each alignment and compared to the reference Sanger sequence using pairwise alignment. Consensus sequences were aligned against all published genome sequences using MAFFT v.761 and manually inspected for errors in the mapping that disrupted the protein coding sequences in BioEdit software v.7.2.562. Sequence pairwise similarities were calculated using Geneious statistics of the MAFFT-aligned whole genome sequences.
Genome-wide SAV phylogenetic analyses
Multiple sequence alignment of 23 SAV genomes (Supplementary Table 2) was done using MAFFT v.7, generating an 11,638 bp alignment (provided in Supplementary Dataset 1), which was uploaded to the IQ-TREE server63 to determine the best-fitting nucleotide substitution model (GTR) and generate a phylogenetic tree with support values gained from 1,000 Ultrafast Bootstrap iterations64. Bayesian phylogenetic analysis was done using the same dataset in BEAST265 employing a relaxed clock model45, a Coalescent Bayesian Skyline tree model66, the GTR substitution model and a Markov Chain Monte Carlo (MCMC) chain of 200 million generations. Tracer67 was used to assess MCMC convergence and estimate effective sample sizes for all sampled parameters (>2,000 in all cases). TreeAnnotator was used to remove the first 10% of sampled trees as burn-in and produce a Maximum Credibility Clade (MCC) tree. RootAnnotator46 was used to estimate posterior support for alternative root positions. MCC trees were visualized using FigTree (http://tree.bio.ed.ac.uk/software/figtree/).
Data Availability
MinION sequences for SAV isolates: SRA BioProject Accession SRP142226. SAV6 consensus genome: NCBI accession MH238448. MinION sequences for ISAV: SRA BioProject Accession SRP155694. ISAV segments 5 and 6: NCBI accessions: MH708654-MH708667. Sanger-sequenced SAV genomes: NCBI accessions MH341514 and MH708650-MH708653.
References
Houldcroft, C. J., Beale, M. A. & Breuer, J. Clinical and biological insights from viral genome sequencing. Nature Reviews Microbiology 15, 183–192 (2017).
Holmes, E. C., Dudas, G., Rambaut, A. & Andersen, K. G. The evolution of Ebola virus: Insights from the 2013–2016 epidemic. Nature 538, 193–200 (2016).
Worobey, M. et al. 1970s and ‘Patient 0’ HIV-1 genomes illuminate early HIV/AIDS history in North America. Nature 539, 98–101 (2016).
Su, Y. C. F. et al. Phylodynamics of H1N1/2009 influenza reveals the transition from host adaptation to immune-driven selection. Nat. Commun. 6, 7952 (2015).
Vijaykrishna, D. et al. The contrasting phylodynamics of human influenza B viruses. Elife 4, e05055 (2015).
Datta, S. et al. Next-generation sequencing in clinical virology: Discovery of new viruses. World J. Virol. 4, 265–76 (2015).
Jones, S., Baizan-Edge, A., MacFarlane, S. & Torrance, L. Viral Diagnostics in Plants Using Next Generation Sequencing: Computational Analysis in Practice. Front. Plant Sci. 8, 1770 (2017).
Qureshi, A., Tantray, V. G., Kirmani, A. R. & Ahangar, A. G. Next-Generation Sequencing (NGS) Based Detection of Viral Pathogens. Expert Biol. 3 (2018).
Rhoads, A. & Au, K. F. PacBio Sequencing and Its Applications. Genomics. Proteomics Bioinformatics 13, 278–289 (2015).
Laver, T. et al. Assessing the performance of the Oxford Nanopore Technologies MinION. Biomol. Detect. Quantif. 3, 1–8 (2015).
Li, J. et al. A comparative study on the characterization of hepatitis B virus quasispecies by clone-based sequencing and third-generation sequencing. Emerg. Microbes Infect. 6, e100 (2017).
Quick, J. et al. Multiplex PCR method for MinION and Illumina sequencing of Zika and other virus genomes directly from clinical samples. Nat. Protoc. 12, 1261–1276 (2017).
Hoenen, T. et al. Nanopore sequencing as a rapidly deployable Ebola outbreak tool. Emerg. Infect. Dis. 22, 331–334 (2016).
Faria, N. R. et al. Mobile real-time surveillance of Zika virus in Brazil. Genome Med. 8, 97 (2016).
FAO. The state of world fisheries and aquaculture. State World Fish. Aquac. 2016 160 doi:92-5-105177-1 (2016).
Lafferty, K. D. et al. Infectious Diseases Affect Marine Fisheries and Aquaculture Economics. Ann. Rev. Mar. Sci. 7, 471–496 (2015).
Aunsmo, A., Valle, P. S., Sandberg, M., Midtlyng, P. J. & Bruheim, T. Stochastic modelling of direct costs of pancreas disease (PD) in Norwegian farmed Atlantic salmon (Salmo salar L.). Prev. Vet. Med. 93, 233–241 (2010).
Karlsen, M. et al. Efficacy and safety of an inactivated vaccine against Salmonid alphavirus (family Togaviridae). Vaccine 30, 5688–5694 (2012).
Garver, K. A., LaPatra, S. E. & Kurath, G. Efficacy of an infectious hematopoietic necrosis (IHN) virus DNA vaccine in Chinook Oncorhynchus tshawytscha and sockeye O. nerka salmon. Dis. Aquat. Organ. 64, 13–22 (2005).
Munang’andu, H. M. et al. Comparison of vaccine efficacy for different antigen delivery systems for infectious pancreatic necrosis virus vaccines in Atlantic salmon (Salmo salar L.) in a cohabitation challenge model. Vaccine 30, 4007–4016 (2012).
OIE. Manual of diagnostic tests for aquatic animals - Infection with Infectious Salmon Anaemic Virus. OIE Aquatic Animal Disease Cards 2.3 (2017).
Nkili-Meyong, A. A. et al. Contribution of Next-Generation Sequencing to Aquatic and Fish Virology. Intervirology 59, 285–300 (2016).
Bayliss, S. C. et al. The Promise of Whole Genome Pathogen Sequencing for the Molecular Epidemiology of Emerging AquaculturePathogens. Front. Microbiol. 8, 1–18 (2017).
Bacharach, E. et al. Characterization of a novel orthomyxo-like virus causing mass die-offs of Tilapia. MBio 7 (2016).
Palacios, G. et al. Heart and skeletal muscle inflammation of farmed salmon is associated with infection with a novel reovirus. PLoS One 5, e11487 (2010).
Løvoll, M. et al. A novel totivirus and piscine reovirus (PRV) in Atlantic salmon (Salmo salar) with cardiomyopathy syndrome (CMS). Virol. J. 7, 309 (2010).
Hammoumi, S. et al. Targeted genomic enrichment and sequencing of CyHV-3 from carp tissues confirms low nucleotide diversity and mixed genotype infections. PeerJ 4, e2516 (2016).
Fringuelli, E. et al. Phylogenetic analyses and molecular epidemiology of European salmonid alphaviruses (SAV) based on partial E2 and nsP3 gene nucleotide sequences. J. Fish Dis. 31, 811–823 (2008).
Weston, J. et al. Comparison of Two Aquatic Alphaviruses, Salmon Pancreas Disease Virus and Sleeping Disease Virus, by Using Genome Sequence Analysis, Monoclonal Reactivity, and Cross-Infection. J. Virol. 76, 6155–6163 (2002).
Hodneland, K., Bratland, A., Christie, K. E., Endresen, C. & Nylund, A. New subtype of salmonid alphavirus (SAV), Togaviridae, from Atlantic salmon Salmo salar and rainbow trout Oncorhynchus mykiss in Norway. Dis. Aquat. Organ. 66, 113–120 (2005).
Karlsen, M., Hodneland, K., Endresen, C. & Nylund, A. Genetic stability within the Norwegian subtype of salmonid alphavirus (family Togaviridae). Arch. Virol. 151, 861–874 (2006).
Matejusova, I. et al. Comparison of complete polyprotein sequences of two isolates of salmon alphavirus (SAV) type I and their behaviour in a salmonid cell line. Arch. Virol. 158, 2143–2146 (2013).
Petterson, E., Stormoen, M., Evensen, Ø., Mikalsen, A. B. & Haugland, Ø. Natural infection of Atlantic salmon (Salmo salar L.) with salmonid alphavirus 3 generates numerous viral deletion mutants. J. Gen. Virol. 94, 1945–1954 (2013).
Bruno, D. et al. Identification of a wild reservoir of salmonid alphavirus in common dab Limanda limanda, with emphasis on virus culture and sequencing. Aquac. Environ. Interact. 5, 89–98 (2014).
Dannevig, B. H., Falk, K. & Namork, E. Isolation of the causal virus of infectious salmon anaemia (ISA) in a long-term cell line from Atlantic salmon head kidney. J. Gen. Virol. 76, 1353–1359 (1995).
OIE. Manual of Diagnostic Tests for Aquatic Animals - Infection with Salmonid Alphavirus. OIE Aquat. Anim. Dis. Cards 2.3.6, 1–14 (2017).
Stagg. The eradication of an outbreak of clinical infectious salmon anaemia from Scotland. In: International Response to Infectious Salmon Anaemia: Prevention, Control and Eradication. US Dep. Agric. 111–124 (2003).
Clouthier, S. C., Rector, T., Brown, N. E. C. & Anderson, E. D. Genomic organization of infectious salmon anaemia virus. J. Gen. Virol. 83, 421–428 (2002).
Markussen, T. et al. Evolutionary mechanisms involved in the virulence of infectious salmon anaemia virus (ISAV), a piscine orthomyxovirus. Virology 374, 515–527 (2008).
Cottet, L. et al. Bioinformatic Analysis of the Genome of Infectious Salmon Anemia Virus Associated with Outbreaks with High Mortality in Chile. J Virol 84, 11916–11928 (2010).
Merour, E. et al. Completion of the full-length genome sequence of the infectious salmon anemia virus, an aquatic orthomyxovirus-like, and characterization of mAbs. J. Gen. Virol. 92, 528–533 (2011).
Toro-Ascuy, D. et al. Development of a reverse genetic system for infectious salmon anemia virus: Rescue of recombinant fluorescent virus by using salmon internal transcribed spacer region 1 as a novel promoter. Appl. Environ. Microbiol. 81, 1210–1224 (2015).
Christiansen, D. H. et al. First field evidence of the evolution from a non-virulent HPR0 to a virulent HPR-deleted infectious salmon anaemia virus. J. Gen. Virol. 98, 595–606 (2017).
Markussen, T. et al. Ultra-Deep Pyrosequencing of Partial Surface Protein Genes from Infectious Salmon Anaemia Virus (ISAV) Suggest Novel Mechanisms Involved in Transition to Virulence. PLoS One 8, e81571 (2013).
Drummond, A. J., Ho, S. Y. W., Phillips, M. J. & Rambaut, A. Relaxed phylogenetics and dating with confidence. PLoS Biol. 4, 699–710 (2006).
Calvignac-Spencer, S., Schulze, J. M., Zickmann, F. & Renard, B. Y. Clock Rooting Further Demonstrates that Guinea 2014 EBOV is a Member of the Zaïre Lineage. PLoS Curr, https://doi.org/10.1371/currents.outbreaks.c0e035c86d721668a6ad7353f7f6fe86 (2014).
Nylund, A., Devold, M., Plarre, H., Isdal, E. & Aarseth, M. Emergence and maintenance of infectious salmon anaemia virus (ISAV) in Europe: A new hypothesis. Dis. Aquat. Organ. 56, 11–24 (2003).
Kibenge, F. S. B., Kibenge, M. J. T., Groman, D. & McGeachy, S. In vivo correlates of infectious salmon anemia virus pathogenesis in fish. J. Gen. Virol. 87, 2645–2652 (2006).
Cárdenas, C., Carmona, M., Gallardo, A., Labra, A. & Marshall, S. H. Coexistence in Field Samples of Two Variants of the Infectious Salmon Anemia Virus: A Putative Shift to Pathogenicity. PLoS One 9, e87832 (2014).
Cottet, L., Rivas-Aravena, A., Cortez-San Martin, M., Sandino, A. M. & Spencer, E. Infectious salmon anemia virus-Genetics and pathogenesis. Virus Research 155, 10–19 (2011).
Kibenge, F. S. B. et al. Mapping of putative virulence motifs on infectious salmon anemia virus surface glycoprotein genes. J. Gen. Virol. 88, 3100–3111 (2007).
Kafetzopoulou, L. E. et al. Assessment of Metagenomic MinION and Illumina sequencing as an approach for the recovery of whole genome sequences of chikungunya and dengue viruses directly from clinical samples. bioRxiv 355560, https://doi.org/10.1101/355560 (2018).
McKinley, T. J., Murcia, P. R., Gog, J. R., Varela, M. & Wood, J. L. N. A bayesian approach to analyse genetic variation within RNA viral populations. PLoS Comput. Biol. 7, e1002027 (2011).
Descloux, E., Cao-Lormeau, V. M., Roche, C. & De Lamballerie, X. Dengue 1 diversity and microevolution. French Polynesia 2001-2006: Connection with epidemiology and clinics. PLoS Negl. Trop. Dis. 3, e493 (2009).
Iqbal, M. et al. Within-host variation of avian influenza viruses. Philosophical Transactions of the Royal Society B: Biological Sciences 364, 2739–2747 (2009).
Hoelzer, K. et al. Intrahost Evolutionary Dynamics of Canine Influenza Virus in Naive and Partially Immune Dogs. J. Virol. 84, 5329–5335 (2010).
Agoti, C. N. et al. Intrapatient Variation of the Respiratory Syncytial Virus Attachment Protein Gene. J. Virol. 84, 10425–10428 (2010).
Vibin, J. et al. Metagenomics detection and characterisation of viruses in faecal samples from Australian wild birds. Sci. Rep. 8, 8686 (2018).
Kearse, M. et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28, 1647–1649 (2012).
Plarre, H. et al. Evolution of infectious salmon anaemia virus (ISA virus). Arch. Virol. 157, 2309–2326 (2012).
Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 30, 772–780 (2013).
Hall, T. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 41, 95–98 (1999).
Trifinopoulos, J., Nguyen, L. T., von Haeseler, A. & Minh, B. Q. W-IQ-TREE: a fast online phylogenetic tool for maximum likelihood analysis. Nucleic Acids Res. 44, W232–W235 (2016).
Minh, B. Q., Nguyen, M. A. T. & Von Haeseler, A. Ultrafast approximation for phylogenetic bootstrap. Mol. Biol. Evol. 30, 1188–1195 (2013).
Bouckaert, R. et al. BEAST 2: A Software Platform for Bayesian Evolutionary Analysis. PLoS Comput. Biol., https://doi.org/10.1371/journal.pcbi.1003537 (2014).
Pybus, O. G. & Rambaut, A. Evolutionary analysis of the dynamics of viral infectious disease. Nat. Rev. Genet. 10, 540–550 (2009).
Rambaut, A., Drummond, A. J., Xie, D., Baele, G. & Suchard, M. A. Tracer v1.7. http://tree.bio.ed.ac.uk/software/tracer/ (2018).
Acknowledgements
This work received support from the Biotechnology and Biological Sciences Research Council (grant BB/M010996/1) and Marine Scotland Science. The authors thank Dr Elena Fringuelli (Agri-Food and Biosciences Institute, Belfast) for providing some of the SAV material used.
Author information
Authors and Affiliations
Contributions
M.G. and D.J.M. devised the study. M.G., I.M. and D.J.M. designed the sequencing work. M.G. and I.M. performed lab-work and analysis, supported by L.N. for the S.A.V. work. I.M. performed Sanger sequencing and provided reagents. N.R. and K.F. provided reagents and isolates included in the study. M.G. drafted the manuscript and figures and tables, barring the phylogenetic tree, done by L.N. All authors contributed to writing, improving the manuscript draft, and approved the final submitted paper.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Gallagher, M.D., Matejusova, I., Nguyen, L. et al. Nanopore sequencing for rapid diagnostics of salmonid RNA viruses. Sci Rep 8, 16307 (2018). https://doi.org/10.1038/s41598-018-34464-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-34464-x
Keywords
This article is cited by
-
Nanopore sequencing provides snapshots of the genetic variation within salmonid alphavirus-3 (SAV3) during an ongoing infection in Atlantic salmon (Salmo salar) and brown trout (Salmo trutta)
Veterinary Research (2024)
-
A multiplexed RT-PCR assay for nanopore whole genome sequencing of Tilapia lake virus (TiLV)
Scientific Reports (2023)
-
Nanopore sequencing: a rapid solution for infectious disease epidemics
Science China Life Sciences (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
