
Abstract
Amplification and sequencing of 16S amplicons are widely used for profiling the structure of oral microbiota. However, it remains not clear whether and to what degree DNA extraction and targeted 16S rRNA hypervariable regions influence the analysis. Based on a mock community consisting of five oral bacterial species in equal abundance, we compared the 16S amplicon sequencing results on the Illumina MiSeq platform from six frequently employed DNA extraction procedures and three pairs of widely used 16S rRNA hypervariable primers targeting different 16S rRNA regions. Technical reproducibility of selected 16S regions was also assessed. DNA extraction method exerted considerable influence on the observed bacterial diversity while hypervariable regions had a relatively minor effect. Protocols with beads added to the enzyme-mediated DNA extraction reaction produced more accurate bacterial community structure than those without either beads or enzymes. Hypervariable regions targeting V3-V4 and V4-V5 seemed to produce more reproducible results than V1-V3. Neither sequencing batch nor change of operator affected the reproducibility of bacterial diversity profiles. Therefore, DNA extraction strategy and 16S rDNA hypervariable regions both influenced the results of oral microbiota biodiversity profiling, thus should be carefully considered in study design and data interpretation.
Similar content being viewed by others


Introduction
Oral microbiota are implicated in the aetiology of many oral and systemic diseases, such as dental caries, periodontal diseases, obesity and cancer1,2,3,4. Therefore, testing the association between oral microbiota structure with diseases is of great interest in clinical diagnosis and treatment1. One vital approach for this purpose is high-throughput sequencing of the 16S rRNA gene amplicons. This approach has allowed characterization of the microbiome diversity from human and environmental microbiomes with an unprecedented depth and coverage5,6,7,8. However, for human intestinal microbiota9,10,11,12,13 and environmental microbiota14,15,16,17, various factors such as DNA extraction method, targeted 16S rRNA hypervariable regions and sample handling environment can all greatly influence the resulted biodiversity profiles.
First of all, DNA extraction can be crucial to the success of microbiome sequencing18,19,20,21,22. The wide variation of cell membrane structures and compositions can pose a significant challenge to the efficient and bias-free extraction of genomic DNA23. In particular, cell lysis is an initial yet crucial step in DNA extraction procedures and typically includes physical, chemical and enzymatic disruption14,24,25,26. Physical disruption can increase the DNA yield, yet at the same time may potentially shear genomic DNA into small fragments which can lead to chimeric products in the final sequencing result27,28. Chemical and enzymatic lysis methods are less likely to damage DNA, but they can introduce bias in DNA extraction, due to the lower general applicability to all target organisms26,29. The oral microbiota are highly complex and heterogeneous, which consist of a wild array of Gram-positive and Gram-negative organisms. However, for oral microbiota, the impacts of these DNA extraction strategies on microbiome sequencing has not been critically assessed. As a result, how to rationally devise an experimental procedure to achieve an accurate and reliable representation of oral microbiota diversity via sequencing is not yet firmly established.
On the other hand, the choice of 16S rRNA hyper-variable regions targeted for sequencing, which have been the most widely used markers to assess the phylogenetic diversity of microbes, is an important decision to make. A frequent choice is the V1–V3 region, but its application has been so far limited to Roche/454 pyrosequencing platform (offering up to 750 bp single-end length reads)30. In addition, the V3–V4 hypervariable region has been targeted via the MiSeq platform (which can produce single-end reads of 350 bp), which can allow for more accurate and cost-effective characterizations of microbiome samples30,31,32. The V4-V5 region is also a traditionally employed hypervariable region in 16S rRNA based microbial biodiversity profiling studies33,34. Previous studies have shown that the choice of particular hypervariable region targeted in 16S rRNA sequencing can significantly alter the perceived structure of microbial community16,17,35,36,37. Therefore, critical assessment of the choice of hyper-variable regions will be important, so as to minimize distortion and conflicts in sequence-based analysis and comparison of oral microbiota.
A number of studies have investigated the effects of various factors on 16S rRNA gene based microbiome profiles (e.g., gut microbiota studies38, human microbiome mock sample30 and saliva sample39), such as sample storage prior to DNA extraction40, DNA extraction procedure22,41,42,43, primers13,35,44 and the sequencing platform used13. However, frequently no consensus conclusions were reached on how particular factors influence the results. For instance, in the case of DNA extraction method, Lazarevic et al.39 and Vesty et al.43 concluded that no significant difference in diversity indices was found between different extraction methods, although the mechanical lysis method revealed higher operational taxonomic unit (OTU) richness. On the contrary, another study found that the global community structure and relative abundance of individual taxa are both affected by DNA extraction method35. Except for such controversy, for human oral microbiome, such investigations have so far been limited to DNA extraction methods22,39,43,45. Therefore, efforts to critically evaluate how individual or combinations of the factors affects the observed structure of oral microbiota are of value.
Here, employing a mock oral bacterial community with a predetermined equal composition, we systematically evaluated the impacts of each or combinations of three factors including DNA extraction method, hypervariable regions and technical reproducibility on oral microbiome sequencing. Our aim is to quantitatively assess the contribution of each factor to the sequencing results. By providing the evidence for rational decision making for each of the factors, the findings should guide the design of experiments and workflows towards a complete, accurate and reliable portrait of the human oral microbiota.
Materials and Methods
Mock community construction
Mock communities were prepared by mixing five representative oral bacteria (Streptococcus mutans UA159, Streptococcus oralis ATCC9811, Actinomyces viscosus C505, Enterococcus faecalis ATCC29212 and Lactobacillus fermentum ATCC9338; Table 1; Fig. 1). Cells cultivated in liquid medium were collected by centrifugation and then re-suspended in PBS to reach a cell density of 109 cells per ml. The cell density was determined by bright-line counting chamber (Hausser Scientific, Horsham, USA). Then a mock community was prepared by mixing equal volumes of cell suspensions of the five bacteria. After centrifugation for 10 minutes at 7,500 rpm, the pellet of cellular mixture was re-suspended in 500 μl TE buffer and frozen at −80 °C.

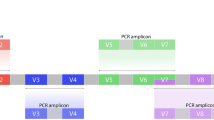
Overall workflow of the study. Mock community samples consisting of five oral strains were sequenced via six DNA extraction methods and followed by amplification of three 16S rRNA gene target regions. The results of subsequent 16S rDNA sequencing on the MiSeq platform were quantitatively compared. For each of the six samples (each of which underwent one of the six DNA extraction method), triple biological replicates (“Bio. replicates”) for each of the amplification regions were sequenced. In addition, for the amplification region of V3-V4 (P2), one technical replicate (“Tech. replicate”) for each of the six samples was sequenced. All the 54 biological samples were processed by the same experimenter (Experimenter A) and sequenced on the same sequencing batch (Batch A), while six technical replicates were processed by the other experimenter (Experimenter B) and on the other batch (Batch A).
DNA extraction
Six DNA extraction methods commonly used in human microbiome studies were chosen for the comparison (Table 2). DNA was extracted from the mock community in triplicates, in parallel with a negative control (PBS buffer). DNA concentrations were estimated by PicoGreen dsDNA quantitation kit (Invitrogen, Carlsbad, USA). Details on those six DNA extraction methods were described as below. It is noted that except for M1, the cell lysis method prior to DNA extraction is the only difference among the five DNA extraction methods from M2 to M6.
Method1- Phenol:chloroform
A 500 μl aliquot of cells was added to a tube containing an equal volume of lysis buffer. Then 30 μl of proteinase K (20 mg/ml, Qiagen, USA) and 75 μl of 10% SDS were added to the mixture, which was then incubated overnight at 53 °C in a shaking water bath. After the addition of 200 μL of 5 M NaCl and then 10 min of incubation on ice, 800 μL of buffer-saturated phenol was added. The tubes were vortexed at the maximum speed and then centrifuged for 10 min at 13,000 rpm. The aqueous phase was transferred to a new sterile centrifuge tube, to which a second 800 μL of buffer-saturated phenol was added. The tubes were vortexed and centrifuged again for 15 min at 13,000 × g. After the upper phase was recovered, an equal volume of chloroform/isoamyl alcohol (24:1) was added. The tubes were vortexed and centrifuged again for 15 min at 13,000 x g. The supernatant from the tube was transferred to a new tube, where 400 μl of isopropanol was added. The tube was then incubated for 10 min at room temperature and centrifuged for 15 min at 13,000 rpm. The supernatant was discarded and the DNA pellets were washed twice with 250 μl of 70% ethanol. Once dried, DNA was dissolved in 100 μl of double-distilled water46,47.
Method2-[DNeasy® Blood + Tissue kit]
The DNeasy® Blood & Tissue DNA kit (Qiagen Valencia, CA) was used based on manufacturer’s instruction. Briefly, cells were harvested from a 500 ul aliquot of culture in a microcentrifuge tube by centrifugation for 10 minutes at 7500 rpm. A buffer was added to re-suspend the pellets and the mixture was incubated for 30 min at 37 °C. Then 20 μl of Proteinase K was added and the mixture was incubated overnight at 56 °C. After these procedures, 200 ml AL buffer and 100 ml of ethanol were added. The rest of isolation protocol was performed based on the DNeasy® Blood & Tissue DNA kit46,48.
Method3-Chemical/enzymatic lysis + [DNeasy® Blood + Tissue kit]
In this method, an extra enzymatic lysis step was added prior to the application of DNeasy® Blood and Tissue kit (Qiagen Valencia, USA). Specifically, samples were mixed with 50 μl of lysozyme (final concentration 20 mg/ml, Sigma-Aldrich) and incubated for 1 hour at 37 °C. After this step, the mixture was supplemented with 20 μl of Proteinase K (20 mg/ml, Qiagen, USA) and incubated overnight at 56 °C. Then 500 μl AL buffer and 500 ml of ethanol were added to the lysate and the genomic DNA was purified according to the manufacturer’s instructions48,49.
Method4-Bead beating + [DNeasy Blood + Tissue kit]
The original sample (500 μl) was transferred into a clean Bead-Beating-Tube (2 ml Eppendorf tube), and 600 mg of 0.1-mm-diameter zirconia-silica beads (BioSpec, Bartlesville, USA) were added to the mixture. Cells were then mechanically disrupted for 3 minutes and 26 Hz at room temperature in a Qiagen Tissue Lyser LT. Then the mixture was centrifuged at 13,000 rpm for 5 min and 360 μl of the crude lysate was transferred into a new tube. The remaining steps were performed using DNeasy® Blood & Tissue Kits according to the manufacturer’s instructions48,50,51.
Method5-Chemical/enzymatic lysis + Bead beating + [DNeasy Blood + Tissue kit]
In this method, an extra step that consists of lysozyme-based enzymatic lysis and beating of zirconia-silica beads (BioSpec, Bartlesville, OK) was applied prior to the usage of DNeasy® Blood and Tissue kit (Qiagen Valencia, USA) as described above. Samples were transferred into clean bead beating tubes (2 ml Eppendorf tube), and 50 μl of lysozyme (20 mg/ml, Sigma-Aldrich, USA) was added to a 500 μl aliquot of cell suspension followed by incubation for 1 hr at 37 °C. Then, 600 mg of 1 mm diameter zirconia-silica beads (BioSpec, Bartlesville, USA) were added to the lysate and the cells were subjected to bead beating using a Qiagen Tissue Lyser LT at 36 Hz for 3 min. Further isolation and purification of the total genomic DNA from lysates were conducted using the DNeasy® Blood and Tissue kit according to the manufacturer’s instructions48,49,51.
Method6-[Chemical/Lytic-Enzyme-Cocktail master-mix lysis] + Bead beating + [DNeasy Blood and Tissue kit]
A two-step cell lysis procedure was employed before the use of DNeasy® Blood and Tissue kit (Qiagen Valencia, USA). Firstly, sample suspensions were kept on ice while a Lytic-Enzyme Cocktail was prepared. Freshly prepared 100 μl of Lytic-Enzyme-Cocktail master mix, including 50 μl of lysozyme (10 mg/ml, Sigma-Aldrich, USA), 6 μl of mutanolysin (5 KU/ml, Sigma-Aldrich, USA) and 3 μl of lysostaphin (4000 U/ml, Sigma-Aldrich), was added to samples and incubated at 37 °C for 45 min. Secondly, 600 mg cleaned and dry 0.1 mm diameter zirconia-silica beads were added to the lysate. Samples were then subjected to bead beating for 3 min at room temperature in a Qiagen Tissue Lyser LT (36 Hz). The remaining DNA extraction procedures were performed using the DNeasy® Blood and Tissue kit according to the manufacturer’s protocol26,48,49.
Evaluation of DNA yield and quality from the mock samples
DNA yield was determined fluorometrically using the High Sensitivity dsDNA kit (Invitrogen, CA, USA) on a Qubit® Fluorometer 1.0. DNA purity was assessed by measuring absorbance ratios spectrophotometrically on a NanoDrop® ND-1000 (NanoDrop Technologies, DE, USA), and the measurement includes A260/280 nm for protein contamination and A260/230 nm for salt and phenol contamination.
DNA standard preparation and quantitative real-time PCR
The known DNA concentration standards were prepared from the PCR products of genomic DNA. Three biological replicates of 10-fold serially diluted DNA standards from the PCR products of Prevotella Veroralis JCM6290 was used for the standard curve generation to quantify total bacteria. Quantitative real-time PCR assays were performed to quantify the total bacteria content of the extracted nucleic acids using Roche LightCycler 480II. Each qPCR was performed in a reaction volume of 20 μl containing 0.5 μM of each primer (Bac-16s-F2: 5′-TTAAACTCAAAGGAATTGACGG, Bac-16s-R2: 5′-CTCACGRCACGAGCTGACGAC)52, 1 μl of genomic DNA, 10 μl of 2 × SYBR Premix EXTaq mix (Takara) and 5 μl sterilized DNase-RNase-free water in MicroAmp fast optical 96-well reaction plates (Applied Biosystems, USA) with adhesive sealing. The qPCR parameters were 95 °C for 10 min, 45 cycles of three amplification steps including: 95 °C for 15 s, 60 °C for 20 s and 72 °C for 15 s, and final cooling at 25 °C for 1 min. Reaction specificities were confirmed by melting curve analysis with a progressive increase in temperature from 65 to 94 °C at a 1 °C/sec transition rate and continuous fluorescence acquisition. Each qPCR reaction was performed in triplicate. A standard curve of each primer was conducted by measuring three 10-fold series diluted DNA standards (i.e., the targeted DNA fragment cloned into the plasmid of pMD 19 T) and three negative controls (ddH2O as the template) were included within every experiment. Gene copy number was calculated automatically based on the standard curve of each primer system using LightCycler 480 Software 1.5 (Roche, USA).
Targeted 16S rRNA amplification and copy number identification
Amplicon libraries of three chosen hyper-variable regions including V1-V3, V3-V4, and V4-V5 in 16S rRNA genes were prepared in triplicates from each extracted DNA sample, and barcode sequenced using the 2 × 300 PE Illumina MiSeq platform23 (Table 3; Fig. 1). Copy numbers of 16S rRNA of the reference bacteria were identified based on the Ribosomal RNA Operon Copy Number Database (https://rrndb.umms.med.umich.edu/ 53; and the NCBI genome database (http://www.ncbi.nlm.nih.gov/sites/genome; Table 1).
Evaluation of technical reproducibility
With respect to the technical reproducibility, we randomly chose six samples that targeted the V3-V4 hypervariable regions, each of which was processed via one of the six DNA-extraction methods tested (Fig. 1). Six mock samples, one for each extraction method, were processed in duplicates by two independent experimenters. Results were compared between these two batches that followed an identical experimental procedure, in order to assess the technical reproducibility of the pipeline.
Bioinformatic analysis
Mothur was used for quality assessment of raw reads54, which were assembled and screened based on the minimum length of 250 bp and a maximum length of 550 bp. The SILVA 16S rRNA database was used for sequence alignment. Taxa were assigned for each sequence to the oral “CORE” reference database55 with a confidence threshold of 80%. Microbial community diversity, including α and β diversity, was analyzed at the species level and the genus level. Simpson and Shannon diversity indices that measure richness and evenness were employed to estimate diversity within each of the experimental protocols above. For β diversity, Bray-Curtis and Euclidean distance measures were applied to assess the dissimilarity among observed microbiome structures. The observed relative abundance of each taxon was estimated by counting the number of reads for each taxon and then normalizing by the total number of reads per sample. Since an equal number of each kind of bacterial cells was present in the mock community, the expected relative abundance of each taxon was calculated based on the observed relative abundances of 16S rRNA gene reads as normalized by the copy number of 16S rRNA gene in each species.
Statistical analysis
To assess the influence of DNA extraction procedures on DNA quantity (i.e. DNA yield), quality (i.e. A260/A280 and A260/A230) and stability (i.e. total bacterial quantification), ANOVA (one-way analysis of variance) was employed with Tukey-Kramer post-hoc test for multiple pairwise comparisons. The p-values were adjusted for multiple testing according to Bonferroni adjustment. Based on the Bray-Curtis metric, PCoA (Principal Component Analysis) was performed to visualize the level of dissimilarity among the observed microbiome structures from the various experimental procedures. Adonis analysis was applied to determine the significance (p-value) and strength (an R2 value) of a given grouping factor in determining the variation of distances, with pairwise Adonis for multiple pairwise comparisons. A hierarchical cluster analysis was performed based on the Bray–Curtis distance for the one expected microbiota and 54 mock communities via the complete linkage method. A dendrogram was employed to visualize the results. To compare the quantitative data in α and β diversity analysis and biomarker selection, the Kruskal-Wallis rank sum test was used with false discovery rate (FDR) adjustment for multiple pairwise comparisons. Regarding α diversity analysis, the Shannon or Simpson indices were compared among the different groups, and between the expected microbiota structure and the observed ones. The extent of changes in α-diversity indices was also assessed among the different groups. Procrustes rotation analysis was performed between two subsets of transformed data to test the degree of difference among distinct batches or experimenters. For Procrustes analysis, p values were generated using 1,000 Monte Carlo simulations.
Results
In this study, based on a mock oral microbiota consisting of five oral bacterial species of equal abundance, we compared the 16S amplicon sequencing results from six frequently employed DNA extraction procedures and three pairs of widely used 16S rRNA hypervariable regions (Tables 1, 2; Fig. 1). There are 60 mock samples in total, which yielded 980,511 bacterial 16S rRNA gene sequences that passed stringent quality control, averaging 16,342 reads per sample.
Influence of DNA Extraction Methods on the Quantity, Quality and Stability of DNA
Using gel electrophoresis, we confirmed the presence of metagenomic DNA from all the six DNA extraction methods. All the DNA extraction methods were able to produce high-quality metagenomic DNA from the mock community samples, but the difference in methods significantly affected the concentration of final extracted DNA (p < 0.01; Fig. 2). Pairwise comparisons showed that the traditional Phenol:chloroform-based method (M1) yielded the lowest amount of metagenomic DNA, which was followed by Kit DNA extraction method (M2) with a significant difference (p < 0.001; Fig. 2). The highest DNA yield was found in lysozyme + kit DNA extraction method (M3; all p values < 0.001; Fig. 2). On the contrary, the addition of beads (M4; p < 0.001) or the combination of [beads + lysozyme] (M5; p < 0.001) or [beads + Lytic- Enzyme Cocktail] (M6; p < 0.001) resulted in a significantly reduction in the final DNA concentration yielded (Fig. 2), although there was no significant difference in the amount of DNA yielded among these three methods (p < 0.05; Fig. 2).

DNA yield of samples processed by the six DNA extraction methods. All the DNA extraction methods produced high-quality metagenomics DNA from the mock community samples, but the difference in methods affected the final extracted DNA yield significantly (p < 0.01, ANOVA). The highest DNA yield was found in the M3 [lysozyme + kit] extraction method (M3, p < 0.001, post-hoc test). The addition of beads (M4, M5, and M6; p < 0.001, post-hoc test) significantly reduced the final concentration of DNA, and no significant difference was found in DNA yield between these three methods (p > 0.05, post-hoc test).
Moreover, the impact of DNA extraction methods on DNA quality was assessed via A260/A280 and A260/A230. There was a significant difference between the mean A260/A280 ratios of DNA extracts between M1 and the other five methods (p < 0.05). In fact, all the A260/A280 ratios were between 1.8 and 2, which are generally indicative of “pure” DNA, except for M1 (Table 4)56. Similarly, the protocols that included commercial kits (M2-M6) had significantly less residual carryover than the non-commercial method (M1; p < 0.01), and the former (M2-M6) all produced DNA with the mean A260/A230 ratio between 1.8 and 2 (Table 4)56. These results suggested that, in this case, the commercial kit methods produced DNA with higher and more consistent purity, while cell lysis methods have no effect on DNA purity.
Furthermore, to compare the stability of DNA derived from the extraction methods, total bacterial DNA copy number was quantified via the real-time qPCR (Table 4). Difference in methods significantly affected the total bacterial DNA copy numbers (p < 0.05, ANOVA). All the five methods using the commercial kit (M2-M6) resulted in increased total bacterial copy numbers, as compared with the non-commercial method (M1; all p < 0.01). Remarkably, the sole addition of lysozyme (M3) produced the highest total bacterial copy numbers. On the other hand, the addition of beads (M4, M5 and M6) appeared to be less effective than M2 (all p < 0.01) in increasing the total bacterial copy numbers (no significant difference in DNA yield among M4, M5 and M6; p > 0.05). Thus the pattern of total bacterial DNA copy numbers across the methods is largely consistent with that of the DNA yields (Table 4), suggesting the stability of DNA produced by the methods tested.
Impact on Observed Microbiota Structure
To identify microbiota features that are associated with the experimental procedures, the observed and expected microbiota structures were clustered via Bray-Curtis-based PCoA (Fig. 3A,B). Both the DNA extraction method and the choice of hypervariable region exhibited significant influence on microbiota community structure (p < 0.001; Fig. 3A), and the effect of the former (R2 = 0.764) was stronger than that of the latter (R2 = 0.210; Fig. 3A). Those two factors collectively explained up to 97.4% variation of microbiota structure (no inter-dependence found between the two factors; Fig. 3A). From the DNA-extraction-method point of view, apart from the similar performance of M5 and M6 (p = 0.513), other extraction methods produced significantly different oral microbiota structures (pairwise comparisons; all p < 0.01; Fig. 3B). With respect to the choice of the hypervariable region, observed structure for oral microbiota from P2 (V3_V4) and P3 (V4_V5) were similar (p = 0.825), and both of them were distinct from P1 (V1_V3) (p < 0.01; Fig. 3B).

Impact of experimental procedures on the variation of mock oral microbiota. Bray-Curtis distance was calculated and compared between all pairs of samples collected from the expected microbiota and the 54 mock ones. (A) The perceived microbiome structure within and between DNA extraction methods and choice of hypervariable regions. DNA extraction method (R2 = 0.764, p < 0.001, Adonis) dominated the effect on oral microbiota structure over choices of primer sets (R2 = 0.21, p < 0.001, Adonis). (B) PCoA plot of the expected and all the 54 mock communities with the Bray-Curtis distance. For the DNA extraction methods, apart from the similar performance between M5 and M6 (p = 0.513, Kruskal-Wallis test), the other extraction methods produced significantly different oral microbiota structures (all p < 0.01, Kruskal-Wallis test). As for the choice of primer sets, results for oral microbiota from P2 (V3-V4) and P3 (V4-V5) were similar (p = 0.825, Kruskal-Wallis test), and both of them were distinct from P1 (V1-V3) samples (p < 0.01, Kruskal-Wallis test).
Impact on Taxonomic Identification
Taxonomic compositions of the observed microbiota were compared with the expected ones to test whether our protocols can accurately measure community composition. Interestingly, none of the procedure tested here was able to perfectly recapitulate the actual composition. In general, Enterococcus faecalis and Streptococcus oralis were over-represented (as compared to the expected relative abundance), while Lactobacillus fermentum and Streptococcus mutans were under-represented at the species level (Fig. 4A). Actinomyces viscosus was not consistently represented as compared to the expected, being underestimated in P1 (V1_V3) and M1 while overestimated in P2 (V3_V4) and P3 (V4_V5) and those DNA extraction methods except M1 (Fig. 4A).

Comparison on the relative abundance of the five bacterial species in the mock community between the observed and the expected microbiome structures. (A) Relative abundance of the five bacterial species from each sample. (B) Comparison among the extraction methods. (C) Comparison among the targeted hypervariable regions. E. faecalis and S. oralis were over-represented as compared to the expected relative abundance, while L. fermentum and S. mutans were under-represented. A. viscosus was not consistently represented as compared to the expected result. M5 and M6 exhibited the best concordance with the expected distribution, followed by M3. P1 (V1_V3) performed better by producing results closer to the expected relative abundance of A. viscosus than V3-V4 and V4-V5, whereas V3-V4 and V4-V5 generated results that are in closer concordance with the expected abundance of S. mutans. Distinct letters denote significant difference between groups (p < 0.05, Wilcoxon sum-rank test).
Specifically, (i) M5 and M6 exhibited the highest concordance with the expected distribution of mock-community members except for Enterococcus faecalis, and they were followed by M3 (Fig. 4B); (ii) Although quantification of Enterococcus faecalis was more accurate in M2 and M3, M2 produced the largest bias on the abundance of Lactobacillus fermentum, Streptococcus oralis and Streptococcus mutans (Fig. 4B); (iii) the largest distortion of the community composition was found in M1 and M4 respectively, i.e., the over-representation of Actinomyces viscosus in M1 and the under-representation of Enterococcus faecalis in M4 (Fig. 4B). The hypervariable region pairs produced different results only for Actinomyces viscosus and Streptococcus mutans (all p values < 0.05; Kruskal-Wallis test). Intriguingly, P1 (V1_V3) offered better performance than P2 (V3_V4) and P3 (V4_V5) for Actinomyces viscosus, whereas P2 (V3_V4) and P3 (V4_V5) exhibited closer concordance with the expected abundance of Streptococcus mutans (p < 0.05, P1 (V1_V3) vs. P2 (V3_V4) and P1 (V1_V3) vs. P3 (V4_V5); Fig. 4C).
Impact on Microbial Diversity Assessment
To quantitatively evaluate which method generates an oral microbiota structure that is the most similar to the expected structure, we calculated the Bray-Curtis distances between expected and observed bacterial profiles for each of the protocols (Fig. 5A). Kruskal-Wallis analysis showed that DNA extraction methods exerted a substantial effect on the β diversity of oral microbiome (p < 0.001), whereas the hypervariable regions had little effects (p = 0.661). Based on pairwise comparisons, the most reliable extraction method for reproducing the original microbiome structure was found in the combination of mechanical and enzymatic-lysis DNA-extraction methods (M5 and M6), followed by M3, M4, M2 and M1 (all p-values < 0.05; Fig. 5A). Moreover, hierarchical clustering of the microbial profiles from different experimental protocols was performed (Fig. S1). The formation of two major clusters suggested again that the difference between the DNA-extraction procedures outweighed the variations due to choice of hypervariable regions (Fig. S1). Moreover, protocols that included enzymatic-mechanical-lysis steps in their extraction procedure (e.g., M5 and M6) were clustered with the expected microbiome structure, thus they produced better microbial diversity representation than those methods without an extra step in the procedure (e.g., M1, M2, and M3; Fig. S1). In terms of the hypervariable regions, samples from P2 (V3_V4) and P3 (V4_V5) tend to cluster closely with the expected microbiota structures (Fig. S1).

Comparison of α- and β- diversity between the observed and the expected community structures from each experimental procedure. (A) Box plots of Bray-Curtis distances between the expected and the observed microbiome diversity. The highest reproducibility of oral microbiota structure was found in those featuring combination of mechanical and enzymatic lysis methods (M5 and M6), followed by M3, M4, M2 and M1. (B) Both Shannon and Simpson indices representing α diversity were significantly different among the DNA extraction methods (p < 0.001, Kruskal-Wallis test), yet no difference was found among hypervariable regions on both indices (p > 0.05, Kruskal-Wallis test). Regardless of the primer sets chosen, α diversity indices from M5, M6 and M3 were the closest to the expected microbiota structure (p > 0.05, Kruskal-Wallis test).
Furthermore, we assessed the impact of the various methods on the α diversity as represented by Shannon and Simpson indices. The results were in agreement with the β diversity analysis: both indices were significantly different among the various DNA extraction methods (p < 0.001, Kruskal-Wallis test; Fig. 5), whereas no difference was found among the hypervariable regions (p > 0.05, Kruskal-Wallis test; Fig. 5). In fact, when compared with the other methods, α diversity indices detected by M5, M6 and M3 were the closest to the expected ones (pairwise comparisons; all p-values < 0.05, Fig. 5).
Reproducibility of the Illumina-sequencing platform
To evaluate the reproducibility of the DNA extraction methods in microbiota sequencing, we performed “replicated” samples of the mock community for those six DNA extraction methods in P2 (V3_V4; Fig. 1), where DNA was extracted by two independent experiments and sequenced on two batches of the Illumina MiSeq platform. PCoA plots of those twelve samples, for which β diversity was estimated using the Euclidean distance matrices, were constructed (Fig. 6). Specifically, one sample for the P2 (V3-V4) region was chosen from each of the six DNA extraction methods, and each of the six samples underwent two experimental procedures. Each procedure involved one distinct technician and one separate sequence batch. Analysis of the data using Procrustes test revealed a good agreement between results from the different experimenters, and also for sequencing performed among different batches (p < 0.001, Monte-Carlo permutation test; Fig. 6).

Validating the reproducibility of bacterial profile between the various experimental and sequencing batches. In the P2 (V3_V4) dataset, twelve mock microbiota were processed by two operators using six DNA extraction methods and then sequenced in the two batches on the Illumina MiSeq platform. Procrustes analysis showed that the agreement of bacterial profiling among technical replicates was excellent based on the Euclidean distances (p < 0.001, Monte-Carlo permutation test).
Discussion
The human body is home to many indigenous microorganisms, with distinct communities at different anatomical sites57. The oral cavity harbored distinctive microbial communities due to its particular ecological and physiological characteristics. Many factors such as the profile of community members, structure of cell wall, physiological state of the cell and relative abundance of cells can affect the representation of specific genomes in the sample preparation and sequencing workflow, which can lead to significant bias in the reconstructed microbiota structure12,16,58,59. In this study, in order to characterize the possible bias-generating steps in a complete procedure and to critically assess data quality and comparability of results, we examined potential sources of variation from DNA extraction methods, targeted 16S rRNA hypervariable regions and technical replicates, to quantify the nature and magnitude of their impacts on oral microbiome diversity profiling.
As is recommended by the Microbiome Quality Control Project, a mock community consisting of known typical oral taxa with a priori determined ratio was employed so as to provide a golden criterion for validating and comparing the methodological protocols60. Adoption of a mock community to assess and calibrate the plethora of experimental and computational methods can be particularly important to microbiome sequencing, as in most circumstances the larger effect size of inter-individual variation than that of methodological variation can hinder identification of those variation specifically attributed to the difference in experimental protocols28,61. In this study, the oral taxa selected as constituents of the mock community were typical strains that are implicated in both healthy hosts62 and oral inflammations such as dental caries63, apical periodontitis64 and periodontitis65. Moreover, all the strains are Gram-positive bacteria, with cell membranes more resistant to lysis than Gram-negative ones. Therefore, we chose a mock microbiota that consists of difficult-to-extract and abundantly represented oral type strains as a model, to validate the efficiency, robust and reproducibility of the potential bias-generating steps.
A principal concern with current experimental protocols is how to quantitatively assess the sources of variation in microbiota profiling. Our result showed that experimental procedures contributed to up to 98% variation of the microbiota structures, and the variation was mainly originated from DNA extraction method (R2 = 0.764), which is in contrast to the hypervariable regions (R2 = 0.210). This result was in accordance with those previously reported39, which supported the DNA extraction procedure’s critical role in shaping the final DNA yield and sequencing results. However, several studies on human gut microbiome reported the opposite results, i.e., the effect of hypervariable region outweighed that of DNA extraction method on the portrayal of mock community structure30,37. The inconsistencies here may arise, firstly, from the similarity in the commercial DNA extraction kits that they have compared (in contrast, sample pretreatment prior to DNA extraction was also tested in our study). Secondly, in studies that used human oral microbiota rather than the mock communities, the inter-method variation could have been masked by the inter-individual variation61.
In our study, all the six extraction methods evaluated offered decent, yet varied, performance on extracting metagenomic DNA from the mock oral microbiota. Higher amounts of DNA yield were obtained from the enzymatic-lysis-only protocol (M3), yet supplementation with [beads beating] or [beads + Lytic- Enzyme Cocktail] yielded a lower amount of DNA. As previous studies reported that the DNA yield was mainly affected by the efficacy of cell lysis, rather than the DNA recovery process20, the improvement in DNA extraction efficiency due to use of lysozyme as shown in our study was likely due to the ability of lysozyme to disrupt peptidoglycans in the cell walls from Gram-positive bacteria. Moreover, to some extent, addition of the bead beating step, despite its tendency to lower the DNA yield due to an inevitable loss by inserting an extra processing step, can improve the accuracy of microbiota portrait, as suggested by the sequencing results. Notably, our results are in agreement with other oral22 and gut23 studies, which observed that the addition of beads plus enzymatic system (M5: lysozyme; M6: a cocktail containing lysozyme, mutanolysin, and lysostaphin) generally produced the most accurate microbial representation. Specifically, a bead-enzyme combination processing step can adequately and accurately identify those Gram-positive bacteria such as Streptococcus mutans, thus ensuring a more accurate portrait of the oral microbiota. These results highlighted the importance of appreciating and tracking the potential bias introduced by DNA extraction methods and sample pre-treatment strategies, during the sequencing of oral microbiome samples.
The various 16S hypervariable regions targeted exhibit different degrees of variability. No individual hypervariable regions by itself can discriminate between all known microbial lineages16. Therefore, in microbiome sequencing studies, the choice of hypervariable regions may significantly affect the estimates of microbial diversity. Several previous studies have documented considerable variations among targeted 16S rRNA gene regions17,66. However, no clear consensus on the hypervariable region choice was reached, as each study has adopted a specific experimental methodology or a variant analysis pipeline, and featured a distinct sample origin, which resulted in the ambiguity in accessing the effect of hypervariable regions on the eventual portrait of oral microbiota. As previously reported67 and reaffirmed in this study, the selection of hypervariable regions had a significant impact on the observed oral microbiota structure, while its impact was relatively minor as compared to the DNA extraction methods. Furthermore, although no “perfect” hypervariable region existed from our experimental result15, we found that V3-V4 and V4-V5 seemed to produce more reproducible results than V1-V3, which is consistent with the studies on the skin and gut microbiota30,67. However, extrapolation of these conclusions to all oral bacteria will require further experiments, as while 16S rRNA gene hypervariable regions exhibited different degrees of variability among species13,35, this study only includes five Gram-positive species. Moreover, selection of hypervariable regions produced no obvious variations on the observed microbial diversity, as represented by either Bray-Curtis-distances-based β diversity or the α diversity index values. In our current study, the synthetic oral microbiota of five Gram-positive species is a relatively simple microbiota (relative to, e.g. saliva samples), thus the subtle difference in observed microbial diversity across the three hypervariable regions may in fact be due to the lack of amplification bias among the particular species tested30. Nevertheless, the selection of a suitable and relative optimal hypervariable region was of great importance, especially when the goal is to track differences across sampling sites, time scales or treatments, or to compare results obtained by different laboratories.
Conclusion
The observed oral microbiota structure is highly affected by the choice of DNA extraction method, while the impact of 16S rRNA hypervariable regions is relatively minor. Among the experimental protocols tested, enzymatic-mechanical-lysis based DNA extraction methods performed best on the characterization of oral microbiota diversity. Moreover, V3-V4 and V4-V5 hypervariable regions appeared to lead to more accurate oral-microbiota structure than V1-V3 hypervariable regions. In addition, magnitude of variation in observed microbial diversity was independent of the operators and sequencing batches. In the current study, as the mock community does not fully represent the complex and heterogeneous oral microbiota, our results on the impacts of bias-generated steps including DNA extraction and 16S rRNA hypervariable region on microbiota structure reconstruction should be interpreted within the context of these tested strains, and extrapolation of them to other oral species (particularly Gram-negative organisms) should be treated with caution. Nevertheless, our findings can serve as one reference for scientists in selecting DNA extraction methods and hypervariable regions for their particular research mission.
References
Teng, F. et al. Prediction of Early Childhood Caries via Spatial-Temporal Variations of Oral Microbiota. Cell Host Microbe 18, 296–306 (2015).
Garcia, R. I., Henshaw, M. M. & Krall, E. A. Relationship between periodontal disease and systemic health. Periodontol 2000 25, 21–36 (2001).
Yang, F. et al. Saliva microbiomes distinguish caries-active from healthy human populations. ISME J 6, 1 (2012).
Lim, Y., Totsika, M., Morrison, M. & Punyadeera, C. Oral microbiome: A new biomarker reservoir for oral and oropharyngeal cancers. Theranostics 7, 4313 (2017).
Werner, J. J., Zhou, D., Caporaso, J. G., Knight, R. & Angenent, L. T. Comparison of Illumina paired-end and single-direction sequencing for microbial 16S rRNA gene amplicon surveys. ISME J 6, 1273–1276 (2012).
Turnbaugh, P. J. et al. The human microbiome project: exploring the microbial part of ourselves in a changing world. Nature 449, 804–810 (2007).
Gill, S. R. et al. Metagenomic analysis of the human distal gut microbiome. Science 312, 1355–1359 (2006).
Consortium, H. M. J. R. S. A catalog of reference genomes from the human microbiome. Science 328, 994–999 (2010).
Costea, P. I. et al. Towards standards for human fecal sample processing in metagenomic studies. Nat. Biotechnol 35, 1069–1076 (2017).
Sinha, R. et al. Assessment of variation in microbial community amplicon sequencing by the Microbiome Quality Control (MBQC) project consortium. Nat. Biotechnol 35, 1077–1086 (2017).
Koren, O. et al. A guide to enterotypes across the human body: meta-analysis of microbial community structures in human microbiome datasets. PLOS Comput. Biol 9, e1002863 (2013).
Brooks, J. P. et al. The truth about metagenomics: quantifying and counteracting bias in 16S rRNA studies. BMC Microbiol 15, 66 (2015).
Schirmer, M., Ijaz, U. Z., D’Amore, R., Hall, N. & Quince, C. Insight into biases and sequencing errors for amplicon sequencing with the Illumina MiSeq platform. Nucleic Acids Res 43 (2015).
Bag, S. et al. An Improved Method for High Quality Metagenomics DNA Extraction from Human and Environmental Samples. Sci. Rep. 6, 26775 (2016).
Baker, G. C., Smith, J. J. & Cowan, D. A. Review and re-analysis of domain-specific 16S primers. J Microbiol Methods 55, 541–555 (2003).
Cruaud, P. et al. Influence of DNA Extraction Method, 16S rRNA Targeted Hypervariable Regions, and Sample Origin on Microbial Diversity Detected by 454 Pyrosequencing in Marine Chemosynthetic Ecosystems. Appl Environ Microbiol 80, 4626–4639 (2014).
Tremblay, J. et al. Primer and platform effects on 16S rRNA tag sequencing. Front Microbiol 6, 771 (2015).
Hwang, C., Ling, F., Andersen, G. L., LeChevallier, M. W. & Liu, W.-T. Evaluation of methods for the extraction of DNA from drinking water distribution system biofilms. Microbes Environ 27, 9–18 (2012).
Momozawa, Y., Deffontaine, V., Louis, E. & Medrano, J. F. Characterization of bacteria in biopsies of colon and stools by high throughput sequencing of the V2 region of bacterial 16S rRNA gene in human. PloS One 6, e16952 (2011).
Willner, D. et al. Comparison of DNA extraction methods for microbial community profiling with an application to pediatric bronchoalveolar lavage samples. PLoS One 7, e34605 (2012).
Wu, G. D. et al. Sampling and pyrosequencing methods for characterizing bacterial communities in the human gut using 16S sequence tags. BMC Microbiol 10, 206 (2010).
Abusleme, L., Hong, B.-Y., Dupuy, A. K., Strausbaugh, L. D. & Diaz, P. I. Influence of DNA extraction on oral microbial profiles obtained via 16S rRNA gene sequencing. J Oral Microbiol 6, 23990 (2014).
Yuan, S., Cohen, D. B., Ravel, J., Abdo, Z. & Forney, L. J. Evaluation of methods for the extraction and purification of DNA from the human microbiome. PloS One 7, e33865 (2012).
Cleaver, L. et al. Application of next generation sequencing for the diagnosis of orthopaedic infection: an evaluation of four DNA extraction techniques. Bone Joint J 98, 32 (2016).
Koshy, L. et al. Evaluating genomic DNA extraction methods from human whole blood using endpoint and real-time PCR assays. Mol Biol Rep 44, 97–108 (2017).
Valentini, A. et al. Next-generation monitoring of aquatic biodiversity using environmental DNA metabarcoding. Mol Ecol 25, 929–942 (2016).
Wintzingerode, V. F., Göbel, U. B. & Stackebrandt, E. Determination of microbial diversity in environmental samples: pitfalls of PCR−based rRNA analysis. FEMS Microbiol Rev 21, 213–229 (1997).
Salonen, A. et al. Comparative analysis of fecal DNA extraction methods with phylogenetic microarray: Effective recovery of bacterial and archaeal DNA using mechanical cell lysis. J Microbiol Methods 81, 127–134 (2010).
Angelakis, E. et al. Glycans affect DNA extraction and induce substantial differences in gut metagenomic studies. Sci Rep 6, 26276 (2016).
Fouhy, F., Clooney, A. G., Stanton, C., Claesson, M. J. & Cotter, P. D. 16S rRNA gene sequencing of mock microbial populations- impact of DNA extraction method, primer choice and sequencing platform. BMC Microbiol 16, 123 (2016).
Loman, N. J. et al. High-throughput bacterial genome sequencing: an embarrassment of choice, a world of opportunity. Nat Rev Microbiol 10, 599–606 (2012).
Caporaso, J. G. et al. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J 6, 1621 (2012).
Claesson, M. J. et al. Comparison of two next-generation sequencing technologies for resolving highly complex microbiota composition using tandem variable 16S rRNA gene regions. Nucleic Acids Res 38, e200 (2010).
Parada, A. E. & Fuhrman, J. A. Marine archaeal dynamics and interactions with the microbial community over 5 years from surface to seafloor. ISME J 11, 2510–2525 (2017).
Walker, A. W. et al. 16S rRNA gene-based profiling of the human infant gut microbiota is strongly influenced by sample processing and PCR primer choice. Microbiome 3, 26 (2015).
Soergel, D. A., Dey, N., Knight, R. & Brenner, S. E. Selection of primers for optimal taxonomic classification of environmental 16S rRNA gene sequences. ISME J 6, 1440 (2012).
Rintala, A. et al. Gut Microbiota Analysis Results Are Highly Dependent on the 16S rRNA Gene Target Region, Whereas the Impact of DNA Extraction Is Minor. J Biomol Tech 28, 19–30 (2017).
Raoult, D. & Henrissat, B. Are stool samples suitable for studying the link between gut microbiota and obesity? Eur J Epidemiol 29, 307–309 (2014).
Lazarevic, V., Gaïa, N., Girard, M., François, P. & Schrenzel, J. Comparison of DNA extraction methods in analysis of salivary bacterial communities. PLoS One 8, e67699 (2013).
Fouhy, F. et al. The effects of freezing on faecal microbiota as determined using MiSeq sequencing and culture-based investigations. PLoS One 10, e0119355 (2015).
Kennedy, N. A. et al. The impact of different DNA extraction kits and laboratories upon the assessment of human gut microbiota composition by 16S rRNA gene sequencing. PLoS One 9, e88982 (2014).
Nechvatal, J. M. et al. Fecal collection, ambient preservation, and DNA extraction for PCR amplification of bacterial and human markers from human feces. J Microbiol Methods 72, 124–132 (2008).
Vesty, A., Biswas, K., Taylor, M. W., Gear, K. & Douglas, R. G. Evaluating the Impact of DNA Extraction Method on the Representation of Human Oral Bacterial and Fungal Communities. PLoS One 12, e0169877 (2017).
Haas, B. J. et al. Chimeric 16S rRNA sequence formation and detection in Sanger and 454-pyrosequenced PCR amplicons. Genome Res 21, 494–504 (2011).
Yamagishi, J. et al. Comparison of Boiling and Robotics Automation Method in DNA Extraction for Metagenomic Sequencing of Human Oral Microbes. Plos One 11, e0154389 (2016).
Dilhari, A. et al. Evaluation of the impact of six different DNA extraction methods for the representation of the microbial community associated with human chronic wound infections using a gel-based DNA profiling method. Amb Express 7, 179 (2017).
Cherepkova, E. V., Maksimov, V. N., Kushnarev, A. P., Shakhmatov, I. I. & Aftanas, L. I. The polymorphism of dopamine receptor D4 (DRD4) and dopamine transporter (DAT) genes in the men with antisocial behaviour and mixed martial arts fighters. World J Biol Psychiatry, 1–14 (2017).
Li, R. et al. Association of keratin 8/18 variants with non-alcoholic fatty liver disease and insulin resistance in Chinese patients: A case-control study. World J Gastroenterol 23, 4047–4053 (2017).
Eckburg, P. B. et al. Diversity of the human intestinal microbial flora. Science 308, 1635–1638 (2005).
Yan, H. et al. Multiplex detection of bacteria on an integrated centrifugal disk using bead-beating lysis and loop-mediated amplification. Sci Rep 7, 1460 (2017).
Ravel, J. et al. Vaginal microbiome of reproductive-age women. Proc Natl Acad Sci USA 108, 4680–4687 (2011).
Sedgley, C. et al. Quantitative real-time PCR detection of oral Enterococcus faecalis in humans. Arch Oral Biol 50, 575–583 (2005).
Lee, Z. M.-P., Bussema, C. & Schmidt, T. M. rrnDB: documenting the number of rRNA and tRNA genes in bacteria and archaea. Nucleic Acids Res 37, D489–D493 (2009).
Schloss, P. D. & Westcott, S. L. Assessing and improving methods used in operational taxonomic unit-based approaches for 16S rRNA gene sequence analysis. Appl Environ Microbiol 77, 3219–3226 (2011).
Griffen, A. L. et al. CORE: a phylogenetically-curated 16S rDNA database of the core oral microbiome. PloS One 6, e19051 (2011).
Olson, N. D. & Morrow, J. B. DNA extract characterization process for microbial detection methods development and validation. BMC Res Notes 5, 668 (2012).
Dethlefsen, L., McFall-Ngai, M. & Relman, D. A. An ecological and evolutionary perspective on human-microbe mutualism and disease. Nature 449, 811–818 (2007).
Kennedy, K., Hall, M. W., Lynch, M. D., Moreno-Hagelsieb, G. & Neufeld, J. D. Evaluating bias of Illumina-based bacterial 16S rRNA gene profiles. Appl Environ Microbiol 80, 5717–5722 (2014).
Brooks, J. P. Challenges for case-control studies with microbiome data. Ann Epidemiol 26, 336–341 (2016).
Wu, G. D. et al. Linking Long-Term Dietary Patterns with Gut Microbial Enterotypes. Science 334, 105–108 (2011).
Lim, Y., Totsika, M., Morrison, M. & Punyadeera, C. The saliva microbiome profiles are minimally affected by collection method or DNA extraction protocols. Sci Rep 7, 8523 (2017).
Naidu, A. S., Bidlack, W. R. & Clemens, R. A. Probiotic spectra of lactic acid bacteria (LAB). Crit Rev Food Sci Nutr 39, 13–126 (1999).
Michalek, S. M., McGhee, J. R., Shiota, T. & Devenyns, D. Virulence of Streptococcus mutans: cariogenicity of S. mutans in adult gnotobiotic rats. Infect Immun 15, 466–471 (1977).
Peciuliene, V., Reynaud, A. H., Balciuniene, I. & Haapasalo, M. Isolation of yeasts and enteric bacteria in root-filled teeth with chronic apical periodontitis. Int Endod J 34, 429–434 (2001).
Tanner, A. C. R., Haffer, C., Bratthall, G. T., Visconti, R. A. & Socransky, S. S. A study of the bacteria associated with advancing periodontitis in man. J Clin Periodontol 6, 278–307 (1979).
Ghyselinck, J., Pfeiffer, S., Heylen, K., Sessitsch, A. & De Vos, P. The effect of primer choice and short read sequences on the outcome of 16S rRNA gene based diversity studies. PLoS One 8, e71360 (2013).
Castelino, M. et al. Optimisation of methods for bacterial skin microbiome investigation: primer selection and comparison of the 454 versus MiSeq platform. BMC Microbiol 17 (2017).
Acknowledgements
We thank Cunpei Bo for technical assistance with sequencing. We appreciate Xia Li for oral sample collection and processing. This work is funded by grant 81430011, 81670979 and 31600099 from National Natural Science Foundation of China, grant 16-5-1-67-jch and 16-5-1-64-jch from Qingdao Natural Science Foundation, and grant KF2016120101 from Open Fund of Guangdong Provincial Key Laboratory of Stomatology, Sun Yat-sen University and Qingdao Exceptional Health Professional Development Fund, Qingdao Outstanding Health Professional Development Fund, and the China Scholarship Council for Visiting Scholarship at UCLA. The funders had no role in study design, data collection, and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
F.T., F.Y. and J.X. envisioned the study. F.T. and F.Y. designed experiments, and P.F., S.L.and X.L. collected and processed samples. F.T. and P.F. prepared and sequenced samples. F.T., S.H. and S.S. analysed data. F.T., F.Y., S.S. and J.X. wrote the paper.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Teng, F., Darveekaran Nair, S.S., Zhu, P. et al. Impact of DNA extraction method and targeted 16S-rRNA hypervariable region on oral microbiota profiling. Sci Rep 8, 16321 (2018). https://doi.org/10.1038/s41598-018-34294-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-34294-x
Keywords
This article is cited by
-
The overlooked evolutionary dynamics of 16S rRNA revises its role as the “gold standard” for bacterial species identification
Scientific Reports (2024)
-
Comparison of four DNA extraction methods for 16s rRNA microbiota profiling of human faecal samples
BMC Research Notes (2023)
-
Benchmarking DNA isolation methods for marine metagenomics
Scientific Reports (2023)
-
The effect of single dose albendazole (400 mg) treatment on the human gut microbiome of hookworm-infected Ghanaian individuals
Scientific Reports (2023)
-
Analysis of microbial composition and sharing in low-biomass human milk samples: a comparison of DNA isolation and sequencing techniques
ISME Communications (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
