
Abstract
We designed a genotyping panel for the investigation of the genetic underpinnings of inter-individual differences in aggression and the physiological stress response. The panel builds on single nucleotide polymorphisms (SNPs) in genes involved in the three subsystems of the hypothalamic-pituitary-adrenal (HPA)-axis: the catecholamine, serotonin and corticoid metabolism. To promote the pipeline for use with wild animal populations, we used non-invasively collected faecal samples from a wild population of Assamese macaques (Macaca assamensis). We targeted loci of 46 previously reported SNPs in 21 candidate genes coding for elements of the HPA-axis and amplified and sequenced them using next-generation Illumina sequencing technology. We compared multiple bioinformatics pipelines for variant calling and variant effect prediction. Based on this strategy and the application of different quality thresholds, we identified up to 159 SNPs with different types of predicted functional effects among our natural study population. This study provides a massively parallel sequencing panel that will facilitate integrating large-scale SNP data into behavioural and physiological studies. Such a multi-faceted approach will promote understanding of flexibility and constraints of animal behaviour and hormone physiology.
Similar content being viewed by others

Introduction
Recent developments in molecular techniques enable researchers to include large-scale investigations of genetic impacts on behavioural or endocrine variables at reasonable costs1,2. In studies on humans, the investigation of genotypic influences on phenotypic characteristics revealed that inter-individual variation can be strongly affected by genotype3,4,5. For example, genotypic variation is responsible for approximately 50% of inter-individual variation in physiological stress levels and aggression6,7. In animal studies, however, the consideration of underlying genotype in behavioural and physiological studies is relatively understudied. Especially regarding studies on wild, non-model populations, several authors have called for a more frequent consideration of genetic impacts on animal behaviour1,8,9. In this study, we provide a multi-locus genotyping pipeline, based on non-invasively sampled material from a population of wild primates, facilitating future research on social and ecological factors driving variation in stress and aggression.
Modulation of the hypothalamic-pituitary-adrenal (HPA)-axis activity is an effective mechanism mediating environmental effects on the organism including its behavioural tendencies. The HPA-axis is a central physiological pathway activated in response to stress and is conserved across vertebrates10,11. In behavioural ecological studies, aggressive behaviour is often linked to HPA-axis activity via quantification of cortisol, the end product of this pathway (fish12, birds13, rodents14, ruminants15, cats and dogs16,17, primates18,19). Links to behaviour have been established in rats where the stimulation of brain areas responsible for aggression causes cortisol release, and similar processes are proposed for other vertebrates, including humans20,21,22. Behavioural ecological studies on aggression often assess how the expression of aggressive behaviour is related to social dominance or affected by characteristics of the competitive situation without conceptually integrating inter-individual variation due to genetic variation.
Three main metabolic circuits contribute to the HPA-axis: the serotonin, the catecholamine and the corticoid. Genes associated with these metabolic circuits have been repeatedly targeted in human clinical stress23,24 and aggression4,25,26 research. Functional polymorphisms in the genes coding for the three subsystems may lead to dysregulations in the HPA-axis pathway and a change in how the organism reacts to external stressors. The serotonin pathway involves the serotonin transporter (SLC6A4), receptor (HTR), tryptophan hydroxylase (TPH) and monoamine oxidase27. The neuropeptide Y (NPY) is a neurochemical that plays a protective role in stress resilience24,28. The catecholamine circuit (e.g. dopamine: DRD, SLC6A3, catechol-O-methyl transferase: COMT) causes general physiological changes that prepare the body for physical activity29. Main components of the corticoid pathway are the corticotropin-releasing hormone (CRH), CRH receptors (CRHR), the glucocorticoid receptor (NR3C1), CRH binding protein (CRHBP), corticosteroid binding globulin (SERPINA6) and the corticotropin receptor (MC2R)30.
For human diagnostics in the field of behavioural genetics, studies acquire large datasets via high-throughput methods such as massively parallel sequencing (i.e. next-generation sequencing)2. Behavioural studies on natural animal populations, however, commonly target one or a few gene loci associated with aggression and HPA-axis activity31,32,33, whereas high-throughput multi-locus approaches are rather rare but see1,34. Due to the high number of genes involved in physiological pathways such as the HPA-axis, the simultaneous assessment of multiple loci known to affect certain traits promises a much more comprehensive understanding of the investigated physiological and behavioural parameters35,36. The introduction of massively parallel sequencing technologies makes a multi-locus approach also feasible in studies on non-model species. The generated data provide high coverage of amplicons or genomes and a large and still growing body of different bioinformatics applications helps to investigate multiple loci in a fast and parallel way (e.g. Genome Analysis Toolkit – GATK37, SAMtools38 and UCSC genome browser39).
Here we report a massively parallel sequencing panel for the assessment of HPA-related SNPs useful for studies investigating the genetics that underlie behavioural and endocrine variation in aggression and the physiological stress response. For this purpose, we targeted loci of previously reported SNPs in 21 candidate genes associated with the HPA-axis. We provide detailed information on all steps from selection of target genes and polymorphisms, via laboratory work to the subsequent bioinformatics analyses of acquired massively parallel sequencing data. We additionally demonstrate the feasibility of application to faecal samples from wild populations, where non-invasive sampling is necessary.
Materials and Methods
Ethical statement
The National Research Council of Thailand (NRCT) and the Department of National Parks, Wildlife and Plant Conservation (DNP) approved (permit numbers: 0004.3/3618, 0002.3/2647, 0002/17, 0002/626, 0002/2424) the data collection at the study site in Thailand and export to Germany as part of a long-term collaboration between the University of Goettingen, the German Primate Center, the DNP and Kasetsart University Bangkok under the agreement of benefit sharing. Faecal samples were collected non-invasively. No animals were harmed or sacrificed for this study. Procedures were in accordance with the American Society of Primatologists’ (ASP) principles for the ethical treatment of non-human primates (https://www.asp.org/society/resolutions/EthicalTreatmentOfNonHumanPrimates.cfm).
Sample collection and storage
Samples were collected at Phu Khieo Wildlife Sanctuary in north-eastern Thailand. The sanctuary is part of the 6,500 km2 protected Western Isaan Forest Complex. Faecal samples were collected from four groups of fully habituated and individually identifiable Assamese macaques. In total, we collected 478 faecal samples from 38 adult males and 41 adult females (1–15 per individual, Ø 6) over the course of the long-term field project between June 2006 and January 2016. 58% of the samples were collected between March 2015 and January 2016. Adult males are involved in reproduction, have fully developed testes and long canines. Females are considered as adult in the mating season that they first conceive in, dating back from observations of their first birth.
For genetic analyses ~5 g of faeces were collected immediately after defecation from the surface of the faecal sample from an identified individual. We applied the two-step storage procedure, which included the collection of faecal samples into 50 ml tubes (62.559.001, Sarstedt, Nümbrecht, North Rhine-Westphalia, Germany) containing 30 ml of 97% ethanol40. After storage for 24 to 36 hours, ethanol was poured off and the faecal samples were dried and stored on 30 ml silica beads (112926-00-2, Intereducation Supplies Co., Ltd., Bangkok, Thailand) in 50 ml tubes in the dark at room temperature41. These samples were exported to Germany within 6 months and then stored at −20 °C until DNA extraction was performed.
SNP selection and amplicon primer design
Target regions were determined by searching the literature for SNPs in candidate genes involved in stress and aggression. The majority of target regions were chosen from literature on humans (for references see Supplementary Table S1), but we also targeted macaque and pig SNPs (for full list see Supplementary Table S1). Further, we chose only target regions located in protein-coding genes that code for receptors, enzymes and transmitter molecules associated with the HPA-axis. Targets were located both in exonic and intronic as well as untranslated regions. In total, we selected 46 target regions in 21 candidate genes. A summary about all genes that were included in the multi-locus genotyping panel can be found in Table 1, for more details about the target regions, including chromosomal position, SNPs, functional consequences and selected references see Supplementary Table S1.
As genome data for our study species, the Assamese macaque (Macaca assamensis), is not available, we designed primers according to the genome sequence of the congeneric rhesus macaque (Macaca mulatta, v8.0.1). For detailed information about amplicon sequences of Macaca mulatta and human see Supplementary Table S1.
Primers were designed using the online-software Primer3Plus42. Due to DNA degradation in faecal samples, primers were designed to amplify short PCR products with a maximum of 380 bp (Ø 207 bp), with the target SNP being as central as possible. Primer annealing temperatures were between 54 and 60 °C, with a maximum difference of 2 °C for each primer pair. Primer annealing temperature was chosen as predicted by Netprimer (Premier Biosoft, Palo Alto, California, USA). Further steps of the primer design included: (i) specificity check: Primer-Blast, NCBI, ‘nr’ database for Mammalia43, (ii) dimerization check: Netprimer (Premier Biosoft, Palo Alto, California, USA), (iii) secondary structure check: The mfold Web Server, DNA Folding Form44. The designed primers were ordered from Metabion (Planegg/Steinkirchen, Bavaria, Germany). In total we designed 41 primer pairs for 46 target loci (Supplementary Table S1).
Laboratory methods
DNA extraction was carried out with the First-DNA all-tissue Kit (D1002000, GEN-IAL GmbH, Troisdorf, North Rhine-Westphalia, Germany), following the manufacturer’s protocol for DNA extraction from faeces. The protocol included an overnight incubation at 37 °C with lysis buffer 1, lysis buffer 2 and proteinase K. After centrifugation, the supernatant was combined with lysis buffer 3, incubated at −20 °C for 5 minutes and centrifuged, followed by washing steps with 70% ethanol stored at −20 °C. Finally, DNA was eluted in 50 µl HPLC water (115333, Merck, Darmstadt, Hesse, Germany) and stored at −20 °C until further processing. All steps of the protocol were carried out with DNA LoBind Tubes (0030108051, Eppendorf AG, Hamburg, Germany). Total genomic DNA concentration was measured with a NanoDrop Spectrophotometer (ND-1000, PEQLAB Biotechnologie GmbH, Erlangen, Bavaria, Germany) and diluted to a final concentration of 100 ng/µl. 78% of the extracted faecal samples were collected between March 2015 and January 2016.
Target regions were amplified in 96-well plates (AB0600, Thermo Fisher Scientific, Waltham, Massachusetts, USA) with 1 U BioThermTaq DNA Polymerase (GC-002-5000, Genecraft, Cologne, North-Rhine Westphalia, Germany) in a 30 μl PCR mix (1 x reaction buffer, 0.16 mM for each dNTP, 0.33 μM for each primer, and 18 ng BSA, 100 ng template DNA), with the following thermocycler (Labcycler, Sensoquest, Göttingen, Lower Saxony, Germany) conditions: 2 minutes at 94 °C, 60 cycles of 30 seconds at 94 °C, 30 seconds at the appropriate annealing temperature (see Supplementary Table S1), 30 seconds at 72 °C, and 5 minutes at 72 °C. To check for PCR contamination, we ran 3 to 7 non-template controls on each 96-well plate. After amplification, aliquots were size-separated on 2% agarose gels along with a size standard (SM0241, Thermo Fisher Scientific, Waltham, Massachusetts, USA) to check for PCR performance and correct amplicon size. PCR products were then purified with Solid Phase Reverse Immobilization (SPRI) technology using 2.5x Ampure Beads (A63881, Beckman Coulter, Brea, California, USA) and again subjected to 2% agarose gel electrophoresis to control for purification performance. DNA concentration was measured with a Qubit 3.0 (Q32854, Thermo Fisher Scientific, Waltham, Massachusetts, USA). To test if our primers are target-specific, all 41 SPRI-purified amplicons of two individuals, acquired via PCR from faecal DNA extracts, were sequenced using Sanger technology. Therefore, we applied both amplification primers (3.3 pmol) and the Big Dye Cycle Sequencing Kit (433776452, Thermo Fisher Scientific, Waltham, Massachusetts, USA), and ran the reactions on an ABI 3130xl genetic analyzer (Thermo Fisher Scientific, Waltham, Massachusetts, USA). Sequence electropherograms were checked with DNA Baser (DNA Sequence Assembler v4, 2013, Heracle Biosoft S.R.L, Mioveni, Argeș, Romania) and compared with the respective target sequences of rhesus macaque and human.
For massively parallel sequencing, the amplicons from each individual were pooled in equimolar amounts to a total of 120 ng. Sequencing libraries were generated following the method described in Rohland et al.45 without uracil-DNA-glycosylase treatment based on Meyer and Kircher46 and Kircher et al.47. To check for performance of library preparation, we ran all libraries on an Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, California, USA) using the High Sensitivity DNA chip (5067-4626). Libraries were then pooled to a library mix with a final concentration of 10 nM. We added an additional step to control for quantity by running a qPCR with a 7500 Fast Real-Time PCR System (Thermo Fisher Scientific, Waltham, Massachusetts, USA) using the Bio-Rad EvaGreen Supermix (1725211, Bio-Rad, Hercules, California, USA) following the manufacturer’s recommendations and using three samples of the library mix and the concentration standards (5 nM, 10 nM, 20 nM). Reactions were run under the following conditions: 2 minutes at 50 °C, 10 minutes at 95 °C, 40 cycles of 30 seconds at 95 °C, 34 seconds at 60 °C and 30 seconds at 72 °C. Sequencing was conducted on an Illumina MiSeq sequencer (paired-end 150 bp) at the Microarray and Deep-Sequencing Core Facility, University Medical Center Goettingen, Lower Saxony, Germany.
SNP calling
After Illumina sequencing, all produced FASTQ reads were quality-checked and trimmed with FastQC48 and Trimmomatic v0.3649. For SNP calling, we used the GATK best practices pipeline for germline SNP discovery50 as well as the SAMtools-bcftools-pipeline38. For both pipelines, all quality-checked reads were mapped against the genome of Macaca mulatta v8.0.1 using BWA MEM v0.7.1251. We followed the GATK best practice pipeline and did not mark duplicates in the bam-file, because it is not possible to distinguish duplicate reads in amplicon sequencing where the major proportion of sequences reads represents PCR duplicates52,53. A first variant call was carried out using GATK’s HaplotypeCaller. Recovered SNPs were filtered using the hard-filtering procedures recommended by GATK’s best practices (VariantFiltration). The following quality filter expression was applied: quality by depth smaller than 2.0 (QD < 2.0), mapping quality smaller than 40.0 (MQ < 40.0), Fisher strand (Phred-scaled p-value using Fisher’s Exact Test) more than 60.0 (FS > 60.0), mapping quality rank sum (the u-based z-approximation from the Mann-Whitney Rank Sum Test for mapping qualities) smaller than −12.5 (MQRankSum < −12.5), and read position rank sum (the u-based z-approximation from the Mann-Whitney Rank Sum Test for the distance from the end of the read for reads with the alternate allele) smaller than −8.0 (ReadPosRankSum < −8.0)50. Afterwards, Base Quality Score Recalibration (BQSR) was performed twice using GATK’s BaseRecalibrator and PrintReads. The final variant calling process was conducted with GATK’s HaplotypeCaller in GVCF mode. The produced GVCF-files were merged using GATK’s GenotypeGVCFs.
SAMtools v1.4 mpileup was also used to generate raw variant calls using Macaca mulatta v8.0.1 as reference genome. The following settings were used: -d (maximal per-file depth) set to 250, -E (recalculate BAQ), –BCF (generate genotype likelihoods in BCF format), and –output-tags set to DP [to get the DP (number of filtered reads covering the corresponding allele) tag in the output file]. Variant calling was done using bcftools call v1.4 with the following settings: –multiallelic-caller (alternative model for multiallelic and rare-variant calling)–variants-only (output variant sites only) -O v (output type: ‘v’ uncompressed VCF)38.
Only variants identified by both the GATK and the SAMtools pipeline lying within the ranges of the genes of interest were used for subsequent analyses. Additionally, we compared the number of variants called without a threshold for the Phred quality (QUAL) score, with QUAL set to be ≥30, and with QUAL set to be ≥100. The Phred quality score gives a logarithmically related prediction-value to the base-calling error. The higher the quality score, the higher the base call accuracy54.
Variant annotation and effect prediction
SnpEff v4.3i55 and Variant Effect Predictor v8756 (VEP) were used for variant annotation and SNP effect prediction. We compared two different applications because variant prediction software can differ in their predicted effects57. With the VEP plugin ‘MaxEntScan’ it was possible to compare scores of the reference and mutant splice sites using a maximum entropy model and to predict splice site effects. Additionally, linkage disequilibrium scores were calculated using vcftools v0.1.1458 with the option ‘geno-r2’. Subsequently, all calculated annotations and effects were analysed in detail by hand.
Results
Sanger sequencing of the 41 amplicons from two macaque individuals revealed that the applied primers are target-specific. From the 79 investigated macaque individuals, we obtained a total of 3066 amplicons, with a minimum of 34 and a maximum of 41 amplicons per individual (Ø 38.6). For 10 of the 79 individuals, the first faecal DNA extracts (3 samples collected in 2007 and 7 samples collected in 2015) did not yield any amplicons (probably due to inhibitors in faeces), however, amplicons were successfully obtained from second extracts derived from other samples from the same individuals in all cases.
After sequencing on Illumina’s MiSeq, we obtained a total of 23,604,930 reads. Around 95% of the reads exhibit a Phred score > 29. About 85% of the reads could be mapped against the reference genome of Macaca mulatta v8.0.1 with a mean depth of 3,219.81 and a mean mapping quality of 59.02. Detailed sequencing statistics can be found in Supplementary Tables S2 and S3.
Variant calling
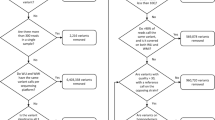
We compared different variant calling approaches in order to use only those variants reproduced by multiple pipelines. Using the complete dataset of shared variants without consideration of any filtering steps (QUAL, QD, MQ, FS, MQRankSum, and ReadPosRankSum), 70.12% of the variants called by SAMtools were also found with the GATK pipeline, whereas only 28.31% of the variants called by GATK were also found by SAMtools (Supplementary Figure S1). In sum, SAMtools called 230 SNPs, whereas GATK detected 373 SNPs. In total, 169 SNPs in 21 genes were verified using both variant calling approaches. 10 out of the 169 SNPs showed homozygosity for the alternate allele and thus represented simply a difference to the rhesus macaque genome, whereas 159 SNPs were identified as polymorphic sites within the study population. General descriptive statistics of the distribution of individuals being homozygous for the reference or alternate allele, or heterozygous, can be found in Table 2.
Using the described filtering steps without consideration of the QUAL scores, the number of detected variants by GATK decreased to 170 SNPs resulting in 144 SNPs in 20 genes shared by both callers. Using different QUAL scores as further selection steps, the number of detected polymorphisms changed again. Extracting only those variants with a QUAL score ≥ 30, GATK still detected 170 SNPs, but SAMtools called only 194 SNPs (Supplementary Figure S1). Only 140 SNPs in 20 genes were shared by both callers. With a QUAL score ≥ 100, the number of variants decreased further to 124 shared SNPs in 20 genes out of 170 SNPs detected with GATK and 162 SNPs detected with SAMtools (Supplementary Figure S1).
Variant effect prediction
The variants detected by both the SAMtools and the GATK pipeline were used to predict possible variant effects. For this purpose, we used two applications: VEP and SnpEff. Comparing the results, almost all variants were classified as the same consequence type in both applications. The only differences are two counts of ‘5’-UTR premature start codon gain variant’ found with SnpEff, but not with VEP and 25 additional ‘intergenic variants’ found by SnpEff (Table 3). Consequently, the predicted variant effects show a high similarity between both methods concerning their impact (Table 4).
Using the VEP plugin MaxEntScan, two SNPs were found to be associated with different entropies at splice sites. These SNPs were located in the serotonin transporter (SLC6A3) and the neuropeptide Y (NPY) genes and changed entropy from 0.40 to −4.66 and 9.38 to 9.43, respectively. These SNPs were identified as ‘splice_region_variant’ and ‘intron_variant’ by VEP and SnpEff. However, not all identified ‘splice_region_variants’ caused differences in splice site entropy, as predicted by MaxEntScan. An analysis of linkage disequilibrium using vcftools revealed that 64 SNPs were linked. Linkage r2 values ranged from 0.28 to 1 (Ø 0.91).
Discussion
Enhancing the approach of behavioural genetics and physiogenetics in wild animals would extend our knowledge of the factors that contribute to the still not completely understood variation within and between populations under natural selection pressures8. Behavioural studies often investigate the impacts of personality, age, sex or external factors such as social environment, group size and dominance hierarchy to explain inter-individual differences in HPA-axis related traits and probably misrepresent the amount of variation to be explained by such factors because they neglect genetic impacts59,60,61,62 or focus only on one or two gene variants31,32,33, but see34,63,64. An extended integration of genotype information in wild populations will facilitate a more comprehensive understanding of the observed phenotypic variation. For example, variant information on multiple loci can be used to generate cumulative genetic risk scores to predict individual variation35,65. Among the best-studied aspects of animal behaviour in the wild are behavioural and physiological reactions to social and environmental stressors66. Behavioural and physiological responses are tightly linked to the HPA-axis, the main physiological pathway activated in response to stressful stimuli67. Thus, genes coding for the components of the HPA-axis, which act in concert to maintain homeostatic balance, are important targets for the investigation of phenotypic variation in stress- and aggression-related traits.
We provide a SNP panel that may serve as a basic tool for future studies investigating the genetics of stress and aggression in behavioural and ecological studies. The offered panel and protocol enables field biologists teaming up with a laboratory to screen entire wild animal populations for multiple highly interesting target regions in a fast and parallel way. This study demonstrates that polymorphisms at purportedly functional sites exist in HPA-linked genes in natural populations. Knowing the samples from individually-recognized individuals, which is usually the case in long-term studies on wild animal groups, allows to accurately determine population frequencies of SNPs. Further, this application is transferable to other species. The HPA-axis is conserved among vertebrates10,11 and orthologous gene regions can be found easily for the species of interest e.g. via BLAST43, PSI-BLAST68, BLAT69, SSEARCH70 (biology.wustl.edu/gcg/ssearch.html) or HMMER3 (hmmer.org) searches. For application to other species, we recommend searching for the primer or amplicon sequences (Supplementary Table S1) using the aforementioned software and choosing the respective sequences to design taxon-specific primers. All subsequent steps can be carried out as outlined in our protocol. With small PCR product sizes, as in our case, allelic dropout is a marginal problem, but to further minimize the risk of allelic dropout we recommend multiple PCRs per sample71 or to perform replicates from different samples of the same individual.
Given that acquired massively parallel sequencing data hold many opportunities for further in-depth analyses, we provide detailed information on the applied bioinformatics pipelines. For example, with the help of GATK37 or SAMtools38 variants can be detected and used for subsequent high-throughput analyses concerning their functionality and possible effects (e.g. VEP56 and SnpEff 55). However, our analyses revealed that GATK called more variants than SAMtools in all conditions of the different quality thresholds and emphasize the importance of comparing pipelines and relying on validated, intersecting sets of SNPs. Further analyses of, e.g. splice site entropy (MaxEntScan) and linkage disequilibrium (vcftools), help to interpret the effects of detected polymorphisms and their potential consequences on physiological pathways.
To promote this application for studies on wild populations of non-model organisms, in which the consideration of genotype is particularly rare, we established our methods based on faecal samples. Studies on protected, free-ranging animals are often confined to the non-invasive collection of genetic material. DNA extracts from such low-quality sources contain only small amounts of host DNA72. The dominance of exogeneous, non-host, e.g. microbiome or food DNA, rules out a massively parallel sequencing-application on the pure DNA extracts, without prior amplification or enrichment of target regions. Sequencing of amplicons with the traditional Sanger method is time- and cost-intensive, particularly when encountering larger sets of target regions and individuals (3066 amplicons in this study). Furthermore, when two SNPs are found in one amplicon, the haplotype structure remains unknown. When applying the classical Sanger sequencing approach, elucidating haplotypes requires additional working steps, such as cloning. Here we have shown that multiple target regions can be easily covered with massively parallel amplicon sequencing from faecal DNA of larger numbers of individuals. Alternatively, target regions could be captured using synthesized or self-made capture probes73,74. While such methods may reach better sequencing uniformity, they are less target-specific and exhibit lower average coverage than amplicon-based technologies75. However, such methods could be applied to calculate additional background population structure74. Another important aspect, especially for studies on wild populations that are often limited to low-quality DNA samples, is that amplicon-based massively parallel sequencing methods allow processing of low-input DNA samples75. Further, due to the large amount of sequence reads produced, the regions of interest (amplicons) exhibit high coverage reducing the detection of false positive variants. A caveat of the study is that it is still ultimately a bottom-up approach needing a priori information to select target regions. As technology will improve in the future, top-down approaches will most likely also become an effective and economical tool for low-quality samples making more data available. These top-down approaches could be applied to generate haplotype data for a multitude of loci across the genome in a fast and parallel way, to calculate relatedness and include kinship relations in behavioural genetics approaches as well as to identify conserved genome regions or gene segments with high mutation rates in the investigated populations.
Numerous field studies have established links between non-invasive measures of HPA-axis activity and the behaviour of animals. Glucocorticoid metabolite levels, the end products of the HPA-axis, increase during reproductive challenges19,76, with increasing aggression given or received76,77 and are often related to social status78. Mostly lab-based studies have established links between HPA-axis activity and genetic variation at individual loci. For example, a mu-opioid receptor polymorphism is associated with cortisol and aggressive threat scores79 and variation in the serotonin transporter gene is associated with increased HPA-axis activity80 in captive primates. Progress is hampered by a lack of (1) integration of both research streams to link genetic variation to HPA-axis activity and behaviour, and (2) studies screening multiple loci involved in HPA-axis regulation at the same time. We propose that our panel can serve as a basis for general behavioural studies aiming to extend their study design on a molecular level and step into the field of behavioural genetics. The simultaneous investigation of genes and behaviour will help to achieve a more comprehensive understanding of individual animal characteristics.
Data Accessibility
Massively parallel sequencing-data were submitted to the Sequence Read Archive (SRA) available via NCBI with the accession number SRP116685. Additional data are available in the Supplementary Information.
References
Ekblom, R. & Galindo, J. Applications of next generation sequencing in molecular ecology of non-model organisms. Heredity. 107, 1–15 (2011).
Perry, G. H. The promise and practicality of population genomics research with endangered species. Int. J. Primatol. 35, 55–70 (2014).
Craig, I. W. & Halton, K. E. Genetics of human aggressive behaviour. Hum. Genet. 126, 101–113 (2009).
Pavlov, K. A., Chistiakov, D. A. & Chekhonin, V. P. Genetic determinants of aggression and impulsivity in humans. J. Appl. Genet. 53, 61–82 (2012).
Plomin, R., DeFries, J. C., Knopik, V. S. & Neiderheiser, J. Behavioral Genetics (Worth Publishers, 2013).
Linkowski, P. et al. Twin study of the 24-h cortisol profile: Evidence for genetic control of the human circadian clock. Am. J. Physiol. Endocrinol. Metab. 264, E173–E181 (1993).
Miles, D. R. & Carey, G. Genetic and environmental architecture on human aggression. J. Pers. Soc. Psychol. 72, 207–217 (1997).
Brent, L. J. N. & Melin, A. D. The genetic basis of primate behavior: Genetics and genomics in field-based primatology. Int. J. Primatol. 35, 1–10 (2014).
Tung, J., Alberts, S. C. & Wray, G. A. Evolutionary genetics in wild primates: Combining genetic approaches with field studies of natural populations. Trends Genet. 26, 353–362 (2010).
Denver, R. J. Structural and functional evolution of vertebrate neuroendocrine stress systems. Ann. NY Acad. Sci. 1163, 1–16 (2009).
Schulkin, J. Evolutionary conservation of glucocorticoids and corticotropin releasing hormone: behavioral and physiological adaptations. Brain Res. 1392, 27–46 (2011).
Øverli, Ø., Kotzian, S. & Winberg, S. Effects of cortisol on aggression and locomotor activity in rainbow trout. Horm. Behav. 42, 53–61 (2002).
Carere, C., Groothuis, T. G. G., Möstl, E., Daan, S. & Koolhaas, J. M. Fecal corticosteroids in a territorial bird selected for different personalities: Daily rhythm and the response to social stress. Horm. Behav. 43, 540–548 (2003).
Huhman, K. L., Moore, T. O., Ferris, C. F., Mougey, E. H. & Meyerhoff, J. L. Acute and repeated exposure to social conflict in male golden hamsters: increases in plasma POMC-peptides and cortisol and decreases in plasma testosterone. Horm. Behav. 25, 206–216 (1991).
Salas, M. et al. Aggressive behavior and hair cortisol levels in captive Dorcas gazelles (Gazella dorcas) as animal-based welfare indicators. Zoo Biol. 35, 467–473 (2016).
Finkler, H. & Terkel, J. Cortisol levels and aggression in neutered and intact free-roaming female cats living in urban social groups. Physiol. Behav. 99, 343–347 (2010).
Rosado, B. et al. Blood concentrations of serotonin, cortisol and dehydroepiandrosterone in aggressive dogs. Appl. Anim. Behav. Sci. 123, 124–130 (2010).
Honess, P. E. & Marin, C. M. Behavioural and physiological aspects of stress and aggression in nonhuman primates. Neurosci. Biobehav. Rev. 30, 390–412 (2006).
Ostner, J., Kappeler, P. & Heistermann, M. Androgen and glucocorticoid levels reflect seasonally occurring social challenges in male redfronted lemurs (Eulemur fulvus rufus). Behav. Ecol. Sociobiol. 62, 627–638 (2008).
Halász, J., Liposits, Z., Kruk, M. R. & Haller, J. Neural background of glucocorticoid dysfunction‐induced abnormal aggression in rats: Involvement of fear‐ and stress‐related structures. Eur. J. Neurosci. 15, 561–569 (2002).
Kruk, M. R., Halász, J., Meelis, W. & Haller, J. Fast positive feedback between the adrenocortical stress response and a brain mechanism involved in aggressive behavior. Behav. Neurosci. 118, 1062–1070 (2004).
Soma, K. K., Scotti, M. A. L., Newman, A. E. M., Charlier, T. D. & Demas, G. E. Novel mechanisms for neuroendocrine regulation of aggression. Front. Neuroendocrinol. 29, 476–489 (2008).
Jabbi, M. et al. Convergent genetic modulation of the endocrine stress response involves polymorphic variations of 5-HTT, COMT and MAOA. Mol. Psychiatry. 12, 483–490 (2007).
Zhou, Z. et al. Genetic variation in human NPY expression affects stress response and emotion. Nature. 452, 997–1001 (2008).
Qayyum, A. et al. The role of the catechol-o-methyltransferase (COMT) GeneVal158Met in aggressive behavior, a review of genetic studies. Curr. Neuropharmacol. 13, 802–814 (2015).
Craig, I. W. The importance of stress and genetic variation in human aggression. Bioessays 29, 227–236 (2007).
D’souza, U. M. & Craig, I. W. Functional polymorphisms in dopamine and serotonin pathway genes. Hum. Mutat. 27, 1–13 (2006).
Kormos, V. & Gaszner, B. Role of neuropeptides in anxiety, stress, and depression: From animals to humans. Neuropeptides. 47, 401–419 (2013).
Molinoff, P. B. & Axelrod, J. Biochemistry of catecholamines. Annu. Rev. Biochem. 40, 465–400 (1971).
Subbannayya, T. et al. An integrated map of corticotropin-releasing hormone signaling pathway. J. Cell Commun. Signal. 7, 295–300 (2013).
Garamszegi, L. Z. et al. The relationship between DRD4 polymorphisms and phenotypic correlations of behaviors in the collared flycatcher. Ecol. Evol. 4, 1466–1479 (2014).
Kalbitzer, U. et al. Insights into the genetic foundation of aggression in Papio and the evolution of two length-polymorphisms in the promoter regions of serotonin-related genes (5-HTTLPR and MAOALPR) in Papionini. BMC Evol. Biol. 16, 121, https://doi.org/10.1186/s12862-016-0693-1 (2016).
Timm, K., Tilgar, V. & Saag, P. DRD4 gene polymorphism in great tits: gender-specific association with behavioural variation in the wild. Behav. Ecol. Sociobiol. 69, 729–735 (2015).
Bergey, C. M., Phillips-Conroy, J. E., Disotell, T. R. & Jolly, C. J. Dopamine pathway is highly diverged in primate species that differ markedly in social behavior. PNAS. 113, 6178–6181 (2016).
Ferguson, B. et al. Genetic load is associated with hypothalamic–pituitary–adrenal axis dysregulation in macaques. Genes Brain Behav. 11, 949–957 (2012).
Pearce, E., Wlodarski, R., Machin, A. & Dunbar, R. I. M. Variation in the β-endorphin, oxytocin, and dopamine receptor genes is associated with different dimensions of human sociality. PNAS. 114, 5300–5305 (2017).
McKenna, A. et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303 (2010).
Li, H. et al. The sequence alignment/map format and SAMtools. Bioinformatics. 25, 2078–2079 (2009).
Kent, W. J. et al. The human genome browser at UCSC. Genome Res. 12, 996–1006 (2002).
Nsubuga., A. M. et al. Factors affecting the amount of genomic DNA extracted from ape faeces and the identification of an improved sample storage method. Mol. Ecol. 13, 2089–2094 (2004).
Roeder, A. D., Archer, F. I., Poinar, H. N. & Morin, P. A. A novel method for collection and preservation of faeces for genetic studies. Mol. Ecol. Notes. 4, 761–764 (2004).
Untergasser, A. et al. Primer3 - new capabilities and interfaces. Nucleic Acids Res. 40, e115, https://doi.org/10.1093/nar/gks596 (2012).
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. J. Mol. Biol. 215, 403–410 (1990).
Zuker., M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 31, 3406–3415 (2003).
Rohland, N., Harney, E., Mallick, S., Nordenfelt, S. & Reich, D. Partial uracil–DNA–glycosylase treatment for screening of ancient DNA. Philos. Trans. R. Soc. Lond. B Biol. Sci. 370, 20130624, https://doi.org/10.1098/rstb.2013.0624 (2015).
Meyer, M. & Kircher, M. Illumina sequencing library preparation for highly multiplexed target capture and sequencing. Cold Spring Harb. Protoc. 6, pdb-prot5448, https://doi.org/10.1101/pdb.prot5448 (2010).
Kircher, M., Sawyer, S. & Meyer, M. Double indexing overcomes inaccuracies in multiplex sequencing on the Illumina platform. Nucleic Acids Res. 40, e3, https://doi.org/10.1093/nar/gkr771 (2012).
Andrews, S. FastQC: A quality control tool for high throughput sequence data. Babraham Institute https://www.bioinformaticsbabrahamacuk/projects/fastqc/ (2010).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics. 30, 2114–2120 (2014).
Van der Auwera, G. A. et al. From FastQ data to high‐confidence variant calls: The genome analysis toolkit best practices pipeline. Curr. Protoc. Bioinformatics. 43, 1–33 (2013).
Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. Preprint at, https://arxiv.org/abs/1303.3997 (2013).
Tata, P. et al. Biologic Basis of Personalized Therapy in Head and Neck Squamous Cell Carcinoma. In Contemporary Oral Oncology: Biology, Epidemiology, Etiology, and Prevention (ed. Kuriakose, M. A.) 461–486 (Springer, 2017).
Ebbert, M. T. et al. Evaluating the necessity of PCR duplicate removal from next-generation sequencing data and a comparison of approaches. BMC Bioinformatics. 17, 239 (2016).
Ewing, B., Hillier, L., Wendl, M. C. & Green, P. Base-calling of automated sequencer traces using Phred. I. Accuracy assessment. Genome Res. 8, 175–185 (1998).
Cingolani, P. et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118, iso-2, iso-3. Fly. 6, 80–92 (2012).
McLaren, W. et al. The ensembl variant effect predictor. Genome Biol. 17, 122, https://doi.org/10.1186/s13059-016-0974-4 (2016).
Wertz. J., Bair, T. B. & Chimenti, M. S. PyVar: An extensible framework for variant annotator comparison. Preprint at, https://www.biorxiv.org/content/early/2016/09/30/078386 (2016).
Danecek, P. et al. The variant call format and VCFtools. Bioinformatics 27, 2156–2158 (2011).
Seyfarth, R. M., Silk, J. B. & Cheney, D. L. Variation in personality and fitness in wild female baboons. PNAS. 109, 16980–16985 (2012).
Young, C., Majolo, B., Heistermann, M., Schülke, O. & Ostner, J. Responses to social and environmental stress are attenuated by strong male bonds in wild macaques. PNAS. 111, 18195–18200 (2014).
Baugh, A. T., Davidson, S. C., Hau, M. & van Oers, K. Temporal dynamics of the HPA axis linked to exploratory behavior in a wild European songbird (Parus major). Gen. Comp. Endocrinol. 250, 104–112 (2017).
Mell, H. et al. Do personalities co-vary with metabolic expenditure and glucocorticoid stress response in adult lizards? Behav. Ecol. Sociobiol. 70, 951–961 (2016).
Song, S. et al. Targeted next-generation sequencing for identifying genes related to horse temperament. Genes & Genomics. 39, 1325–1333 (2017).
Madlon‐Kay, S. et al. Weak effects of common genetic variation in oxytocin and vasopressin receptor genes on rhesus macaque social behavior. Am. J. Primatol. e22873, https://doi.org/10.1002/ajp.22873 (2018).
Belsky, D. W. & Israel, S. Integrating genetics and social science: Genetic risk scores. Biodemography Soc. Biol. 60, 137–155 (2014).
Reeder, D. M. & Kramer, K. M. Stress in free-ranging mammals: Integrating physiology, ecology, and natural history. J. Mammal. 86, 225–235 (2005).
Del Rey, A., Chrousos, G. & Besedovsky, H. The hypothalamus-pituitary adrenal axis. (Elsevier, 2008).
Altschul, S. F. et al. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402 (1997).
Kent, W. J. BLAT—the BLAST-like alignment tool. Genome Res. 12, 656–664 (2002).
Smith, T. F. & Waterman, M. S. Comparison of biosequences. Adv. Appl. Math. 2, 482–489 (1981).
Goodrich, J. K. et al. Conducting a microbiome study. Cell. 158, 250–262 (2014).
Perry, G. H., Marioni, J. C., Melsted, P. & Gilad, Y. Genomic‐scale capture and sequencing of endogenous DNA from feces. Mol. Ecol. 19, 5332–5344 (2010).
Maricic, T., Whitten, M. & Pääbo, S. Multiplexed DNA sequence capture of mitochondrial genomes using PCR products. PloS One. 5, e14004, https://doi.org/10.1371/journal.pone.0014004 (2010).
Snyder-Mackler, N. et al. Efficient genome-wide sequencing and low-coverage pedigree analysis from noninvasively collected samples. Genet. 203, 699–714 (2016).
Samorodnitsky, E. et al. Evaluation of hybridization capture versus amplicon‐based methods for whole‐exome sequencing. Hum. Mutat. 36, 903–914 (2015).
Ostner, J., Heistermann, M. & Schülke, O. Dominance, aggression and physiological stress in wild male Assamese macaques (Macaca assamensis). Horm. Behav. 54, 613–619 (2008).
Wittig, R. M., Crockford, C., Weltring, A., Deschner, T. & Zuberbühler, K. Single aggressive interactions increase urinary glucocorticoid levels in wild male chimpanzees. PloS One. 10, e0118695, https://doi.org/10.1371/journal.pone.0118695 (2015).
Goymann, W. & Wingfield, J. C. Allostatic load, social status and stress hormones: The costs of social status matter. Anim. Behav. 67, 591–602 (2004).
Miller, G. M. et al. A mu-opioid receptor single nucleotide polymorphism in rhesus monkey: association with stress response and aggression. Mol. Psychiatry. 9, 99–108 (2004).
Barr, C. S. et al. Rearing condition and rh5-HTTLPR interact to influence limbic-hypothalamic-pituitary-adrenal axis response to stress in infant macaques. Biol. Psychiatry. 55, 733–738 (2004).
Acknowledgements
We thank the National Research Council of Thailand (NRCT) and the Department of National Parks, Wildlife and Plant Conservation (DNP) for permission to conduct this study and for all the support granted. We are grateful to J. Prabnasuk, K. Nitaya, T. Wongsnak, M. Pongjantarasatien and K. Kreetiyutanont, M. Kumsuk, W. Saenphala (Phu Khieo Wildlife Sanctuary) for their cooperation and permission to carry out this study. We thank A. Koenig and C. Borries, who developed the field site at Huai Mai Sot Yai. Special thanks go to S. Jumrudwong, W. Nueorngshiyos, N. Juntuch, J. Wanart, R. Intalo, T. Kilawit, N. Pongangan, B. Klaewklar, N. Bualeng, A. Ebenau, P. Saisawatdikul, K. Srithorn, M. Swagemakers, and T. Wisate for their excellent help in the field. We acknowledge C. Schwarz, N. Westphal, F. Aron and L. Walter for their support in the genetics lab. We thank our colleagues, particularly F. Ludewig, from the Microarray and Deep-Sequencing Core Facility, University Medical Center Göttingen for sequencing. We also thank T. S. Gutleb for valuable comments on the manuscript. We thank the members of the research unit ‘Sociality and Health in Primates’ (DFG FOR 2136) for stimulating discussions. This research was funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) – Project Number OS201/8-1; 254142454/GRK 2070.
Author information
Authors and Affiliations
Contributions
D.R.G., J.O., O.S. and C.R. designed research, D.R.G., J.O., O.S., W.W. and M.S. collected data in the field, D.R.G. performed research, A.N. performed bioinformatics analyses, D.R.G. and A.N. analysed data, D.R.G. wrote the paper, J.O., O.S., W.W., M.S., C.R. and A.N. commented on the paper.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Gutleb, D.R., Ostner, J., Schülke, O. et al. Non-invasive genotyping with a massively parallel sequencing panel for the detection of SNPs in HPA-axis genes. Sci Rep 8, 15944 (2018). https://doi.org/10.1038/s41598-018-34223-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-34223-y
Keywords
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
