
Abstract
Since it is known that cancer cells exhibit a preference for increased glycine consumption, the respective glycine metabolizing enzymes are in focus of many research projects. However, no cancer associated studies are available for the Glycine Cleavage System Protein H (GCSH) to date. Our initial analysis revealed a GCSH-overexpression of the protein-coding transcript variant 1 (Tv1) in breast cancer cells and tissue. Furthermore, a shorter (391 bp) transcript variant (Tv*) was amplified with an increased expression in healthy breast cells and a decreased expression in breast cancer samples. The Tv1/Tv* transcript ratio is 1.0 in healthy cells on average, and between 5–10 in breast cancer cells. Thus, a GCSH-equilibrium at the transcript level is likely conceivable for optimal glycine degradation. A possible regulative role of Tv* was proven by Tv1-Tv*-RNA-binding and overexpression studies which consequently led to serious physiological alterations: decreased metabolic activity, release of the lactate dehydrogenase, increased extracellular acidification, and finally necrosis as a result of impaired plasma membranes. In contrast, Tv1-overexpression led to an additional increase in cellular vitality of the tumor cells, primarily due to the acceleration of the mitochondrial glycine decarboxylation activity. Ultimately, we provide the first evidence of a sensitive GCSH-antisense regulation which determines cancerous cell viability.
Similar content being viewed by others


Introduction
Glycine metabolism, particularly the glycine-to-serine conversion by the glycine decarboxylase (GLDC) and the three other proteins of the glycine cleavage system (GCS), in conjunction with serine hydroxymethyltransferase (SHMT), are functionally associated with one-carbon (C1)-metabolism for the generation of purines in nearly all organisms. With respect to cancer cell metabolism, recent studies revealed the glycine-to-serine conversion as another capability for energy generation and as a precursor for one-carbon metabolism in tumorigenic, highly proliferative cells1,2,3,4,5,6. Therefore, many cancer cells are not exclusively dependent on glycolysis (Warburg effect) for the supply with reducing equivalents. Rather, the glycolytic flux can be redirected from the intermediate product 3-phosphoglycerate to form serine, the precursor of glycine, cysteine and a variety of C1 compounds1,3,4. These novel investigations also proved that enzymes involved in the glycine-to-serine conversion are overexpressed in cancer cells to match the high demand of C1-bodies for proliferation purposes3. For example, the metabolic dependency on glycine is impressively illustrated by Jain et al.5, showing a preference for increased glycine consumption in 60 tumor cell lines (NCI-60 panel, CORE profiles of 219 metabolites), and Zhang et al.6 identified enzymes of the GCS as a central switching site for tumor initiating cells (TICs) in the non-small cell lung carcinoma. In this context, abnormal activation of the glycine decarboxylase correlates with a poor survival rate of lung cancer patients. In addition, glycine-dependent rapid cancer cell proliferation via upregulation of mitochondrial glycine biosynthesis is closely linked to the high mortality rate of breast cancer patients3,5. Factors such as lymph node status and tumor grade were congruent, and thus confirmed the conclusion for a poor prognosis. In summary, increased expression of mitochondrial GCS promotes the metabolism of glycine and thus tumorigenesis1,3,5,6. Interestingly, plant physiologists focusing on glycine metabolism in Arabidopsis thaliana (Arabidopsis), could proof that elevated GCSH protein (alternatively named H-protein) concentrations increase GCS activity7. Consequently, GCSH overexpression improved plant growth, which could likely be explained due to alleviated feedback inhibition of photosynthesis from photorespiratory intermediates8. However, up to now we can only hypothesize that the GCSH protein content might be involved in the proliferation behavior in plants as well as in human cells, and therefore should be studied in more detail. So far, no GCSH expression studies in correlation with tumor progression are available.
The GCS is a ubiquitously occurring multi-enzyme system consisting of four proteins: GLDC (EC 1.4.4.2, glycine decarboxylase, P-protein, 113 kDa), AMT (EC 2.1.2.10, aminomethyl transferase, T-protein, 44 kDa), DLD (EC 1.8.1.4, dihydrolipoyl dehydrogenase, L-protein, 112 kDa) and GCSH (glycine cleavage system protein H, H-protein, 19 kDa)9. This mitochondrial multi-enzyme system converts glycine, NAD+ and tetrahydrofolate (THF) to serine, NADH and N5,N10-methylene-THF, thereby releasing CO2 and ammonia10,11. The lipoamide-containing H-protein (GCSH) is an enzymatically inactive cofactor of the other three GCS enzymes and functions as oxidizing agent and shuttle of the methylamine group remaining after oxidative glycine decarboxylation from the GLDC to the AMT. Defects in the GCSH gene could be a cause of nonketotic hyperglycinemia in human beings12. The single human GCSH gene is located on chromosome 16 at q23.2 and composed of 5 exons spanning 13.5 kb. So far, two GCSH transcript variants are annotated: Tv1 encodes GCSH while Tv2 is a non-coding variant that lacks an alternate exon because of a frame shift13. Interestingly, the GCSH protein tends to dimerize and complex formation14. According to in silico data, GCSH expression is elevated in brain (e.g. cerebral cortex) and endocrine tissues (e.g. thyroid gland), as well as in kidney, liver, colon and sexually specific organs such as breast and prostate in human beings. In contrast, GCSH expression in cancer seems to be often very high in thyroid, lung, colon and breast cancer tissues (https://www.proteomicsdb.org; http://www.proteinatlas.org).
Here, we present the first expression analysis of the GCSH gene in human healthy and cancerous breast cells which directed us to the identification of a previously not described regulatory mechanism. This is an antisense mechanism that regulates cellular GCSH dimerization and possibly the flux via the glycine cleavage system by a newly identified transcript variant, namely Tv*. This evaluation could be of major interest since breast cancer has the highest incidence rate of all cancers in women worldwide15. Treatment strategies depend on the histological subtype and receptor expression status. Positive testing for estrogen, progesterone, and the human epidermal growth factor receptor-2 allows effective therapy with Tamoxifen, Anastrozole, or Trastuzumab16,17. No detectable expression of these three receptors is associated with poor prognosis and difficult treatment options. To include both, receptor-positive and -negative breast cancer cells, we used in this study two triple negative breast cancer cell lines (BT-20, MDA-MB-231) and one luminal receptor positive cell line MCF-7. MCF-10A and MCF-12A were used as healthy control breast cells18.
Results
GCSH protein is overexpressed in both, breast cancer tissue and breast cancer cell lines
To evaluate GCSH expression in vivo as well as in vitro, paraffin-embedded normal breast and breast cancer tissue (Fig. 1A,B) as well as three commercially available breast cancer cell lines (MCF-7, MDA-MB-231 and BT-20; Fig. 1C) in comparison with non-tumorigenic controls were immunolabeled with a monoclonal, full length GCSH-specific antibody. In general, non-tumorigenic, resting mammary tissue revealed a low GCSH expression. Only in metabolic active regions e.g. ducts, small amounts of GCSH protein (score 1+) could be verified. In contrast, the GCSH protein could be detected equally distributed in the whole mammary carcinoma tissue, with an expression score ranging from 2+ to 3+. In all samples, the GCSH protein is associated with mitochondria as visualized by a hematoxylin-eosin counterstaining. Some of the detected GCSH protein in the non-tumorigenic tissue appear to be also associated with the nucleus (Personal communication Prof. Erbersdobler, Department of Pathology, University of Rostock, Rostock, Germany). Similar results were obtained by GCSH-immunofluorescence labeling of breast cancer cell lines (Fig. 1C). A GCSH-overexpression could be confirmed for the breast cancer cell lines MCF-7 and BT-20. Interestingly, MDA-MB-231 showed low GCSH-expression, comparable with that of non-tumorigenic control cell lines (MCF-12A and MCF-10A). Association between GCSH and mitochondria was confirmed by co-localization studies, too.

GCSH expression analysis. (A) GCSH expression in vivo. Representative images of GCSH immunohistochemistry staining in normal and breast cancer tissue samples in comparison with high (liver) or low (thyroid) GCSH-expressing organs. Staining intensity grade is scored from 0 to 4+. Higher magnifications are shown in boxes in the right upper corner of every image. (B) Relative frequency of GCSH scores of IHC stained normal and breast cancer tissues. Higher GCSH scores and thus also higher GCSH expression occurs significantly more frequently in breast cancer. Normal breast tissue n = 9. Breast cancer tissue n = 14. (C) GCSH expression in vitro. GCSH immunofluorescence staining of two non-tumorigenic, epithelial breast cell lines (MCF-10A, MCF-12A) in comparison with three established breast cancer cell lines (MCF-7, MDA-MB-231, BT-20). A co-localization of GCSH and mitochondria was found. Notably, breast cancer tissue and all cancer cell lines revealed a strong GCSH overexpression compared to normal breast tissue and non-tumorigenic cell lines.
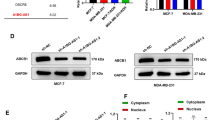
These expression results were verified by qRT-PCR and semi quantitative RT-PCR (Fig. 2A,B), as well as immunoblotting (Fig. 2C). Indeed, increased GCSH expression levels of the breast cancer cell lines MCF-7 and BT-20 were also calculated on transcript and protein levels, with MDA-MB-231 as an exception. A higher GCSH protein content could also be verified by immunoblotting, but on a higher molecular weight level (~60 kDa). As internal controls PCNA (proliferating nuclear antigen), ß-actin (intended as housekeeping protein), and AMT (aminomethyltransferase: a direct GCSH interacting protein, also names T-protein) were used (Fig. 2E). All three breast cancer cell lines displayed approximately 10-fold higher GCSH protein expression as well as significantly boosted PCNA contents and moderate lowered ß-actin levels. Again, MDA-MB-231 exceptionally showed a higher AMT-protein content. However, identical loading of soluble proteins was guaranteed by stain-free technology (Fig. 2D). Protein expression factors were determined densitometrically and normalized to the non-tumorigenic control cell line MCF-10A, which was arbitrarily set to 1. Given the existence of two GCSH transcript variants exist, primers flanking the full-length GCSH transcript were used to confirm qRT-PCR results by reverse transcriptase PCR (Fig. 2B). All cell lines expressed the full-length transcript variant Tv1 (522 bp) to a comparable extent as detected by qRT-PCR. Additionally, a second, shorter variant could be amplified (~350 bp), further designated as Tv*. Unexpectedly, Tv* was significantly downregulated in all cancer cell lines.

GCSH transcript and protein analysis. (A) qRT-PCR analysis using GCSH Tv1 specific primers showed an increase in GCSH Tv1 expression in all three breast cancer cell lines (red) as well as in the non-tumorigenic control cell line MCF-12A compared to the normal breast cells of MCF-10A (green). MCF-7 and BT-20 revealed the highest GCSH Tv1 transcript levels. Mean ± SD, n = 5, ***P < 0.001, **P < 0.01, *P < 0.05, significantly different compared to MCF-10A, unpaired t-test. (B) RT-PCR analysis using GCSH Tv1 full length primers confirmed the qRT-PCR results. A 2nd transcript variant (Tv*) with 391 bp in size was amplified and significantly lower expressed in all cancer cell lines. Tv1/Tv* ratios ranged from 5–10 for the cancer cells. (C) GCSH protein content in all 5 cell lines was visualized by western blotting. Native GCSH protein (19 kDa) content was increased for MCF-7 and BT-20, as analogous to the transcript analysis. However, further signals could be detected at ~58 kDa and ~65 kDa. The densitometrically calculated sum factor revealed an approximately 10-fold higher GCSH protein content in all breast cancer cell lines. (D) Stainfree imaging to guarantee identical loaded protein contents, used for the protein content normalization additionally. (E) Control Western blots of PCNA (proliferating nuclear antigen), ß-actin (intended as housekeeping protein) and AMT (aminomethyltransferase, direct GCSH interacting enzyme). Protein contents were calculated densitometrically and set in relation to MCF-10A protein levels.
Transcript variants Tv1 and Tv* display an antisense regulation mechanism
DNA sequencing and the usage of basic local alignment search tools (BLAST) annotated the transcript variant Tv* as the homo sapiens GCS H-protein (aminomethyl carrier) pseudogene 3 (GCSHP3), non-coding RNA (NCBI accession no.: NR_033248.1) with 100% nucleotide sequence identity to Tv1. Tv* sequencing revealed a transcript length of 391 bp (Fig. 3A). Nucleotide identity between Tv* and Tv1 was calculated to 97%. Obviously, Tv* lacks 30 bp of the N-terminal mitochondrial transit peptide region which makes mitochondrial localization unlikely. However, based on the altered transcriptional expression between non-tumorigenic and cancerous cells, a regulative function for Tv* would be possible. Speculating on an antisense regulating mechanism, we analyzed whether Tv1 and Tv* RNA sequences can bind each other. Using northern blotting analysis, a stable Tv1-Tv*-RNA-binding was observed, and, moreover, revealed that Tv* is present in antisense orientation in the breast cancer cell line MCF-7 (Fig. 3B).

Sequence alignment and RNA-binding study. (A) Nucleotide sequence alignment between the GCSH transcript variant 1 (Tv1, coding for the GCSH protein), the newly identified transcript variant (Tv*). Tv1 to Tv* is 97% identical. (B) Northern blotting demonstrated a positive antisense binding of the Tv* to isolated RNA of the cancer cell line MCF-7. (C) Model of Tv1-Tv* RNA antisense binding. Original figure ‘Transcrpition’ by Servier Medical Art (https://smart.servier.com/smart_image/transcription/) is licensed under a Creative Commons Attribution 3.0 Unported License. This modified figure is licensed under CC BY by Nadja Engel.
Transient Tv* elevation reduces Tv1 transcript and protein content
The assumed antisense regulation mechanism was analyzed by nanoparticle driven Tv*-overexpression in all three cancer cell lines and in both non-tumorigenic control cell lines (see SFig. 1). All further experiments were conducted with the breast cancer cell line MCF-7, presenting the most common breast cancer subtype (positive for estrogen and progesterone receptor expression). For transient Tv*-transfection purposes, silica and magnetic, red fluorescent nanoparticles were chosen. These nanoparticles mediated mitochondrial localization after cellular internalization, visualized by yellowish co-localization with green mitochondrial tracker (Fig. 4A). Stable binding of Tv*-DNA with nanoparticles was verified by DNA-agarose electrophoresis (Fig. 4B), and therefore its suitability for transfection is guaranteed. Hence, breast cancer cells were next transfected with GCSH-Tv*-loaded nanoparticles. A considerable increase of Tv* PCR products was detected by RT-PCR, with a simultan decrease in the amount of Tv1 PCR products (Fig. 4C). This result represents the first indication for an antisense-regulative mechanism. In parallel, GCSH protein expression of Tv*-transfected cells was analyzed by confocal laser-scanning microscopy (Fig. 4D). Mock transfected cells revealed a mitochondria-associated localization of red fluorescent nanoparticles. Also, GCSH protein distribution in mock transfected cells was not altered in comparison to the non-transfected MCF-7 cells. In contrast, Tv*-transfected MCF-7 cells showed an accumulation of red nanoparticles around the nucleus and a lowered GCSH protein content in general.

Nanoparticle-mediated transient Tv*-overexpression. (A) Co-localization (yellow) between mitochondria (green) and nanoparticles (red) was found, indicating a direct association of the nanoparticles with mitochondria. (B) Evidence of the stable binding between Tv* and nanoparticles. All of the 50 ng Tv*-DNA was completely bound to the nanoparticles which cannot pass the agarose gel. (C) RT-PCR of GCSH transcripts Tv1 and Tv* of non-transfected (n.t.), mock-transfected (C), Tv*-transfected or GAPDH-transfected MCF-7 cells. Results were similar with silica and magnetic nanoparticles: Tv*-overexpression decreased Tv1 expression, and Tv* was found as the most prominent band. In general, control transfection (nanoparticles, w/o DNA) did not alter the GCSH transcript profile. (D) Immunofluorescent visualization and fluorescence intensity histograms of the endogenous GCSH protein content using a monoclonal GCSH antibody (green), fluorescence of the nanoparticles (red) and cell nuclei (blue) in non-, control- and Tv*-transfected MCF-7 cells. Tv*-overexpressed cells and GCSH-siRNA knockdown cells. Notably, GCSH fluorescence intensity was obviously reduced in Tv* transfected cells and GCSH-siRNA knockdown cells.
Stable transfection with Tv* decreases cell viability
Nanoparticle mediated Tv*-overexpression was used as a transient, primary test system, while physiological alterations can only be monitored with stably transfected cells. Thus, the GCSH transcript variant Tv1 as well as Tv* were cloned in frame with the GFP-ORF of the vector pCMV6-AC-GFP (SFig. 2) and transfected into the cells (Fig. 5A). As positive control, the GFP-vector was used, known to cause cytoplasmic green fluorescence in MCF-7 cells. The Tv*-GFP-construct revealed a minor cytoplasmic fluorescence, which indicates the absence of the full-length mitochondrial transit peptide. However, Tv1-GFP overexpressing cells showed the expected mitochondrial distribution (Fig. 5A, right). Notably, mitochondria showed a round, swollen shape in comparison with non-treated MCF-7 cells (Fig. 5A, left). Tv*-overexpression modulated the Tv1/Tv* ratio to a similar extent as transient particle-driven Tv*-expression, that reduced Tv1 and increased Tv* transcript (Fig. 5B, compare with Fig. 4C). In contrast, overexpression of Tv1-GFP did not significantly increase the Tv1 transcript content but induced formation of a third transcript variant (~300 bp). Simultaneous visualization of the endogenous GCSH protein (red) and the GCSH-GFP-constructs (green) confirmed low cytosolic expression of Tv* and high mitochondrial expression of Tv1 (Fig. 5C), too.

Stable overexpression of Tv1 and Tv*. (A) Plasmid mediated overexpression of Tv1 and Tv* in comparison with the GCSH-labeled, non-transfected (n.t) or GFP-transfected (C) MCF-7 cells. Notably, Tv*-overexpression mediated a weak, cytosolic expression whereas Tv1-overexpressors displayed a strong, mitochondrial GFP-signal. Those mitochondria are much bigger and located around the cell nucleus. (B) RT-PCR of GCSH transcripts Tv1 and Tv* of non- (n.t.), control- (C), Tv*- and Tv1-transfected MCF-7 cells. Tv*-overexpression revealed comparable results to nanoparticle mediated transfection: low Tv1 and high Tv* signals. Surprisingly, Tv1-overexpression did not increase the Tv1 content. Furthermore, no Tv* amplificat could be detected but instead a third ~300 bp transcript variant (Tv?) appeared. (C) Simultaneous visualization of Tv1- or Tv*-GFP-expression (green) and endogenous GCSH content (red) by confocal laser-scanning microscopy.
The physiological impact of Tv1 and Tv*-overexpression was examined in GFP-sorted MCF-7 cells. First, the impact on the general cell viability was checked (Fig. 6A). Whilst overexpression of Tv1 induced a significant increase (+30%), overexpression of Tv* mediated a decrease (−40%) in the metabolic viability in comparison with corresponding control treatments. This observation was verified by lactate dehydrogenase (LDH) release measurements, accounting for a first indication of plasma membrane permeabilization, a measure of necrosis or apoptosis induction (Fig. 6B). Tv1-transfected cells harbored a significantly reduced extracellular LDH activity while Tv* showed an upregulation of up to 400%. Metabolic real-time monitoring of viable MCF-7 breast cancer cells confirmed the rise of the extracellular acidification over time (Fig. 6C). 24–30 h after Tv*-transfection, tumor cells are no longer viable. In general, membrane impairment leads to leakage of intracellular molecules and enzymes like the abundant LDH. MCF-7 cells first lost their normal shape and subsequently died by damaged plasma membrane (Fig. 6D). By immunoblotting it was shown that Tv* reduces the multimerized GCSH bands and increases the monomeric GCSH band at 19 kDa. This result clearly proves that Tv* influences more the GCSH expression mainly by impairing multimerization events. Next, a conclusion can be drawn between the GCSH expression ratio of the multimerized upper bands and the monomeric 19 kDa bands. The lower ratio between those bands is the higher is the impact on apoptosis induction (cleaved caspase 7) and proliferation inhibition (PCNA-proliferating nuclear antigen). Further, immunoblotting showed that Tv1 overexpression, here fused with GFP-protein, also enhanced AMT and lowered the glycine N-methyltransferase protein expression (Fig. 6E). Expression of the mitochondrial serine hydroxymethyltransferase (mSHMT) was not altered at any cells. The cytosolic SHMT (cSHMT) was not expressed although co-expression was predicted for GLDC, GNMT and both SHMT variants (Fig. 6F). In contrast, Tv* overexpression induced caspase-7 cleavage and a slight reduction of PCNA (Fig. 6F). Loading control was achieved by stain free imaging prior and after blotting as well as detection of ß-actin. Finally, the impact on GCS activity was examined by determination of the glycine-serine ratio (Table 1). Notably, reduction of the GCSH content, achieved either by the transfection of Tv* or siRNAs, resulted in an approximately 20% lowered glycine levels. On the contrary, overexpression of GCSH variant Tv1 elevated the glycine levels up to 25%. The glycine-to-serine ratio revealed a shift in glycine-to-serine equilibrium after Tv* transfection, since the ratio between both amino acids decreased from 2.4 to 1.7 on average.

Physiological alterations of Tv1- and Tv*-overexpressors. (A) Metabolic cell viability was significantly increased in Tv1- and significantly reduced in Tv*-overexpressors in comparison to the non- and control-transfected cells; n = 8. (B) Cell membrane impairment was measured by the release of the cytosolic lactate dehydrogenase (LDH). The non-transfected control was set to 100%. Triton X-100 (T.X-100) incubation functioned as positive control. Tv1-overexpressors showed a slight decrease and Tv*-overexpressors a 4fold increase in extracellular LDH activity; n = 8. (C) The extracellular acidification was measured by real-time online monitoring of living MCF-7 cells over 24 h. Also, a 4fold increase was detected. (D) Brightfield images of non- (n.t.), control- (C), Tv*- and Tv1-transfected MCF-7 cells. Only Tv*-overexpressors showed a necrotic phenotype. (E) Immuno blots of GCSH, AMT, CSHMT, mSHMT, GNMT, and cleaved caspase-7, PCNA, ß-actin proteins in non-treated (n.t.), mock-treated (C), three individually transfected Tv1- and Tv*-overexpressor MCF-7 cell populations. (F) Stain-free image functioning as loading control. (G) Network of protein interactions created with, https://string-db.org shows evidence and data-based co-expression (black line) of GLDC, GNMT, cSHMT and mSHMT (framed in red). All data and download files in STRING are freely available under a ‘Creative Commons BY 4.0’ license.
Discussion
Given the fact that cancer cells are characterized by a high demand for C1 bodies to ensure continuous growth, enzymes of glycine metabolism, such as GLDC, are overexpressed in tumorigenic tissue6,19. In addition, a combination of low-GLDC/negative-HIF-1α expression has emerged as early prognostic factor for long-term survival in non-small lung cell cancer20. However, GLDC is only one out of four protein components of the GCS, which is ubiquitously essential for one-carbon metabolism9. Another protein of this system is the GCS H-protein (GCSH) which forms the core of the entire GCS, since it functionally connects all GCS enzymes via its lipoyl arm9. Under in vitro conditions, GCS activity is stimulated by external GCSH supply. Consequently, a ~50-fold stimulation of the glycine–bicarbonate exchange rate, relative to the rate measured in the absence of exogenous H-protein, could be reached7. In vivo this phenomenon was also verified in the model plant Arabidopsis. Transgenic GCSH overexpressors showed accelerated glycine turnover, yielding higher biomass production8. This fundamental finding raises the question, if higher GCSH protein amounts have an impact on the proliferation of breast cancer cells?
Our first result confirmed that the GCSH protein is overexpressed in breast cancer tissue and breast cancer cells. Unexpectedly this overexpression does not depend on the histological subtype. Both, luminal, hormone receptor positive, and, basal, triple negative breast cancer cells exhibited a 10-fold increased GCSH content (Fig. 2C), which is congruent with the reported higher GCS in various cancer types6. However, we here demonstrate for the first time that additional GCSH, which very likely results in higher overall GCS activity, strengthens the viability of the breast cancer cells and therefore forces tumorigenesis (Fig. 6). This finding is further supported by the fact that higher GSCH contents correlate with a poorer long-term and relapse free survival rate (SFig. 3).
Secondly, due to the identification of a second transcript variant (Tv*), which is downregulated in all tested breast cancer cell lines, we hypothesized that Tv* could be important for the regulation of the cellular GCSH content, perhaps via antisense repression (Fig. 2). This hypothesis was verified by RNA-binding studies and Tv*-overexpression. Antisense binding of Tv* to Tv1 was shown by northern blotting (Fig. 3). Physiologically, transfection with Tv* mediated a weak cytosolic expression that causes reduced cell viability and cell membrane impairment of MCF-7 cells. Excess of Tv* reduces the expression of Tv1 and induces necrosis of the breast cancer cells within 24–30 h, which was verified by caspase-7 cleavage, an executioner protein of apoptosis and necrosis (Fig. 6). This result implies that the Tv1/Tv* ratio decisively determines GCSH expression and subsequently the capacity for providing C1-units to several biosynthetic pathways. Furthermore, measurement of the cellular glycine and serine levels implicate that the GCSH content significantly affects the cellular glycine-serine equilibrium (Table 1). To our surprise Tv* overexpression did not significantly change glycine levels, and the small change is in the opposite direction as expected - one expects increased glycine levels due to decreased glycine degradation. On explanation is that the amount and/or activity of the glycine producing serine hydroxymethyltransferase (SHMT) is increased. The cytoplasmic and mitochondrial SHMTs catalyze the conversion of serine to glycine with the transfer of β-carbon from serine to tetrahydrofolate (THF) to form 5, 10-methylene-THF. SHMTs are directly interacting with proteins of the glycine decarboxylating system and thus with GCSH, too (Fig. 4G). By western blotting we showed that the amount of the mitochondrial SHMT (mSHMT) was not altered and the cytosolic SHMT protein was not expressed. Therefore, we checked several glycine degrading and converting enzymes. On protein level, we found that the glycine N-methyltransferase (GNMT) expression is repressed in Tv1 overexpressors. Thus, one could speculate that glycine is not further converted into N-methylglycine in Tv1 overexpressors and therefore accumulates intracellularly. Tv* cells overexpress GNMT protein, as a result, more glycine is metabolized and thus measured in reduced concentrations by LC-MS. However, the effects of such a shift in the Gly-Ser balance on cell vitality and its consequences must be determined in the future studies.
Nevertheless, Jain et al.5 proved that glycine metabolism plays an essential role for high cell proliferation rates. In addition, metabolic studies showed that fast-growing cells take up more glycine. On the other hand, non-proliferative active cells even released glycine into the medium. Radiolabeling of the ingested glycine has been shown to be used directly for purine synthesis. And here we are, glycine degradation by the glycine decarboxylation system is the primary C1 unit donor for purine synthesis.
Collectively, our results suggest that the cellular GCSH content is a key factor that determines the metabolic state and viability of cells, including tumorigenesis, and thus is a potent tumor marker for highly proliferative breast cancers. The cellular GCSH content itself is sensitively regulated by antisense binding of a 391 bp GCSH transcript variant Tv* to the coding transcript variant Tv1. We hypothesize that overexpressed Tv* directly binds to the coding transcript variant Tv1 whereby GCSH translation is restrained. No GCSH protein could be imported into the mitochondria whereby overall mitochondrial GCS activity is lowered over time leading to plasma membrane impairment and necrosis. Thus, it is likely to argue that the GCSH content could be a key player in tumorigenesis, and, therefore, a potent metabolic tumor marker.
Material and Methods
Cell lines and culture conditions
Experiments were performed as reported in our previous work21. Briefly, the non-tumorigenic mammary epithelial control cell lines MCF-10A (ATCC® CRL-10317™) and MCF-12A (ATCC® CRL-10782™) were grown in Dulbecco’s modified Eagle’s medium Ham’s F12 without phenol red (Invitrogen, Darmstadt, Germany) containing 10% horse serum (PAA Laboratories GmbH, Munich, Germany), the Mammary Epithelial Cell Growth Medium Supplement Pack (Promo Cell, Heidelberg, Germany) including 0.004 ml/ml bovine pituitary extract, 10 ng/ml epidermal growth factor (recombinant human), 5 mg/ml insulin (recombinant human), 0.5 mg/ml hydrocortisone and 1% gentamycin (Ratiopharm GmbH, Ulm, Germany). The breast cancer cell lines MCF-7 (ATCC® HTB-22™; estrogen and progesterone receptor positive), BT-20 (ATCC® HTB-19™) and MDA-MB-231 (ATCC® HTB-26™), both triple negative cancer cell lines were cultured in Dulbecco’s modified Eagle’s medium (Gibco, Paisly, UK) with 10% fetal bovine serum (PAN Biotech GmbH, Aidenbach, Germany) and 1% gentamicin (Ratiopharm GmbH, Ulm, Germany). Confluent cells were treated with 0.05% trypsin – 0.02% EDTA. The medium was changed every 2 days. Cell line authentication was performed by Seqlab Sequencing Laboratories (Göttingen, Germany).
Expression analysis
Total RNA was extracted with NucleoSpin® RNA II (Machery-Nagel, Düren, Germany) and cDNA was produced with the First Strand cDNA Synthesis Kit (Thermo Fisher Scientific Inc., Rockford, IL, USA). GCSH full length coding sequence was amplified with primer pair fw: 5′-ATGGCGCTGCGAGTGG-3’ and rv: 5′-TCACTCCTCAATAGATTTTATG-3′ using Dream TaqTM Green PCR Master Mix (Thermo Fisher Scientific Inc., Vilnius, Lithuania). qRT-PCR was performed with primer pair fw: 5′-GTCTCCCTGAAGTTGGGACA-3′ and rv: 5′-TCTGAAGGGTTACTCAGTGTCA-3′ using iTaqTM SYBR® Green Supermix (Bio-Rad Laboratories Inc., Hercules, USA) in the iQTM5 Multicolor Real-Time PCR Detection System (Bio-Rad, München, Germany). Relative gene expression was normalized to the GAPDH housekeeping gene: fw: 5′-CAAGGTCATCCATGACAACTTTG-3′ and rv: 5′-GTCCACCACCCTGTTGCTGTAG-3′. Sequencing of both transcript variants was done by Seqlab Sequencing Laboratories (Göttingen, Germany).
Protein expression analysis
Western blotting procedure was performed as already described22. For protein detection, primary antibodies anti-GSCH (#16726-1-AP; Proteintech Europe, Manchester, UK), anti-GAPDH (#D16H11; Cell Signaling, USA), anit-PCNA (#sc-56; Santa Cruz, USA); anti-β-Actin (#4970; Cell Signaling, USA), anti-AMT (#sc-99267; Santa Cruz, USA), anti-cSHMT (#sc-365203; Santa Cruz, USA), anti-mSHMT (#sc-390641; Santa Cruz, USA), anti-GNMT (#sc-166834; Santa Cruz, USA) were incubated overnight at 4 °C followed by labeling with a horseradish peroxidase (HPR)-conjugated secondary antibody (Dako, Glostrup, Denmark) for 1 h at room temperature. Band intensity was analyzed densitometrically with the Molecular Imager ChemiDoc XRS and Image Lab 3.0.1 software (Bio-Rad, München, Germany).
Immunofluorescence and immunohistochemistry (IHC)
Experiments were performed as reported in our previous work21. Briefly, the cells were grown on Menzel-Gläser coverslips (Thermo Fisher Scientific Inc., Schwerte, Germany) or in Ibidi dishes/ slides (Ibidi GmbH, Martinsried, Germany), fixed in 4% paraformaldehyde (Santa Cruz, Dallas, USA), permeabilized with 0.1% Triton X-100 (Santa Cruz, Dallas, USA) and labeled with anti-GCSH primary antibody (#16726-1-AP; Proteintech Europe, Manchester, UK) and Alexa Fluor 488 dye/Alexa Fluor 594 dye secondary antibody (Thermo Fisher Scientific Inc., USA). Mitochondria were labeled with MitoTracker® RedCMXRos or Green FM (Life Technologies Europe BV, Bleiswijk, Netherlands), counter-stained with Hoechst (PanReacAppliChem, Darmstadt, Germany) and investigated with a confocal laser-scanning microscope (LSM780, Carl Zeiss, Jena Germany). Notably, images were taken at identical device settings to guarantee comparable results. The expression intensity and distribution of GCSH protein was determined in healthy (GTX24324, GeneTex, Irvine, USA) and tumorigenic (GTX24701, GeneTex, Irvine, USA) human breast tissue. Murine liver and human thyroid tissue served as controls. Tissues were formalin-fixed, paraffin-embedded, cut, labeled with a GCSH primary antibody (#16726-1-AP; Proteintech Europe, Manchester, UK) in a 2% BSA solution followed by a secondary antibody−labeled, polymer−horseradish peroxidase (HRP) (Dako Envision+ Kit; Dako, Glostrup, Denmark). AEC (3-amino-9-ethylcarbazol) served as chromogenic agent. All sections were counterstained with Mayer’s hemalum solution (Merck KGaA, Darmstadt, Germany). Each section was digitalized by using an AxioImager.M2 equipped with an integrated XY-scanning device and a MRc camera (all from Carl Zeiss Microscopy GmbH, Göttingen, Germany). Statistical evaluation of in vivo GCSH protein expression was performed by semi quantitative scoring (0 = no, 1+ = low, 2+ = medium, 3+ = strong and 4 + = very strong GCSH expression).
GCSH overexpression plasmids and GCSH-siRNAs
A GCSH-cDNA-GFP-tagged clone (transcript variant 1; NCBI accession no: NM_004483) was purchased from OriGene (#RG217886, Rocville, USA). Transcript variant Tv* of MCF-12A was cloned in frame into the same vector by using the restriction sites SgfI and MluI. GCSH full length coding sequence for the GCSH cloning vector was amplified with primer pair fw: 5′-AAGCGATCGCCATGGCGCTGCGAGTG-3′ and rv: 5-AAACGCGTCTCCTCAATAGATTTTATG-3′ using Universe Hot Start High-Fidelity DNA polymerase Mastermix (Biotool, Munich, Germany) and MyCyclerTM thermal cycler (Bio-Rad, München, Germany) to visualize full length of GCSH Tv1 and GCSH Tv*. The GCSH-Tv* fragment and GCSH-Tv1-pCMV6-AC-GFP vector were cut on SgfI and MluI restriction sites (Thermo Fisher Scientific Inc., Waltham, USA). Positive transformed DH5α E.coli cells were selected by restriction and sequencing (Seqlab Sequencing Laboratories, Göttingen, Germany). 3 × 106 cells of breast cancer cell lines MCF7, BT20, MDA-MB-231 as well as control cell lines MCF10-A and MCF12-A were transfected with 1 μg/ml GCSH (Tv1 or Tv*) plasmid DNA using the CLB-Transfection System (Amaxa Nucleofection 6097129; Lonza, Cologne, Germany). As positive control the Pmax GFPTM vector (Lonza, Cologne, Germany) was used. Transfection efficiency was controlled with a confocal laser-scanning microscope. To generate stable transfected cells, GFP negative cells were eliminated by fluorescence-based cell sorting (BD FACSAria, Heidelberg, Germany). 3 unique 27mer GCSH-siRNAs (GCSH Human siRNA Oligo Duplex (Locus ID 2653) were purchased from Origene (#SR301770, Rocville, USA) and were used according the manufacture’s protocol.
Nanoparticle mediated transfection
Sicastar®-redF and BNF-Starch-redF (micromod Partikeltechnologie GmbH, Rostock, Germany) are amorphous silica nanoparticles and magnetic bionized nanoferrite nanoparticles, respectively, both 100 nm in size, precoated with amino groups (NH2), and labeled with a red fluorophore (Sicastar: ex. λ = 569 nm, BNF: ex. λ = 552 nm). The cell-internalization of magnetic BNF nanoparticles was stimulated by a conventional permanent magnet. To prevent nanoparticle aggregation, all nanoparticle associated experiments were performed in serum-free medium Dulbecco’s modified Eagle’s medium with 1% gentamicin23,24. 50 µg/ml nanoparticle working solutions were prepared at room temperature, protected from light, and coated with 4.5 µg/ml of a poly-D-lysine solution (Sigma-Aldrich, St. Louis, USA) to ensure DNA binding. Nanoparticles were incubated with 50 ng/ml GCSH (Tv1 or Tv*) cDNA for 1 h. Cells were transfected with labeled nanoparticles for 4 h, washed twice with serum-free medium and cultivated up to 24 h in cell culture medium. Following controls were used: 1. Non-transfection control (w/o DNA, w/o nanoparticles); 2. mock control (nanoparticles, w/o cDNA), and 3. GAPDH-control (GAPDH-cDNA, nanoparticles). Transfection efficiency, nanoparticle internalization, and transient expression of GCSH variants were determined by confocal laser-scanning microscopy and flow cytometry.
Cell viability and cytotoxicity
Cell viability was calculated by colorimetric measurements of metabolic activity with the CellTiter 96®AQueous One Solution Cell Proliferation Assay (Promega Corp., Madison, USA)25. Lactate dehydrogenase release was calculated with the Cytotoxicity Detection Kit (LDH) according to the manufacturer´s instructions (Roche Diagnostics GmbH, Mannheim, Germany)26,27. At least, eight replicates with corrected background absorbance were conducted.
Live-cell metabolic monitoring
A real-time live-cell monitoring of three metabolic parameters (impedance, mitochondrial respiration and extracellular acidification) was performed with the Bionas Discovery™ 2500 system (Bionas GmbH, Rostock, Germany) as described previously28. Cells were applied to the Bionas DiscoveryTM SC1000 measurement chips 24 h before measurement in a cell density of 200,000 cells/sensor chip in 450 μl medium. The software CSv1.47 Controlling was used for the raw data acquisition and further analyzed by Bionas15002 Data analyzerV1.07. Controls were normalized and set to 100%.
DNA binding studies
The northern blotting technique was already described29. DIG-labeled GCSH-Tv* and -Tv1 RNA-probes were synthesized by in vitro transcription with the DIG RNA Labeling Kit (SP6/T7) (Roche Diagnostics, Mannheim, Germany) using T7 RNA polymerase GCSH-Tv* and -Tv1 plasmids were linearized by the restriction enzyme MluI (Thermo Fisher Scientific Inc., Waltham, USA). RNA probes (1.5 µg) and 0.5–10 kb RNA Ladder (Life Technologies Europe, Bleiswijk, Netherlands) were denatured for 5 min at 65 °C. After 1% agarose gel electrophoresis with 6.5% formaldehyde solution (Sigma-Aldrich, Munich, Germany) blotting on positively charged nylon membranes (Boehringer Mannheim, Mannheim, Germany) was performed overnight. Thereafter, RNA probes and positive controls (100 ng/µl drop on membrane) were fixed by UV irradiation for 5 min. The prehybridization was carried out in DIG Easy Hyb (Roche Diagnostics GmbH, Mannheim, Germany) at 60 °C for 30 min. 25 ng/ml DIG-labeled RNA probes were denatured at 95 °C and hybridized overnight at 50 °C in DIG Easy Hyb (Roche Diagnostics GmbH, Mannheim, Germany). After 5 min washing in (2×) Saline-sodium-citrate buffer containing 0.1% SDS and 15 min in 0.1x Saline-sodium-citrate buffer containing 0.1% Sodium-dodecyl-sulfate at 55 °C, membranes were blocked (2% FCS, 0.3% Triton, 150 mM NaCl, 0.1 M Tris, pH 7.5). For RNA detection anti-Digoxigenin-AP Fab fragments (Roche Diagnostics GmbH, Mannheim, Germany) were applied in the dilution of 1:5000 and incubated for 1 h at room temperature. The RNA-RNA binding signals were visualized with the ELF®97 Endogenous Phosphatase Detection Kit (Invitrogen, Eugene, USA).
Quantification of glycine and serine
For the extraction of glycine and serine we followed the protocol of Lisec et al.30. Briefly, 24 h after transfection 1*106 MCF-7 cells were washed three times with ice-cold 0.9% NaCl and were extracted by lysing cells in 500 µl ice-cold methanol/acetonitrile/H2O (50:30:20). Additional 500 μl extraction buffer were used to remove all cell remnants. Samples were shaken at 4 °C for 10 min, then centrifuged for 15 min at 16,000 g, and the supernatant was collected and analyzed by LC-MS. The final aqueous supernatant was dried by lyophilization. The dry extracts were dissolved in 200 µl of A. dest. (ROTISOLV® LC-MS-grade, Roth, Germany) and filtered through 0.2 µm filters (Omnifix®-F, Braun, Germany). The cleared supernatants were analyzed using the high-performance liquid chromatograph mass spectrometer LCMS-8050 system (Shimadzu, Japan) and the incorporated LC-MS/MS method package for primary metabolites (version 2, Shimadzu, Japan)31. In brief, 1 µl of each extract was separated on a pentafluorophenylpropyl (PFPP) column (Supelco Discovery HS FS, 3 µm, 150 × 2.1 mm) with a mobile phase containing 0.1% formic acid. The compounds were eluted at 0.25 ml min−1 using the following gradient: 1 min 0.1% formic acid, 95% A. dest., 5% acetonitrile, within 15 min linear gradient to 0.1% formic acid, 5% A. dest., 95% acetonitrile, 10 min 0.1% formic acid, 5% A. dest., 95% acetonitrile. Aliquots were continuously injected in the MS/MS part and ionized via electrospray ionization (ESI). The compounds were identified and quantified using the multiple reaction monitoring (MRM) values given in the LC-MS/MS method package and the LabSolutions software package (Shimadzu, Japan). Authentic glycine and serine standard substances (Merck, Germany) at varying concentrations were used for calibration.
Statistical analysis
All experiments were replicated at least three times with individually passaged cells, and data sets were expressed as means ± standard deviations (SD). Statistical significance was determined by the unpaired student’s t-test or one-way ANOVA (***P < 0.001; **P < 0.01; *P < 0.05) using the software Graphpad Prism Version 5 (http://www.graphpad.com/scientific-software/prism/).
References
Amelio, I., Cutruzzolá, F., Antonov, A., Agostini, M. & Melino, G. Serine and glycine metabolism in cancer. Trends in biochemical sciences 39, 191–198 (2014).
Go, M. K., Zhang, W. C., Lim, B. & Yew, W. S. Glycine decarboxylase is an unusual amino acid decarboxylase involved in tumorigenesis. Biochemistry 53, 947–956 (2014).
Tedeschi, P. M. et al. Contribution of serine, folate and glycine metabolism to the ATP, NADPH and purine requirements of cancer cells. Cell death & disease 4, e877 (2013).
Locasale, J. W. Serine, glycine and one-carbon units: cancer metabolism in full circle. Nature reviews. Cancer 13, 572–583 (2013).
Jain, M. et al. Metabolite profiling identifies a key role for glycine in rapid cancer cell proliferation. Science (New York, N.Y.) 336, 1040–1044 (2012).
Zhang, W. C. et al. Glycine decarboxylase activity drives non-small cell lung cancer tumor-initiating cells and tumorigenesis. Cell 148, 259–272 (2012).
Hasse, D., Mikkat, S., Hagemann, M. & Bauwe, H. Alternative splicing produces an H-protein with better substrate properties for the P-protein of glycine decarboxylase. The FEBS journal 276, 6985–6991 (2009).
Timm, S. et al. Glycine decarboxylase controls photosynthesis and plant growth. FEBS letters 586, 3692–3697 (2012).
Kikuchi, G., Motokawa, Y., Yoshida, T. & Hiraga, K. Glycine cleavage system: reaction mechanism, physiological significance, and hyperglycinemia. Proceedings of the Japan Academy. Series B, Physical and Biological Sciences 84, 246–263 (2008).
Douce, R., Bourguignon, J., Neuburger, M. & Rébeillé, F. The glycine decarboxylase system: a fascinating complex. Trends in plant science 6, 167–176 (2001).
Oliver, D. J., Neuburger, M., Bourguignon, J. & Douce, R. Interaction between the Component Enzymes of the Glycine Decarboxylase Multienzyme Complex 1. Plant Physiology 94, 833–839 (1990).
Kure, S. et al. Chromosomal localization, structure, single-nucleotide polymorphisms, and expression of the human H-protein gene of the glycine cleavage system (GCSH), a candidate gene for nonketotic hyperglycinemia. Journal of human genetics 46, 378–384 (2001).
Zay, A., Choy, F. Y. M., Patrick, C. & Sinclair, G. Glycine cleavage enzyme complex: molecular cloning and expression of the H-protein cDNA from cultured human skin fibroblasts. Biochemistry and cell biology = Biochimie et biologie cellulaire 89, 299–307 (2011).
Okamura-Ikeda, K., Fujiwara, K. & Motokawa, Y. Purification and characterization of chicken liver T-protein, a component of the glycine cleavage system. The Journal of biological chemistry 257, 135–139 (1982).
Ferlay, J. et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. International journal of cancer 136, E359–86 (2015).
Maxmen, A. The hard facts. Nature 485, S50–1 (2012).
den Hollander, P., Savage, M. I. & Brown, P. H. Targeted therapy for breast cancer prevention. Frontiers in oncology 3, 250 (2013).
Neve, R. M. et al. A collection of breast cancer cell lines for the study of functionally distinct cancer subtypes. Cancer cell 10, 515–527 (2006).
Sun, W. Y., Kim, H. M., Jung, W.-H. & Koo, J. S. Expression of serine/glycine metabolism-related proteins is different according to the thyroid cancer subtype. Journal of translational medicine 14, 168 (2016).
Berezowska, S. et al. Glycine decarboxylase and HIF-1α expression are negative prognostic factors in primary resected early-stage non-small cell lung cancer. Virchows Archiv: an international journal of pathology 470, 323–330 (2017).
Engel, N. et al. First evidence of SGPL1 expression in the cell membrane silencing the extracellular S1P siren in mammary epithelial cells. PloS one 13, e0196854 (2018).
Engel, N. et al. Synergistic Action of Genistein and Calcitriol in Immature Osteosarcoma MG-63 Cells by SGPL1 Up-Regulation. PloS one 12, e0169742 (2017).
Kasper, J. et al. Flotillin-involved uptake of silica nanoparticles and responses of an alveolar-capillary barrier in vitro. European journal of pharmaceutics and biopharmaceutics: official journal of Arbeitsgemeinschaft fur Pharmazeutische Verfahrenstechnik e.V 84, 275–287 (2013).
Wiogo, H. T. R. et al. Insight into serum protein interactions with functionalized magnetic nanoparticles in biological media. Langmuir: the ACS journal of surfaces and colloids 28, 4346–4356 (2012).
Engel, N. et al. Pro-apoptotic and anti-adhesive effects of four African plant extracts on the breast cancer cell line MCF-7. BMC complementary and alternative medicine 14, 334 (2014).
Riss T. L et al. Cell Viability Assays. Eli Lilly & Company and the National Center for Advancing Translational Sciences (2013 May 1 [Updated 2016 Jul 1].).
Smith, S. M., Wunder, M. B., Norris, D. A. & Shellman, Y. G. A simple protocol for using a LDH-based cytotoxicity assay to assess the effects of death and growth inhibition at the same time. PloS one 6, e26908 (2011).
Engel, N. et al. Antitumor evaluation of two selected Pakistani plant extracts on human bone and breast cancer cell lines. BMC complementary and alternative medicine 16, 244 (2016).
Gandrillon, O., Solari, F., Legrand, C., Jurdic, P. & Samarut, J. A rapid and convenient method to prepare DIG-labelled RNA probes for use in non-radioactive in situ hybridization. Molecular and cellular probes 10, 51–55 (1996).
Lisec, J., Schauer, N., Kopka, J., Willmitzer, L. & Fernie, A. R. Gas chromatography mass spectrometry-based metabolite profiling in plants. Nature protocols 1, 387–396 (2006).
SHIMADZU. LC-MS/MS method package for primary metabolites version 2., P/N 225-24862-92. Available at, https://www.shimadzu.eu.com/lcmsms-method-package-primary-metabolites.
Acknowledgements
We wish to thank Petra Seidel (Dept. of Cell Biology, Rostock University Medical Center) and Daniel Wolter (Dept. of Oral and Maxillofacial Surgery, Facial Plastic Surgery, Rostock University Medical Center) for technical assistance and Dr. Robby Engelmann (Institute of Immunology, Rostock University Medical Center) for cell sorting. We thank Prof. Dr. Erbersdobler (Institute for Pathology, Rostock University Medical Center) for scoring of the IHC staining’s, Dr. Alexander Rebl (Leibnitz Institue for Farm Animal Biology FBN, Institute for Genome Biology) for providing of the DIG Easy Hyb Solution, and Christoph Prohaska (Department of Genetics) for assistance in the planning of the Northern blots. This work was financially supported by the Federal Ministry of Education and Research Germany (Women Professors Program). The LC-MS/MS equipment was purchased through a grant of the HBFG program (GZ: INST 264/125-1 FUGG).
Author information
Authors and Affiliations
Contributions
N.E. conceived the project. A.A., P.M., B.N., A.K., S.T. and N.E. performed the research and analyzed the data. A.A. drafted the article. N.E., H.B., S.T., and G.S. wrote the article, with additions and revisions from all authors. All authors have read and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Adamus, A., Müller, P., Nissen, B. et al. GCSH antisense regulation determines breast cancer cells’ viability. Sci Rep 8, 15399 (2018). https://doi.org/10.1038/s41598-018-33677-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-33677-4
Keywords
This article is cited by
-
Cuproptosis-related gene subtypes predict prognosis in patients with head and neck squamous cell carcinoma
Journal of Otolaryngology - Head & Neck Surgery (2023)
-
Comprehensive analysis of cuproptosis-related genes in prognosis, tumor microenvironment infiltration, and immunotherapy response in gastric cancer
Journal of Cancer Research and Clinical Oncology (2023)
-
System analysis based on the cuproptosis-related genes identifies LIPT1 as a novel therapy target for liver hepatocellular carcinoma
Journal of Translational Medicine (2022)
-
Vincetoxicum arnottianum modulates motility features and metastatic marker expression in pediatric rhabdomyosarcoma by stabilizing the actin cytoskeleton
BMC Complementary Medicine and Therapies (2021)
-
SGPL1321 mutation: one main trigger for invasiveness of pediatric alveolar rhabdomyosarcoma
Cancer Gene Therapy (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
