
Abstract
We studied the core bacterial communities of 19 sponge species from Nha Trang Bay (Central Vietnam), with particular emphasis on the contribution of planktonic seawater bacteria to the sponge core microbiomes. To ensure consistent sponge-microbe associations and accurate identification of planktonic bacteria transmitted from seawater, we were very restrictive with the definition of the sponge core microbiomes (present in all the replicates), and with the identification of valid biological 16S rRNA gene sequences (100% sequence identity) that belonged to potentially different bacterial taxa. We found a high overlap (>50% relative abundance) between the sponge species core microbiome and the seawater bacterial core in ca. a half of the studied species, including representatives of both, HMA and LMA sponges. From our restrictive analysis, we point to horizontal transmission as a relevant way of symbiont acquisition in sponges. Some species-specific recognition mechanisms may act in sponges to enrich specific seawater bacteria in their tissues. These mechanisms would allow the maintenance of bacterial communities in a species across geographical ranges. Moreover, besides contrasting preferences in bacteria selection from seawater, divergent physiological traits may also account for the different microbiomes in species of HMA and LMA sponges.
Similar content being viewed by others

Introduction
The first step to study multi-microbial symbionts within animals is to focus on permanent symbionts by ruling out the background noise produced by transient microbes1. In other words, to concentrate on those bacteria, which have established tight associations with the host through several evolutionary time scales2, independently of potential, mutual benefits or costs for the partners involved3. In this context, the core microbiota concept was adopted to ascertain the consistent associations of a metaorganism4,5, but also allows to study the core metabolic functions provided by the host–microbe interaction to the system6. The core concept was first applied to differentiate host–microbe interactions of the mammalian gut and in plant root systems5,6. Further, it was extended to marine animals with the aim of understanding the consistent contributions of the microbial symbionts to the host ecology, success, or decay1.
In marine habitats, studies on coral and sponge microbiomes have proliferated in the last 10 years. For instance, the persistent microbial symbionts (core) of several coral species from a reef were identified7 through spatial and temporal scales, concluding that the complexity of the reef habitat, and the life coral history traits likely influence the coral core microbiomes. Thus, more in deep research to explore accurately the core microbiome of many invertebrates is needed to overcome the constraints associated to their complex habitats. Although there is some controversial in the literature about how the core microbiota should be defined1,8, the most reasonable definition always depends on the question approached9. For instance, more or less restrictive criteria (i.e. present from 7% to 100% of the replicates) have been used for marine invertebrates1,5,10,11.
Sponges are a diverse group of sessile filtering invertebrates that play important ecological roles in benthic marine ecosystems12,13. They harbour the highest diversity of microsymbionts among marine invertebrates14,15. A wide range of studies using massive sequencing methods have retrieved thousands of microbial 16S rRNA gene sequences for each targeted sponge species11, which stressed the difficulty to unveil the mechanisms underlying these associations due to the high diversity of the partners involved. Indeed, some of the 16S rRNA gene sequences belonged to transient bacteria captured from the environment by the sponges while filtering seawater, and do not represent permanent symbionts11. Consequently, indirect analytical methods have been implemented trying to split stable symbionts from transient planktonic bacteria in sponge-microbial systems. The core microbiota concept was adopted to focus on permanent bacteria in a sponge species11 or phylum10, notwithstanding the host geographical and ecological origins, or temporal scales16. The rationale underlying the core concept is that stable symbionts should represent tight biological associations and thus, they are expected to be present in most, or all, host individuals.
Substantial differences in the resulting diversity metrics related to the core definition applied (from 12% to 100% occurrence), have been reported recently9. Several studies have proposed that stable sponge microsymbionts should be present in the sponges but not (at least not in high abundance) in the surrounding seawater11. But, recording the presence of a sponge bacterium in seawater greatly depends on the abundance threshold used to include or exclude sequences from downstream analysis, the sequencing depth and the number of samples. Moreover, it has been proposed that stable symbionts are mainly inherited by the progeny from their parent sponges17,18,19,20. However, vertical and horizontal transmission of the same bacteria have been described21 and more recent investigations reported different microbiomes in adults and larvae of the same species, which highlights the role of seawater bacteria in the structural composition of sponge microbiomes17,22.
Differences in microbial diversity between High Microbial Abundance (HMA) and Low Microbial Abundance (LMA) sponges have been reported23,24. These differences have been related to contrasting structural and physiological traits of the two species groups25. HMA sponges have a denser mesohyle and a more complex aquiferous system, with smaller choanocyte chambers, than LMA sponges25. Also a partial trophic niche separation for HMA and LMA sponges has been proposed26. Comparisons of the core microbiomes of these two groups, which consisted in bacterial T-RFLPs that were present in all species replicates across seasons and study years, showed that HMA sponges had a larger number of core bacterial groups with a higher overlap with seawater than LMA hosts27.
In this study, we attempted to cast some light on the acquisition modes of microbial symbionts in sponges by i) unveiling the permanent microbiomes within a large number of sponge species, and ii) estimating the contribution of seawater bacteria to the sponge core microbiomes in representatives of both HMA and LMA sponges. To address these goals, we analysed the sponge microbiomes of the 19 most abundant sponge species inhabiting a small geographical area in Nha Trang Bay (Central Vietnam), as well as the bacterial assemblages of the surrounding seawater. We applied a restrictive approach to core community concept in terms of bacteria occurrence across species replicates and to OTU definition. Sequence identity thresholds <99% for the 16S rRNA V4 region have been proved to be inaccurate for bacterial species delimitation28, in particular for short reads obtained from Next Generation Sequencing (NGS). Instead, a 100% of sequence identity has been proposed to obtain true biologically informative sequences, which can underlie metabolic and ecological particularities28. To ensure that the 16S rRNA gene sequences recovered from the seawater samples were identical to those recovered from the sponges, we clustered OTUs (Operational Taxonomic Units) at 100% identity (Zero radius OTUS or ZOTUs28), which has only been recently applied in a couple of studies of sponge microbiomes29,30. Clustering sequences at 100% identity and restricting the core to microbes present in 100% of the analysed samples seem particularly relevant when trying to elucidate horizontal symbiont acquisition.
Results
Specificity of sponge bacterial communities
The main factor structuring the sponge microbiomes was the sponge species (Permanova: R2 0.56, p-value < 0.01), which means that replicates from the same species were more closely related to each other than to any other species. However, dispersion within replicates greatly varied depending on the sponge species (Permutest F: 6.9 p-value < 0.01). A dichotomy (Permanova: R2 0.11, p-value < 0.01) between the so-called HMA and LMA sponges regarding their bacterial composition was detected (Supplementary Fig. S1). Moreover, dispersion within the three HMA species (Aaptos suberitioides, Neofibularia hartmani and Suberea cf. laboutei) was much lower than the dispersion within the 16 LMA species (Permutest F: 60.8 p-value < 0.01, Supplementary Fig. S1).
Core communities and species specific ZOTUs
The ZOTU richness of the core communities (ZOTUs present in all replicates of the same sponge species) varied from 54 in Clathria reinwardti to 600 in Thrinacophora cf. raphidophora (Table 1). The number of species replicates influences the number of ZOTUs forming the core community, the more replicates taken into account, the lowest the number of core ZOTUs (RS = −0.83, p-value < 0.01, Supplementary Fig. S2A). However, ZOTU abundance of the sponge species core did not depend on the number of species replicates since no correlation was found between both variables (RS = −0.21, p-value > 0.05, Supplementary Fig. S2B
). This means that the abundant ZOTUs are the major contributors to the core of a species, and that the variable fraction is represented by the low abundance ZOTUs. Thus, we have considered the comparisons based on the relative abundances of the core ZOTUs.
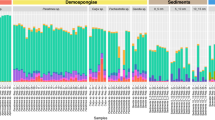
The core community represented more than 75% of the reads from the total microbiome in most species (Fig. 1). N. hartmani was the species with the largest core microbiome, representing up to 94% of relative abundance (Table 1) and Gellioides sp. had the smaller core microbiome representing <50% of relative abundance.

Mean relative abundances for the core (green) and the variable fraction (yellow) of the sponge microbiomes. Blue lines delimit the percentage of the sponge bacterial core shared with the seawater bacterial community. H = High Microbial Abundance sponge, L = Low Microbial Abundance sponge.
The number of species-specific ZOTUs (those present in all replicates of a species and absent from the core of any other study sponge species) varied from 0 (in C. reinwardti) to 195 (in T. cf. raphidodophora). However, the abundance of species–specific ZOTUs did not surpass 25% in any case, and most often were below 5% of relative abundance in the respective core communities. Gellioides sp. and S. cf. laboutei were the species with the highest relative abundances of species-specific ZOTUs (25% and 20%, respectively).
Similar values of core communities and species-specific bacteria were obtained for the dataset analysed with OTUs clustered at 97% sequence similarity (Supplementary Table S1).
Composition of the bacterial core communities
Specific associations between certain bacteria (phylum and class level) and HMA or LMA species were detected with the Indval analysis (Supplementary Fig. S3). These indicator bacteria are present in the core communities of the studied sponges (Fig. 2). The three HMA species presented an alike core microbial composition and abundances at phylum level with specific bacterial phyla overrepresented, compared to LMA species and seawater (SW). Phyla associated with LMA sponges also showed a similar core microbial composition but with contrasting abundances in the several species. Seawater samples had their own core bacterial composition with representatives of bacterial phyla shared with either HMA or LMA sponges. Mean Shannon diversity indices of the species core communities were significantly higher (Kruskal-Wallis <0.01) for HMA species (3.8 ± 0.24) than for LMA species (2.3 ± 0.61) (Supplementary Fig. S4).

Mean relative abundance of core microbial taxa at a phylum level (class for Proteobacteria) within each sponge species and seawater samples. Bacterial taxa with significant Indval values (Supplementary Fig. S3) associated to HMA species are marked in reddish colours and the ones associated to LMA species are marked in bluish colours.
Seawater (SW) ZOTUs
The relative abundances of the shared core SW ZOTUs (detailed in the Experimental procedures section) with the sponge core microbiomes varied between 9% and 99% depending on the sponge species (Fig. 3A). The sponges with the highest contribution of core SW ZOTUs to their core microbiome were A. paraviridis (99.5%), Gellioides sp. (93.2%) and C. reinwardti (90.4%), while Antho sp. (9.9%) and Pseudosuberites sp. (10.9%) showed the lowest overlap between the sponge and the SW core bacteria (Fig. 3A, Table 1). In some species (i.e.: A. suberitoides, C. reinwardti, Mycale sp.), rare SW ZOTUs (>0.01% of relative abundance) were abundant in the core of the sponge species. This is particularly visible in the example of C. reinwardti, which harboured ZOTUs that represented just a 5% of the SW core community and 90% of the sponge core. The opposite occurred in other sponge species such as Pseudosuberites sp. and T. cf. raphidophora, which had highly abundant (>1% of relative abundance) SW ZOTUs poorly represented in their core.

Mean relative abundances of shared bacteria between the SW and the core microbiomes for each sponge species when comparisons were made with the (A) SW core (cosmopolitan bacteria) and (B) the abundant (>0.01%) SW bacteria. Bars represent percentages of relative abundances of the shared bacteria in both, the sponge (green colour) and the SW (blue colour). H = High Microbial Abundance sponge, L = Low Microbial Abundance sponge.
In addition, when the most abundant SW ZOTUs were considered (abundances higher than 0.01%), instead of the SW core ZOTUs, the relative abundances of bacteria shared with SW were drastically reduced in most of sponge core microbiomes, with most values below 40% (Fig. 3B). This reduction was notably relevant in some species, such as Gellioides sp. N. hartmani, Mycale sp. A. suberitioides, C. reinwardti, and S. cf. labutei (Fig. 4).

Variation in relative abundance of SW ZOTUs in the sponge cores according to the method used for comparisons. Light green bars show ZOTUs shared with the SW core. Dark green bars represent ZOTUs shared with the abundant SW ZOTUs. Negative bars (yellow) represent the differences in shared ZOTUS between methodologies. H = High Microbial Abundance sponge, L = Low Microbial Abundance sponge.
Results of both comparisons for OTUs at 97% sequence identity are shown in Table S2. Differences between OTUs and ZOTUs were more remarkable when considering the abundant SW ZOTUs. For instance, in the case of C. reinwardti, the proportion of SW OTUs changed from 10% (for ZOTUs) to 73% (for OTUs). Also remarkable were the differences for A. paraviridis, Gellioides sp., and Phorbas sp.
The abundance and distribution of the SW core ZOTUs in the sponge core microbiomes is shown as a heatmap representation (Fig. 5). Two sponge clusters (A and B) were differentiated in the dendrogram. Cluster A contained sponge species that shared two highly abundant SW ZOTUs (ZOTU1, ZOTU2 at mean abundances of ~10%), and some ZOTUs (10, 37) at abundances higher than 1%. Cluster B comprised species harbouring different SW bacteria at contrasting abundances. Two different ZOTUs of Candidatus Branchiomonas (Betaproteobacteria) accounted for more than 30% and 50% of the Mycale sp. and A. paraviridis core microbiomes, respectively. Similarly, ZOTUs belonging to several Endozoicomonas (Gammaproteobacteria) were present at different relative abundances in the four species with the highest proportion of SW ZOTUs (Gellioides sp, A. paraviridis, Mycale sp. and C. reinwardti). Two main groups (C, D) were also differentiated according to the bacterial classes. The first group (C) corresponded to bacterial classes associated to LMA sponges, with high abundances of Alpha-, Beta-, and Gamma- Proteobacteria. The second group (D) showed bacterial classes associated to HMA sponges. ZOTUs belonging to PAUC34f, Chloroflexi, Acidobacteria, Actinobacteria, and Nitrospinae were almost exclusively found in the three HMA species (N. hartmani, S. cf. laboutei and A. suberitioides).

Heatmap showing core (cosmopolitan) SW ZOTUs with relative abundances higher than 1% across sponge species (listed on the right side). On the left, hierarchical clustering using Bray-Curtis dissimilarity matrix of the sponge species (colours on the vertical stripe represent the relative abundance of SW ZOTUs). On top, hierarchical clustering using Bray-Curtis dissimilarity matrix of the bacterial ZOTUs (colours on the horizontal stripe indicate ZOTU taxonomy at a Phylum level (class level for Proteobacteria). ZOTUs abundance is represented in the colour temperature bar on the left. Letters A, B, C, D indicate the different clusters. ZOTU numbers between parenthesis means: (1) ZOTUs shared by cluster A, (2) Candidatus Branchiomonas ZOTUs, (3) Endozoicomonas ZOTUs. H = High Microbial Abundance sponge, L = Low Microbial Abundance sponge.
Discussion
The sponge-associated bacteria were species specific for the 19 study species, as previously reported for sponges from other geographical areas10,17,25,31,32,33,34. Selection of specific bacteria and competition among the selected bacteria31 may converge to shape the species-specific patterns of sponge microbiomes, which used to be similar in geographically distant individuals of the same genus35.
However, differentiated patterns of bacterial composition are observed according to the affiliation to the HMA or LMA groups, as previously reported36,37. Indicator bacteria at class level for both groups are found, which match the indicator classes inferred from differential abundance analysis38. It has been reported that HMA sponges show higher microbiome similarity among species than LMA sponges27. Moreover, bacterial diversity is also claimed to be higher in HMA14,23,27,39,40. These aspects are confirmed in our study when considering the core members of both HMA and LMA sponges, although the number of representatives of each group is unbalanced.
It has been assumed40,41, but also questioned24,25, that LMA sponges contain mainly transient seawater bacteria. Representatives of both HMA and LMA study sponges contained high percentages of SW core bacteria (>50% of core relative abundance). However, species of each group acquire different SW bacteria that are indicators of either HMA or LMA sponges (Fig. 2 and Supplementary Fig. S3), suggesting contrasting bacteria selection mechanisms in each group. Differences might also be enhanced by particular traits of the respective physiology of the two sponge types42.
To define species core and species-specific bacteria, we clustered sequences at 100% identity (ZOTUs). Only recently, the sponge microbiomes have been analysed at the ZOTU level29,30. By recording ZOTUs, we were able to identify closely related bacteria28, which may inhabit sponge and seawater biomes making comparisons more reliable.
Analysing the bacterial core at species level provides information about stable, purportedly fixed associations, which may be involved in sponge-bacteria interaction patterns. To define the species core, an appropriate percentage of bacteria occurrence across species replicates has to be selected depending on the study aims9. For example, 85% occurrence was used for a species with more than 47 replicates to conduct an interaction network analysis11. In our study, as we wanted to focus on the truly symbiotic bacterial community of each sponge species, we choose a restrictive approach to the species core by only considering bacteria that were present in all the replicates of each species (100% occurrence), disregarding whether they were present among the SW core or not. In this way we ensured that we were focusing on persistent symbionts rather than on transient microbes.
The size of the bacterial core, which represents the permanent part of the sponge microbiome, seems to be intrinsic of each sponge species16. A high stability of the microbiome across sponge replicates can be indicative of the strength of the sponge-bacteria associations, whereas the opposite would indicate the presence of facultative /transient bacteria11. Most of the associations among bacteria and the studied species appeared to be highly constant, thus, suggesting a strongly fixed relationship, although we are aware that this might depend on the number of replicates analysed.
The contribution of the species-specific ZOTUs to the core communities, in terms of relative abundance, was surprisingly low. Those values may be influenced by the number of sponge species taken into account in the study, the similarity between their microbiomes, and, more strongly, by the restrictions associated to the way species-specific OTUs are defined, whether being part of the species core community (our study) or not10,32. Schmitt et al.10 suggested that the species-specific bacteria would probably be vertically inherited. If this assumption is true, the percentage of vertically inherited symbionts in our sponges would be rather low, since we have found a few species-specific microbes and the majority of the sponge bacterial taxa are found in more than one species.
Both, sponge microbiomes and seawater communities are in close contact because of the filtering activity of sponges, which can result in occasional bacteria transfer from one source to another24. To avoid this potential contamination, we used the 100% occurrence core approach in both the sponges and seawater, because it is unlikely that a ZOTU contaminates all the replicates of the same source. Moreover, we took into account the differential abundances that a ZOTU can be present in both, the sponge and the seawater. In this way, to be conservative, we could only suspect of SW bacteria contamination in the cases in which abundant SW bacteria are in low abundance in a sponge for which only few replicates are available. These cases would merit further investigation.
Our results comparing the bacterial core of sponges and seawater showed that all the studied sponges contained SW bacteria, as previously reported for other sponge species24,32. However, the relative abundance of SW bacteria in the sponge microbiomes was species dependent, ranging from almost 100% in A. paraviridis to less than 10% in Antho sp.
The quantification of the relative abundance of SW bacteria in the sponge microbiomes is particularly relevant to address seasonal or geographical changes and species specific traits24, but also allows inference about bacterial transmission modes. Thus, we estimated the relative abundance of the SW- sponge shared bacteria in each biome core trying to differentiate SW microbes that may represent stable symbionts from transient contaminant bacteria. We postulated that enrichment of seawater bacteria in the sponge occurred when low abundance SW core bacteria were found as the main components of the sponge microbiomes (Fig. 3). This can be proposed for 11 out of 19 sponge species analysed and in particular, for C. reinwardti, A. paraviridis, Mycale sp., Gellioides sp. N. hartmani, and A. suberitoides. No pattern related to the HMA and LMA dichotomy could be withdrawn here, as representatives of both groups showed enrichment of seawater bacteria in their microbiomes. The proportion of the sponge core bacteria shared with SW is reduced drastically in many species when comparisons are performed with the abundant SW bacteria, instead of with the SW core bacteria. Among these species, the three HMA species (N. hartmani, A. suberitioides, and S. cf. laboutei), reduced their relative abundance of SW ZOTUs from ~50%, ~70% and ~25%, respectively, to values below 5% (see Fig. 4). Reduction occurs in sponges that harbour low-abundance SW bacteria that would be ignored when only abundant SW bacteria are used for comparisons. Therefore, the study HMA sponges contain in their microbiomes SW bacteria that are at low-abundance in the water. For species that harbour both abundant and SW core bacteria in their microbiomes, similar percentages of SW bacteria were obtained with both methodologies. With this comparison, we like to point out how different approaches may influence the results on the overlap between the sponge and SW microbiomes. In particular, we emphasize the importance of the approach used for studies aiming to elucidate the dichotomy between HMA and LMA sponges.
We considered relevant the contribution of SW bacteria to the formation of the sponge microbiome when they were present across all species replicates. In contrast, some authors11 considered that abundant (>0.01%) SW OTUs likely represent environmental contaminants and should be removed from the sponge samples, independently of their abundance in the sponge. Conversely, we propose, that only highly abundant SW bacteria that are rare in the sponge-species core are potential candidates to represent SW bacteria contamination, especially when only few replicates are available. Two sponge species from our dataset (e.g. Pseudosuberites sp. and T. raphidophora) are examples of possible contamination.
Overall, we consider the comparisons using the SW core bacteria as a more accurate way to assess the true sponge-SW shared bacteria, as it considers bacteria that would be available across locations to be incorporated in the sponge microbiome. Our results support that “sponge-specific” bacteria are rather “sponge-enriched” bacterial clusters, and that seawater acts as a seed bank for sponge microbiomes, as suggested by Webster et al.17, Webster and Thomas43 and Moitinho-Silva et al.24. We detected widespread but rare44 SW bacteria forming part of sponge core microbiomes. Whether these taxa are metabolically active in the water column or represent dormant stages that reactivate after being incorporate to the sponge24 remains to be elucidated.
We find a high specificity of the associations between sponges and seawater bacteria. Each sponge species seems to incorporate different bacteria from the seawater in its microbiome. This suggests that some species-specific mechanisms have been fixed in the sponges to select some seawater bacteria and not others. Recognition mechanisms have been proposed to explain horizontal acquisition of microbes from the surrounding environment22. Taking into account the high percentages of seawater bacteria detected in some of our sponge species and the species-specificity of many of them, we propose that environmental acquisition would play a major role in the establishment of species-specific sponge microbiomes.
To summarize, sponge species is the main factor structuring microbiomes of the most common sponges from Nha Trang bay (Vietnam). By using a very restrictive approach of the “core species” concept and ZOTUs with 100% sequence identity for defining bacterial species, we proved that intra-species microbiome stability is the rule for most sponges. A high percentage of SW bacteria shaped the core microbiome in many study species. Our results point to horizontal transmission, as an ubiquitous mechanism of symbiont acquisition in sponges, while vertical transmission would represent a rather complementary acquisition way. Apparently, some highly specific recognition mechanisms may be acting in sponges to specifically enrich some SW bacteria in their tissues, and not others. Moreover, contrasting preferences in bacteria selection may account for differences in the microbiomes of HMA and LMA sponges and some physiological traits such as contrasting filtration rates might also contribute to enhance the differences. These mechanisms would allow the maintenance of stable bacterial communities disregarding environment conditions and geographical distance and merits to be confirmed by analysing in the same way as in the current study a larger number of sponge and water samples from different geographical regions.
Experimental Procedures
Sponge and seawater sampling and DNA extraction
Sponge samples were collected in April 2015 by SCUBA diving along 13 transects, 25 m long each, randomly placed between 3 and 9 m deep in three neighbouring locations ~2 km apart (i.e.: Dambay, Hun Mun and Nock Island) within Nha Trang Bay (central Vietnam). This quantitative sampling method allowed us to detect the most representative sponges in the study area but not to collect the same number of replicates for all the species. For instance, only three HMA sponges were found in the whole sampling but two of them were present at high abundances. Overall, we collected 203 sponge samples, from which we only considered for this study the ones that were found at least twice. Thereby, 98 samples belonging to 19 sponge species with between 2 and 15 replicates each (Table 1) were analysed.
Each sponge individual fitting within a transect was photographed and a piece of ca. 3 cm2 was collected in a 50 mL Falcon tube in seawater. Seawater was immediately replaced by 100% ethanol once on board. Back in the lab, the ethanol was replaced twice again with fresh absolute ethanol for a good sample preservation. DNA from those sponges was extracted following the protocol of DNeasy Blood &Tissue Kit (Qiagen).
Triplicate plankton samples were taken from the three sampling locations where the ecological transects were performed (i.e.; Dambay, Hun Mun and Nock Island). Two litres of water were collected and sequentially filtered throughout 5 μm, to remove undesired plankton components, and then throughout 0.22 μm polycarbonate membranes. The size fraction between 5 and 0.22 μm was processed for DNA extraction. Membranes were enzymatically digested with lysozyme, proteinase K and sodium dodecylsulfate and afterwards, DNA was extracted with phenol:chloroform-isoamyl alcohol (25:24:1, vol/vol/vol) and chloroform:isoamyl alchohol (24:1, vol/vol). Purification and concentration of the DNA was carried out with Amicon® Ultra 4 Centrifugal Filter Units – 100000 NMWL (Millipore). The extraction procedures used for sponge samples and SW were the most appropriated according to their respective preservation.
Sponge identification
We identified sponge species to the best possible taxonomic resolution by molecular markers and morphological features. Fragments of the nuclear genes encoding the 18S rRNA (~1700 bp) and 28S rRNA (~650 bp), as well as the cytochrome c oxidase subunit I (COI ~680 bp) were amplified and sequenced. Primers 1 F and 1795R45 were employed to amplify 18S rRNA, Por28S-830F and Por28S-1520R46 primers were used to amplify the D3-D5 partition of the 28S rRNA and LCO1490 and HCO219847 were used for COI. PCR amplifications were conducted in 50 μl reactions containing 1 ng of template genomic DNA, 5 μl of 10x PCR buffer (containing 1.5 mM MgCl2), 2 μl of dNTP mix (10 mM), 2 μl of bovine serum albumin, 1 μl pf each primer (10 mM) and 0.4 μl of Taq DNA polymerase (5 U μl−1). The temperature profile for the 18S rRNA was as follows: 94 °C/5 min; (94 °C/1 min, 50 °C/1 min, 72 °C/1 min) × 35 cycles; 72 °C/5 min; for 28S rRNA: 94 °C/5 min; (94 °C/30 s, 53 °C/30 s, 72 °C/30 s) × 30 cycles; 72 °C/5 min; and for COI: 94 °C/2 min; (94 °C/1 min, 45 °C/1 min, 72 °C/1 min) × 35 cycles; 72 °C/7 min. Purification and sequencing were carried out by an external service (Macrogen, Netherlands). The obtained sequences were manually edited in Geneious v 9.0.2 and blasted against NCBI databaste (https://blast.ncbi.nlm.nih.gov/Blast.cgi) to confirm the morphological identification of the sponges at the lowest taxonomic level possible.
Preparation of spicules and histological sections were made from specimens’ subsamples and observed under both, light and scanning electron microscopes. Morphological characters such as spicule types, shape, length, and width, as well as skeletal arrangement48 were used in combination with individual sequences and phylogenetic reconstructions to obtain, the most accurately possible, taxonomic identifications. Sponges were classified as HMA or LMA on the basis of their pertinence to genera already known to belong to any of these two groups, what was additionally confirmed by looking to the characteristics and structure of the sponge aquiferous system: small, relatively few (HMA) vs. large abundant choanocyte chambers (LMA), and mesohyle (dense vs. lax mesohyle, respectively)25.
16S rRNA gene amplification, sequencing and analysing
PCR and high-speed multiplexed SSU rRNA gene Illumina MiSeq sequencing (NGS) were carried out following the genomic core facilities and methods of the MrDNA Lab (Texas, USA) (http://www.mrdnalab.com/). The variable V4 region of the 16S rRNA gene (c.a. 250 nt) was amplified using the primers 564F (5′AYTGGGYDTAAAGNG3′) and 785R (5′TACNVGGGTATCTAATCC3)49. Raw rRNA gene sequences were processed using the UPARSE pipeline50. A quality check and de-replication were applied to our dataset. Denoising (error-correction) of amplicons was performed to identify all correct biological sequences following the UNOISE pipeline51. This algorithm removed chimeras, reads with sequencing errors, PhiX, and low complexity sequences due to Illumina artifacts, and generates ZOTUs (“zero-radius” OTUs) consisting of sequences of 100% identity. For comparison purposes, sequences were also clustered at 97% threshold (Supplementary information). For this analysis, reads were dereplicated and clustered into operational taxonomic units (OTUs) at cut-off 0.03% identity after chimera removal (UCHIME) and excluding the singletons.
Taxonomic assignment was done with SINA v1.2.1152 using SILVA 128 database. SINA uses Lowest Common Ancestor method (LCA). We configurated a “Min identity” of 0.7 and a maximum number of search results of 1 per sequence results in “best match” type. Sequences with low alignment quality (<75%) and sequences identified as mitochondria or chloroplasts were removed from the analysis. In order to minimize biased effects for differences in sampling effort, the original ZOTU table was rarefied (Supplementary Fig. S5) at a minimum reads threshold of 4100053.
Raw sequences are available in the SRA archive under the project number PRJNA453898.
Defining core and species-specific ZOTUs
We identified the ZOTUs that were present in all replicates to define the core microbiome of each sponge species. The ZOTUs that did not meet this requirement were assigned to the variable community. Moreover, we considered species-specific ZOTUs those belonging to a single core microbiome for a particular sponge species, compared to the remaining collected sponges.
Seawater (SW) ZOTUs
We combined two approaches to estimate the real contribution of the seawater (SW) bacteria to the sponge core microbiomes. First, we looked for bacteria in each sponge species that were already present in the core SW community. We considered the core community of the SW as the community formed by the ZOTUs present in all water replicates. With this approach, we attempted to identify bacteria that commonly inhabited in the SW and that its presence was not merely circumstantial. Therefore, they could represent a potential source for the formation of the sponge microbiome over time. In the second approach, we made the comparison with the most abundant bacteria of the SW. We identified the ZOTUs with relative abundances higher than 0.01% in average across all water samples. This threshold was chosen since it has been used previously to remove from the sponge microbiome samples the OTUs that were likely to represent environmental contaminants11. With this approach, we aimed to gain insight on the SW bacteria that are more likely detected in the sponge microbiomes just because of their high abundance in SW and may represent transient (environmental) contaminants11. On the other hand, we considered rare ZOTUs those with a relative abundance <0.01% and highly abundant ZOTUs those with relative abundance >1%32.
Statistical analyses
We carried out a distance-based multivariate analysis at ZOTU level of the microbial communities of the sponges and seawater samples using the vegan package54 in R. A cluster dendrogram was built using the Bray-Curtis dissimilarity distance matrix of samples to visualize patterns of bacterial community structure in sponges and seawater. We tested the effect of host identity (species) as well as the effect of the HMA/LMA identity, on the structure of microbial communities with non-parametric Permutational Analysis of Variance (PERMANOVA). PERMUTEST was applied to detect differences in the dispersion between groups. A bias correction55 for the unequal sample size of HMA and LMA groups was applied in the betadisper function of the vegan package54. P-values of PERMANOVA and PERMUTEST were calculated using 999 permutations and significance cut-off for p-values was 0.05.
The mean relative abundance of the bacterial phyla and classes was calculated for both, HMA and LMA groups. We applied an IndVal analysis using the labdsv package56 in R to detect potential associations of certain bacterial phyla and classes to any of these two groups. We fixed an IndVal threshold of 0.6 (p-value < 0.01) to consider a bacterial taxon strongly associated to (or Indicator of) HMA or LMA sponges.
Data Availability Statement
The raw prokaryotic sequences analysed during the current study are available in the SRA archive under the project number PRJNA453898 (https://www.ncbi.nlm.nih.gov/sra/?term = PRJNA453898). SSU and LSU Sponge sequences are available under the accession numbers MH731279 to MH731308. COI sponge sequences are available under the accession numbers MH784603 to MH784613. Moreover, the results of the dataset analysed with OTUs clustered at 97% sequence similarity are available in the Supplementary material of the current paper.
References
Ainsworth, T. D. et al. The coral core microbiome identifies rare bacterial taxa as ubiquitous endosymbionts. ISME J. 9, 2261–2274 (2015).
Douglas, A. E. Symbiosis as a general principle in eukaryotic evolution. Cold Spring Harb. Perspect. Biol. 6, 1–14 (2014).
Moran, N. A. & Sloan, D. B. The Hologenome Concept: Helpful or Hollow? PLoS Biol. 13, 1–10 (2015).
Bäckhed, F. et al. Defining a healthy human gut microbiome: Current concepts, future directions, and clinical applications. Cell Host Microbe 12, 611–622 (2012).
Shade, A. & Handelsman, J. Beyond the Venn diagram: The hunt for a core microbiome. Environ. Microbiol. 14, 4–12 (2012).
Shafquat, A., Joice, R., Simmons, S. L. & Huttenhower, C. Functional and phylogenetic assembly of microbial communities in the human microbiome. Trends Microbiol. 22, 261–266 (2014).
Hernandez-Agreda, A., Leggat, W., Bongaerts, P. & Ainsworth, T. D. The microbial signature provides insight into the mechanistic basis of coral success across reef habitats. MBio 7, 1–10 (2016).
Hernandez-Agreda, A., Gates, R. D. & Ainsworth, T. D. Defining the Core Microbiome in Corals’ Microbial Soup. Trends Microbiol. 25, 125–140 (2017).
Astudillo-García, C. et al. Evaluating the core microbiota in complex communities: A systematic investigation. Environ. Microbiol. 19, 1450–1462 (2017).
Schmitt, S. et al. Assessing the complex sponge microbiota: Core, variable and species-specific bacterial communities in marine sponges. ISME J. 6, 564–576 (2012).
Thomas, T. et al. Diversity, structure and convergent evolution of the global sponge microbiome. Nat. Commun. 7, 1–12 (2016).
Goeij, J. M. D. et al. Surviving in a Marine Desert: The Sponge Loop retains resources within coral reefs. Science (80-.). 342, 108–110 (2013).
Bell, J. J. The functional roles of marine sponges. Estuar. Coast. Shelf Sci. 79, 341–353 (2008).
Taylor, M. W., Radax, R., Steger, D. & Wagner, M. Sponge-Associated Microorganisms: Evolution, Ecology, and Biotechnological Potential. Microbiol. Mol. Biol. Rev. 71, 295–347 (2007).
Simister, R. L., Deines, P., Botté, E. S., Webster, N. S. & Taylor, M. W. Sponge-specific clusters revisited: A comprehensive phylogeny of sponge-associated microorganisms. Environ. Microbiol. 14, 517–524 (2012).
Bjork, J. R., O’Hara, R. B., Ribes, M., Coma, R. & Montoya, J. M. The dynamic core microbiome: Structure, dynamics and stability. bioRxiv 137885, https://doi.org/10.1101/137885 (2018).
Webster, N. S. et al. Deep sequencing reveals exceptional diversity and modes of transmission for bacterial sponge symbionts. Environ. Microbiol. 12, 2070–2082 (2010).
Schmitt, S., Weisz, J. B., Lindquist, N. & Hentschel, U. Vertical transmission of a phylogenetically complex microbial consortium in the viviparous sponge Ircinia felix. Appl. Environ. Microbiol. 73, 2067–2078 (2007).
Lee, O. O., Chiu, P. Y., Wong, Y. H., Pawlik, J. R. & Qian, P. Y. Evidence for vertical transmission of bacterial symbionts from adult to embryo in the Caribbean Sponge Svenzea zeai. Appl. Environ. Microbiol. 75, 6147–6156 (2009).
Enticknap, J. J., Kelly, M., Peraud, O. & Hill, R. T. Characterization of a culturable alphaproteobacterial symbiont common to many marine sponges and evidence for vertical transmission via sponge larvae. Appl. Environ. Microbiol. 72, 3724–3732 (2006).
Sipkema, D. et al. Similar sponge-associated bacteria can be acquired via both vertical and horizontal transmission. Environ. Microbiol. 17, 3807–3821 (2015).
Fieth, R. A., Gauthier, M.-E. A., Bayes, J., Green, K. M. & Degnan, S. M. Ontogenetic Changes in the Bacterial Symbiont Community of the Tropical Demosponge Amphimedon queenslandica: Metamorphosis Is a New Beginning. Front. Mar. Sci. 3, 1–20 (2016).
Hentschel, U. et al. Molecular Evidence for a Uniform Microbial Community in Sponges from Different Oceans Molecular Evidence for a Uniform Microbial Community in Sponges from Different Oceans. Appl. Environ. Microbiol. 68, 4431–4440 (2002).
Moitinho-Silva, L. et al. Specificity and transcriptional activity of microbiota associated with low and high microbial abundance sponges from the Red Sea. Mol. Ecol. 23, 1348–1363 (2014).
Blanquer, A., Uriz, M. J. & Galand, P. E. Removing environmental sources of variation to gain insight on symbionts vs. transient microbes in high and low microbial abundance sponges. Environ. Microbiol. 15, 3008–3019 (2013).
Morganti, T., Coma, R., Yahel, G. & Ribes, M. Trophic niche separation that facilitates co-existence of high and low microbial abundance sponges is revealed by in situ study of carbon and nitrogen fluxes. Limnol. Oceanogr. 62, 1963–1983 (2017).
Erwin, P. M., Coma, R., López-Sendino, P., Serrano, E. & Ribes, M. Stable symbionts across the HMA-LMA dichotomy: Low seasonal and interannual variation in sponge-associated bacteria from taxonomically diverse hosts. FEMS Microbiol. Ecol. 91, 1–11 (2015).
Edgar, R. C. Updating the 97% identity threshold for 16S ribosomal RNA OTUs. bioRxiv 192211, https://doi.org/10.1101/192211 (2017).
Moitinho-Silva, L. et al. The sponge microbiome project. Gigascience 6, 1–7 (2017).
Glasl, B., Smith, C. E., Bourne, D. G. & Webster, N. S. Exploring the diversity-stability paradigm using sponge microbial communities. Sci. Rep. 8, 1–9 (2018).
Easson, C. G. & Thacker, R. W. Phylogenetic signal in the community structure of host-specific microbiomes of tropical marine sponges. Front. Microbiol. 5, 1–11 (2014).
Reveillaud, J. et al. Host-specificity among abundant and rare taxa in the sponge microbiome. ISME J. 8, 1198–1209 (2014).
Lee, O. O. et al. Pyrosequencing reveals highly diverse and species-specific microbial communities in sponges from the Red Sea. ISME J. 5, 650–664 (2011).
De Mares, M. C. et al. Host specificity for bacterial, archaeal and fungal communities determined for high- and low-microbial abundance sponge species in two genera. Front. Microbiol. 8, 1–13 (2017).
Montalvo, N. F. & Hill, R. T. Sponge-associated bacteria are strictly maintained in two closely related but geographically distant sponge hosts. Appl. Environ. Microbiol. 77, 7207–7216 (2011).
Schmitt, S., Deines, P., Behnam, F., Wagner, M. & Taylor, M. W. Chloroflexi bacteria are more diverse, abundant, and similar in high than in low microbial abundance sponges. FEMS Microbiol. Ecol. 78, 497–510 (2011).
Giles, E. C. et al. Bacterial community profiles in low microbial abundance sponges. FEMS Microbiol. Ecol. 83, 232–241 (2013).
Moitinho-Silva, L. et al. Predicting the HMA-LMA status in marine sponges by machine learning. Front. Microbiol. 8, 1–14 (2017).
Schmitt, S., Angermeier, H., Schiller, R., Lindquist, N. & Hentschel, U. Molecular microbial diversity survey of sponge reproductive stages and mechanistic insights into vertical transmission of microbial symbionts. Appl. Environ. Microbiol. 74, 7694–7708 (2008).
Gerçe, B., Schwartz, T., Syldatk, C. & Hausmann, R. Differences Between Bacterial Communities Associated with the Surface or Tissue of Mediterranean Sponge Species. Microb. Ecol. 61, 769–782 (2011).
Weisz, J. B., Hentschel, U., Lindquist, N. & Martens, C. S. Linking abundance and diversity of sponge-associated microbial communities to metabolic differences in host sponges. Mar. Biol. 152, 475–483 (2007).
Ribes, M. et al. Functional convergence of microbes associated with temperate marine sponges. Environ. Microbiol. 14, 1224–1239 (2012).
Webster, N. S. & Thomas, T. The sponge hologenome. MBio 7, 1–14 (2016).
Taylor, M. W. et al. Sponge-specific bacteria are widespread (but rare) in diverse marine environments. ISME J. 7, 438–443 (2013).
Medlin, L., Elwood, H. J., Stickel, S. & Sogin, M. L. The characterization of enzymatically amplified eukaryotic 16S-like rRNA-coding regions. Gene 71, 491–499 (1988).
Morrow, C. C. et al. Congruence between nuclear and mitochondrial genes in Demospongiae: A new hypothesis for relationships within the G4 clade (Porifera: Demospongiae). Mol. Phylogenet. Evol. 62, 174–190 (2012).
Folmer, O., Black, M., Hoeh, W., Lutz, R. & Vrijenhoek, R. DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Mol. Mar. Biol. Biotechnol. 3, 294–299 (1994).
Hooper, J. N. A. & Van Soest, R. W. M. Order Poecilosclerida Topsent, 1928. in Systema Porifera: A Guide to the Classification of Sponges (eds Hooper, J. N. A., Van Soest, R. W. M. & Willenz, P.) 403–408, https://doi.org/10.1007/978-1-4615-0747-5_49 (Springer US, 2002).
Klindworth, A. et al. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 41, 1–11 (2013).
Edgar, R. C. UPARSE: Highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 10, 996–998 (2013).
Edgar, R. C. UNOISE2: improved error-correction for Illumina 16S and ITS amplicon sequencing. bioRxiv 081257, https://doi.org/10.1101/081257 (2016).
Pruesse, E., Peplies, J. & Glöckner, F. O. SINA: Accurate high-throughput multiple sequence alignment of ribosomal RNA genes. Bioinformatics 28, 1823–1829 (2012).
Caporaso, J. G. et al. QIIME allows analysis of high- throughput community sequencing data. Nat. Methods 7, 335–336 (2010).
Oksanen, J. et al. vegan: Community Ecology Package. R package version 2, 5–1 (2018).
Stier, A. C., Geange, S. W., Hanson, K. M. & Bolker, B. M. Predator density and timing of arrival affect reef fish community assembly. Ecology 94, 1057–1068 (2013).
Roberts, A. D. W. & Roberts, M. D. W. labdsv: Ordination and Multivariate Analysis for. Ecology. R package version 1, 8–0 (2016).
Acknowledgements
This research was partially funded by projects MarSymbiOmics (CTM2013-43287-P) and PopCOmics (CTM2017-88080, MINECO/AEI/FEDER/UE) from the Spanish Government. JC and EOC were supported by the project BRIDGES (CGL2015-69043-P). We thank Daniel Martin (CEAB, Blanes), Temir A. Britayev (RAS, Russia) and Elena S. Mekhova (RAS, Russia) for the help provided in the sponge sampling.
Author information
Authors and Affiliations
Contributions
M.T. and M.J.U. conceived the study. M.T. and J.C. performed the analyses. M.T. and M.J.U. wrote the manuscript. L.G., E.O.C. and J.C. commented on later versions of the manuscript.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Turon, M., Cáliz, J., Garate, L. et al. Showcasing the role of seawater in bacteria recruitment and microbiome stability in sponges. Sci Rep 8, 15201 (2018). https://doi.org/10.1038/s41598-018-33545-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-33545-1
Keywords
This article is cited by
-
Microbiome species diversity and seasonal stability of two temperate marine sponges Hymeniacidon perlevis and Suberites massa
Environmental Microbiome (2023)
-
Profiling Prokaryotic Communities and Aaptamines of Sponge Aaptos suberitoides from Tulamben, Bali
Marine Biotechnology (2023)
-
New Negombata species discovered: latrunculin mystery solved
Coral Reefs (2023)
-
Archaeal communities of low and high microbial abundance sponges inhabiting the remote western Indian Ocean island of Mayotte
Antonie van Leeuwenhoek (2021)
-
Microbial symbionts and ecological divergence of Caribbean sponges: A new perspective on an ancient association
The ISME Journal (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
