
Abstract
The capture spiral of web from N. clavipes spider consists of a single type of spidroin - the flagelliform silk protein, a natural material representing a combination of strength and high elasticity. Flagelliform spider silk is the most extensible silk fibre produced by orb weaver spiders and the structure of this remarkable material is still largely unknown. In the present study we used a proteomic approach to elucidate the complete sequence and the post-translational modifications of flagelliform silk proteins. The long sequence of flagelliform silk protein presents 45 hydroxylated proline residues, which may contribute to explain the mechanoelastic property of these fibres, since they are located in the GPGGX motif. The 3D-structure of the protein was modelled considering the three domains together, i.e., the N- and C-terminal non-repetitive domains, and the central repetitive domain. In the resulting molecular model there is a predominance of random structures in the solid fibres of the silk protein. The N-terminal domain is composed of three α-helices and the C-terminal domain is composed of one small helical section. Proteomic data reported herein may be relevant for the development of novel approaches for the synthetic or recombinant production of novel silk-based spider polymers.
Similar content being viewed by others


Introduction
Produced by flagelliform gland, the flagelliform silk protein (FSP) forms the silk fibres of the orbital web’s capturing spiral. This part of the silk is highly elastic and in conjunction with the silk properties of the frame and the ray of the orbital web, dissipates the energy of prey impacting on the web. The enormous resistance of these fibres is important for the capture and arrest of prey that is sometimes larger than spiders themselves1,2.
The FSP has the sequence repeat GPGGX, GGX and a spacer sequence composed of residues of charged amino acids and is highly conserved. According to Becker et al.3 the crosslinks between silk proteins may come from spacer sequences in the flagelliform silk protein. Adrianos et al.4 described a series of mechanical tests of the recombinant FSP, in which the GGX motif contributes to extensibility, while the spacer motif contributes to the strength of the recombinant fibres - according to the authors likely a primary contributor of strength, with the GGX motif supplying mobility to the protein network of native N. clavipes flagelliform silk fibres.
Up to now no complete structural model was determined/proposed for FSP; since there is a lack of data regarding to the real silk protein structures in their native state. Unlike the other protein constituents of the silk, the FSP does not undergo conformational changes during the spinning process2. There is limited information about the structures constituted by the GPGGX and GGX repeated sequences in the FSP; however, some studies describe these regions as amorphous with no crystalline structures in the fibres, and conformations without preferential orientation1,5,6; the abundance of proline residues regularly distributed throughout the repeat GPGGX sequence, prevents the formation of crystalline β-sheet structures in the fibre7,8. In contrast, in other studies have been proposed, for the GPGGX motif, the formation of a 310-helical structure or β-spiral structures; for this last one is suggested that can act as molecular nanosprings providing elasticity to the fibres6. In addition, studies by Perea et al.9 demonstrated the presence of polyglycine II nanocrystals in Argiope trifasciata flagelliform silk. According to the authors, the polyglycine dominant motifs GGX and GPGGX can contribute to increase toughness and achieve the ability of flagelliform silk to supercontract; this is possible due the conformational changes induced by the stretching step that leads the protein to a molecular reorganization of spatially close chains. As we can note, despite of these studies regarding to the FSP structure, still it is not clear the presence or absence of ordered regions in their fibres; it is necessary more chemical information and structural data for a better comprehension of FSP mechanoelastic properties.
It is known that spider silk can be used for many applications due of mechanical and physicochemical properties of spidroins; thus, the spider silk has been produced by recombinant DNA technology10,11,12. However, there exists limited information about post-translational modifications (PTMs), for example. So far, all work on mechanical properties of the silk fibres has been carried out with no consideration of the presence of PTMs in the spidroin sequences. Modifications such as phosphorylation13,14,15,16 and glycosylation17,18 have been reported in silk from spiders. Available studies have used mass spectrometry with different experimental approaches for the identification and location of PTMs in most diverse biological samples14,15,16,19,20,21,22,23. In the present study, an experimental approach was adopted combining 2-DE gels with multiple proteolytic in-gel digestion, followed by mass spectrometry (MSn) analysis using two different fragmentation methods, collision-induced dissociation (CID) and electron-transfer dissociation (ETD) for the characterization of FSP. Phosphorylation at tyrosine residues, for example, is considered relatively stable during MSn fragmentation, whereas at serine and threonine residues it has a moderate stability. Nitrotyrosine is considered to be unstable to moderately stable, and hydroxyproline is considered stable24,25,26. Therefore, all these modifications are detectable also by CID as observed in the current study.
Considering that the sequence as well as the identification of the post-translational modifications (PTMs) of FSP produced by orb weaver spiders are not completely known, the web silk protein produced by N. clavipes spider (Fig. 1a,b) was submitted to proteomic analysis for its structural characterization. The complete sequence and the PTMs assigned in the present study were used to simulate the 3-D structure of the FSP from the capture spiral (Fig. 1c) under natural conditions.

Scanning electron microscopy analysis of the web silk produced by (a) N. clavipes spider; (b) flagelliform gland of the N. clavipes spider; (c) in detail the yellowish colored fibres, secreted by the flagelliform gland, that form the capture spiral of the orbital web; (d) Ultrastructure of the flagelliform silk (spiral) highlighting the droplets (white arrow) on the fibre. (e,f) Solubilization in lithium thiocyanate highlighting the layer structures (white arrow) of silk. (g) Representative 2-DE profile of the flagelliform silk stained with Coomassie Colloidal Blue (From original image demonstrated in Supplementary Information - Figure S7).
Results
Morphological and structural characterization of silk fibres
Scanning electron microscopy (SEM) analysis of the web silk protein demonstrated the fibres ultrastructure at differentiated capture spiral positions (Fig. 1d–f) and showed that these fibres comprise compacted forms of silk layers. Figure 1d shows an SEM image of several droplets on the capture spiral fibre. As the flagelliform silk fibres are expelled by the flagelliform gland (Fig. 1b) a highly viscous coating is simultaneously deposited by the aggregate gland on these fibres, forming droplets27,28. The solubilization of silk fibres in lithium thiocyanate was a key step for the dissolution of the silk proteins prior to performing the 2-DE electrophoresis, used as strategy for proteomic analysis of this very large protein.
Flagelliform silk protein sequencing
The electrophoretic profile of the solubilized web silk (Fig. 1g) revealed six spots of spidroins (labelled 1 to 6) with apparent molecular weights higher than 250 kDa. The proteins in these spots were sequenced using mass spectrometry and identified in the gel as follows: FSP (spots 1 and 2) (GenBank ID accession numbers O44358 and O44359); spidroin-1A (spot 6) (GenBank ID accession numbers B5SYS5 and P19837); and spidroin-1B (spots 3, 4 and 5) (GenBank ID accession numbers B5SYS6 and P19837). The spidroins-1A and -1B, produced in the major ampullate glands, are present only in the frame and radial threads of the web; while the FSP, produced by the flagelliform gland, is present in the capture spiral. The analyses were carried out with the FSP identified in the spots 1 and 2; all the sequence obtained are shown in Tables S1 and S2. The sequence assignments of spidroins-1A and -1B are not presented in this study, since they were reported in a previous study15.
A gel-based mass spectrometry strategy with collision-induced dissociation (CID) fragmentation method was applied to sequence flagelliform silk protein (including the N- and C-terminal non-repetitive domains and the central repetitive domain) and to check the presence/position of PTMs within the FSP sequence. The spidroins (spots 1 and 2) were cleaved using different proteases and subjected to tandem mass spectrometry analysis. This allowed the identification and sequence assignment of 292 proteolytic peptides related to the FSP sequence: 5 tryptic peptides, 164 chymotryptic peptides, 29 proteolytic peptides were detected after digestion with Glu-C/V8 protease, 9 proteolytic peptides from digestion with subtilisin protease and 75 proteolytic peptides from digestion with proteinase 10. All peptide fragments (Tables S1 and S2) were identified by only two Accession Numbers: O44358 (a large fragment containing the N-terminal and central domains sequences) and O44359 (a large fragment containing the central and C-terminal domains sequences). The Table S3 shows the list of the 185 non-redundant proteolytic peptides in the chromatograms generated by the digestion of flagelliform silk protein with trypsin, chymotrypsin, Glu-C/V8 protease, proteinase 10 and subtilisin, presented according to the domain region to which they belong to (non-repetitive N-terminal, central repetitive, and non-repetitive C-terminal domains) along the sequence of N. clavipes.
The sequences of these peptides were aligned with the sequences of three large FSP fragments, corresponding to different domains of N. clavipes FSPs deposited in UniProtKB (http://www.uniprot.org/help/uniprotkb) (Fig. S1): (i) accession number O44358 - fragment corresponding to the non-repetitive N-terminal domain (NR-NTD); (ii) accession number Q9NHW4 - fragment corresponding to the most of central repetitive domain (CRD); and (iii) accession number O44359 - fragment corresponding to the central repetitive domain (CRD), as well the non-repetitive C-terminal domain (NR-CTD). The overall alignments revealed an identity of 89% of the experimental sequence of N. clavipes FSP determined in the present investigation, in relation to the overall sequence of the three domains deposited in UniProtKB.
Semi-quantitative analysis of peptide sequences
There are some peptide sequences that occurred many times all over N. clavipes FSP, while other peptides occurred only once; the Table S3 shows the exact origin of each peptide along FSP sequence. Thus, in order to verify if the relative abundance of each one of the 185 non-redundant proteolytic peptides were proportional to their occurrence in the FSP sequence, the peptides were semi-quantified using the spectral counting strategy. Technically, the absolute quantification of these peptides, is difficult to be performed; for that it would be necessary to isolate each peptide for individual quantification; thus, it was used a semi-quantitative method based on spectral counting strategy. It is important to emphasize that the amount of each peptide in the chromatogram is reflecting its occurrence along the FSP sequence; and it makes the N. clavipes FSP sequencing highly probable. Table S3 shows the integer value of the normalized spectral counting during mass spectrometry analysis of each peptide, which is reflecting directly the number of peptides occurrence in N. clavipes FSP sequence. This table also shows the m/z values of the respective molecular ions, charge state, and the position of each peptide along the sequence. Thus, the FSP sequencing results seem to be trustworthy, and apparently, the proteolytic digestion used for generating the peptides (tools of sequencing) reached the fullness. As a consequence of FSP sequencing, it was possible to identify, between the N- and C-terminal domains, three modules of the central repetitive domain in tandem, (Fig. S1). A careful observation of alignments of all experimental sequences and those of three large FSP fragments revealed that the most of these sequences are complementary to each other. The calculated MW for N. clavipes FSP sequence as shown in figure S1, is 195 kDa; considering that the apparent MW values observed for protein spots in 2-DE gel profile is about 266 kDa (Fig. 1g), it is possible to speculate that the FSP present in electrophoresis sample presented four CRD modules, while that analysed by a proteomic approach presented three CRD modules.
Post-translational modifications assignment
High-sequence coverage of FSP alllowed the identification of a series of post-translational modifications (PTMs) and amino acid substitutions. The MASCOT protein engine search and Modiro® were used to analyse all generated spectral data allowing the assignment of PTMs and amino acid substitutions such as S244Y, V396L, A444V, E503A, S512P, Y513D, G676R, S677Y, V819L, S979Y, V1131L, A1179V, E1238A, S1247P, Y1248D, G1411R, S1412Y, V1554L, S1714Y, V1866L, A1914V, E1973A, S1982P, Y1983D, G2146R, S2147Y, V2289L, G2341S, Y2346N and N2411D in the sequence of N. clavipes FSP (Tables S2 and S3). The detection of amino acid substitutions may be due to single nucleotide polymorphism, mutations or just mean the change in the amino acid composition of FSP in response to prey variation. Some studies show that spider silk proteins expression can be altered due to the interaction among the location, diet and type of prey29,30 and furthermore that these substitutions may change protein conformation and possibly even the mechanical properties of silk fibres. Tables, S1 and S2, are showing detailed proteomic data including all of PTMs assignments. During sample preparation or analytical procedures, some modifications such as oxidation, deamidation and methylation might have occurred due to various artefacts However, 45 sites of hydroxyproline, 9 sites of phosphotyrosine and 3 sites of nitrotyrosine were the major PTMs assigned to the FSP sequence (Table 1).
The Fig. 2a,b shows two representative spectra that present the assignment of hydroxyproline and phosphotyrosyne sites. Thus, Fig. 2a shows the CID spectrum of the chymotryptic peptide GP*GGSGPGGY (occurring at the positions 135–144, 145–154, 155–164, 165–174, 175–184, 190–199, 558–567, 578–587, 588–597, 598–607, 608–617, 628–637, 638–647, 708-617, 870–879, 880–889, 890–899, 900–909, 910–919, 925–934, 1293–1302, 1313–1322, 1323–1332, 1333–1342, 1343–1352, 1363–1372, 1373–1382, 1443–1452, 1605–1614, 1615–1624, 1625–1634, 1635–1644, 1645–1654, 1660–1669, 2028–2037, 2048–2057, 2058–2067, 2068–2077, 2078–2087, 2098–2107, 2108–2117, 2178–2187) used to assign the hydroxyproline sites on *P136, *P146, *P156, *P166, *P176, *P191, *P559, *P579, *P589, *P599, *P609, *P629, *P639, *P709, *P871, *P881, *P891, *P901, *P911, *P926, *P1444, *P1294, *P1314, *P1324, *P1334, *P1344, *P1364, *P1374, *P1606, *P1616, *P1626, *P1636, *P1646, *P1661, *P2029, *P2049, *P2059, *P2069, *P2079, *P2099, *P2109 and *P2179) (Table S3), while Fig. 2b shows the CID spectrum of the chymotryptic peptide GPGGAGGPYGPGGAGGPY* (occurring at the positions 481–498, 1216–1233, 1951–1968) (Table S3), used to assign the phosphotyrosine sites on *Y498, *Y1233 and *Y1968. Since there is no sufficient space to present all spectra used for each PTM site assignment, others representative spectra are also shown in figures S2–5.it is important to emphasize that in the current study we are using a numbering sequence based on the protein fragments used for alignment of N. clavipes FSP sequence (Table S3), while in protein databases the numbering standard refers to each partial sequence.

Representative mass spectra of N. clavipes flagelliform silk protein. (a) CID spectrum of the chymotryptic peptide GP*GGSGPGGY (135–144, 145–154, 155–164, 165–174, 175–184, 190–199, 558–567, 578–587, 588–597, 598–607, 608–617, 628–637, 638–647, 708–617, 870–879, 880–889, 890–899, 900–909, 910–919, 925–934, 1293–1302, 1313–1322, 1323–1332, 1333–1342, 1343–1352, 1363–1372, 1373–1382, 1443–1452, 1605–1614, 1615–1624, 1625–1634, 1635–1644, 1645–1654, 1660–1669, 2028–2037, 2048–2057, 2058–2067, 2068–2077, 2078–2087, 2098–2107, 2108–2117, 2178–2187), which was acquired by selecting the m/z 411.210 [M + 2 H]2+ as a precursor ion and showing the hydroxyproline sites at (P136, P146, P156, P166, P176, P191, P559, P579, P589, P599, P609, P629, P639, P709, P871, P881, P891, P901, P911, P926, P1444, P1294, P1314, P1324, P1334, P1344, P1364, P1374, P1606, P1616, P1626, P1636, P1646, P1661, P2029, P2049, P2059, P2069, P2079, P2099, P2109 and P2179). (b) CID spectrum of the chymotryptic peptide GPGGAGGPYGPGGAGGPY* (481–498, 1216–1233, 1951–1968), which was acquired by selecting the m/z 763.411 [M + 2 H]2+ as a precursor ion and showing the phosphorylation sites at Y498, Y1233 and Y1968.
Phosphotyrosine sites were also detected and confirmed by immunoblotting, using IgG anti-phosphotyrosine for staining. Figure 3a is showing an intense staining observed for the protein without any treatment, in its turn the protein treatment with alkaline phosphatase removes the staining totally; as well as nitrotyrosine sites were also detected and confirmed by immunoblotting, using IgG anti-nitrotyrosine for staining. In this case, Nitro-BSA (Nitrated bovine serum albumin MW 68 kDa, Sigma) was used as positive control (Fig. 3b). Figure 4a,b shows the N. clavipes FSP sequence, indicating the N- and C-terminal domains as well as three modules of the repetitive central domain with the assignment of hydroxyproline positions for the FSP.

Proteomic analysis of N. clavipes web silk. (a) Western blotting showing phosphotyrosine immunoreactivity (lane 1). Lane 2 shows no immunoreactivity after phosphatase treatment. (b) Western blotting showing nitrotyrosine immunoreactivity (lane 4). Lane 3 shows control sample Nitro-BSA (Nitrated bovine serine albumin, Sigma) (From original images demonstrated in Supplementary Information - Figures S8 and S9).

Representative sequence of N. clavipes FSP, highlighting the Non-Repetitive N-terminal Domain (NR-NTD), the three modules of the Central Repetitive Domain (CRD), and the Non-Repetitive C-Terminal Domain (NR-CTD). The hydroxyproline sites are assigned in yellow and marked with red “h” letter; the “spacer” domain is assigned in green.
3D structure of the flagelliform silk protein
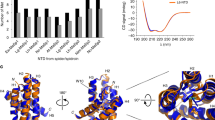
Given the results provided by the identification and extensive sequencing of FSP, molecular modelling and molecular dynamics simulations for this protein were performed to obtain structural information to support the understanding of mechanical and physicochemical properties of silk fibres from N. clavipes. FSP was modelled using the following proteins as templates: PDB ID: 2LPJ (major ampullate spidroin 1 N-terminal domain)31, 3HQV and 3HR2 (type I collagen)32, and 2MFZ (C-terminal domain of minor ampullate spidroin)33. Considering that FSP may get direct contact with large amounts of water from air humidity and/or rain under natural conditions, the protein was submitted to molecular dynamics (MD) simulations in presence of water. Thus, our simulations results are more adequate for FSP in solution, than in the fibres, where could occur interaction among the proteins. For simulation of protein-protein interaction, it would be necessary to perform the MD using a specific force field for this purpose, in a system with very high computation power to complete all analyses. The MD simulations were run for 10 ns; after this time, all model validation parameters (protein backbone RMSD, radius of gyration, potential energy analysis, and number of hydrogen bonds) revealed that the molecular model was reliable (Fig. S6a–d). The secondary structure analysis of N. clavipes FSP was performed using the JNET Secondary Structure Prediction program and PROCHECK, which indicated that FSP exhibits a heterogeneous, disordered and randomly oriented structural conformation. There is predominance of random structures in the solid fibres of the silk; the N-terminal domain is composed of 3 α-helices and the C-terminal domain is composed of 1 small helical section. The proposed 3D structure of the molecular model of the FSP was generated from the PyMol program34 and is shown in Fig. 5.

Representative three-dimensional molecular model of FSP, constituent of the silk fibres from N. clavipes spider. In details the N- and C-terminal non-repetitive domains, and the central repetitive domain, composed of the three modules of the primary sequence, as shown in Fig. 4.
Discussion
The sequence of N. clavipes FSP has been analyzed by mRNA and cDNA analysis35,36 and it is known that spidroins in general contain a large repetitive core sequence, which is flanked by non-repetitive N- and C-terminal domains37. Considering this, the aim of the present study was to characterize the FSP structure (including the non-repetitive N- and C-terminal domains and the central core) by a proteomic approach.
The protein databases GenBank (http://www.ncbi.nlm.nih.gov/genbank/submit) and Uniprot (http://www.uniprot.org/help/uniprotkb) contain many entries for different spidroin fragments that were virtually translated from DNA sequences and deposited in databases. Therefore, our strategy for sequencing FSP was to cover as much as possible the sequence, based on the proteolytic fragments from spots 1 and 2 (Fig. 1g); and overlapping the peptide sequences using the sequence informations deposited in GenBank and UniProt to achieve the highest possible coverage.
Our sequencing results demonstrated that the MW calculated for the sequence of N. clavipes FSP is 195 kDa; studies have shown that the spidroins of silk fibres have MW around 250–300 kDa after being secreted by the spinning duct forming the solid fibres38,39. The apparent MW of 266 kDa obtained in the denaturing gels is consistent with N. clavipes FSP presenting NR-NTD and NR-CTD flanking four modules of CRDs, while the sequence shown in Fig. 4 is showing the presence of three modules of CRDs. The number of CRD modules was previously reported to change from three to six for the spidroins15,16, which may explain the results reported above.
Another aspect that must be emphasized is that all the PTMs detected in the present investigation occurred in the CRDs (Fig. 4 and Table S2). Denaturing gels revealed by phosphotyrosine-immunoreactivity (Fig. 3) demonstrated a band around 85 kDa, which was not detected in a 2-DE (possibly because it is present in minimal quantities in the silk fibres). This MW value seems to be consistent with an FSP fragment corresponding either to NR-NTD or NR-CTD associated to one module of CRD. In the present investigation, a total of 9 phosphotyrosine sites were observed on FSP that were initially assigned by using CID fragmentations in mass spectrometric analysis. The assignments of phosphotyrosine residues were confirmed in N. clavipes FSP by immunoblotting, in absence and presence of treatment with alkaline phosphatase (Fig. 3a). It is important to note that, some proteomic studies have demonstrated the presence of phosphorylation on some known silk proteins including the fibroin from Bombyx mori (i.e. the heavy and light chain of fibroins, and the P25 protein)19,40,41 and the spidroin-1 protein from N. clavipes, N. madagascariensis and N. edulis spiders14. In our own studies, published recently15,16, in N. clavipes spider silk a total of 15 and 16 phosphorylation sites were identified on spidroin-1A and −1B, respectively; and a total of 36 phosphorylation sites on spidroin-2. So far, it is not clear the biological significance of many phosphorylation sites observed on the spider silk proteins. In spite that, based on literature data suggests that such modifications might be related to the structural conformation domains which are responsible for the mechanoelastic properties of spider silk fibres.
Nitrotyrosination sites were also observed on FSP which was confirmed by immunoblotting (Fig. 3b). The tyrosine oxidations were reported to form oxidation-mediated cross-links from two tyrosine radicals42. The functional relevance of nitrotyrosinations observed on FSP in the present study remains unclear, but previous work revealed the effects of tyrosine nitration on mechanoelastic properties and on protein-protein interactions21,43.
Hydroxyproline was the major PTM reported in the present study, with a total of 45 sites assigned to the FSP sequence. This modification consists of the addition of hydroxyl groups to the proline residues catalyzed by prolyl hydroxylase (PHD), warranting protein stability44. The N. clavipes web-silk is known to be highly elastic and the observation of a large amount of hydroxylated proline residues could potentially explain some elastic properties presented by the web fibres, just as it occurs in collagen found in the mussel Mytilus edulis45,46. Hydroxyproline is an essential amino acid residue present in collagen and studies indicate the presence of structural domains in the collagen of M. edulis, similar to the domains observed in spider silk; these domains may increase strength and extensibility of collagen45,46,47. Sutherland et al.48 have also demonstrated the presence of the structural motif of collagen in the insect silk willow sawfly, Nematus oligospilus (Hymenoptera) and has been characterized as collagen silk proteins.
In the case of FSP hydroxyprolines are located in the GPGGX motif, probably representing the formation of β-spiral structures that can act as molecular nanosprings providing elasticity to the fibres5. Previous work has shown that FSP does not undergo conformational changes during the spinning process, the vast abundance of proline residues regularly distributed throughout the repeat GPGGX sequence, prevents the formation of crystalline β-sheet structures in the fibre7,8. Previous studies demonstrating the proline and the glycine content of elastomeric and amyloids proteins, indicate that elastin-like behaviour occurs above a threshold level of combined proline and glycine content; with proline as the primary determinant of elastin-like behaviour49,50.
Based upon results obtained by the extensive sequencing of FSP we are presenting here a 3D model for FSP, which was submitted to MD simulations in presence of water, since that the fibres are exposed to air humidity and/or rain under natural conditions. In Guinea et al.51 studies with Argiope trifasciata spider silk, it was demonstrated that high humidity conditions are required for mechanoelastic property response of flagelliform fibre; and that this fibre shares with major ampullate fibre many aspects of their mechanical behaviour.
Modelling of N. clavipes FSP was performed for the complete protein, i.e., in the presence of the Non-Repetitive N-terminal Domain (NR-NTD), the three modules of the Central Repetitive Domain (CRD) and the Non-Repetitive C-Terminal Domain (NR-CTD) (Fig. 5). So far only parts of 3D structures of the N- and C-terminal regions of spidroin-1 have been reported33,39,52,53. In our own previous studies with N. clavipes spider silk, we presented a structural model for spidroin-1 consisting of the N- and C-terminal regions and the repetitive central region composed of a single module of the primary sequence of this protein15. The proposed 3D model of FSP indicates the predominance of stretched structures in the solid fibres of the silk protein (like the type I collagen, molecule used as template), presenting a heterogeneous, disordered and randomly oriented structural conformation, even after the simulations by molecular dynamics. In this protein model, the N-terminal region exhibits three complete helices despite the absence of the segment of the sequence at position 110–180 in the N-terminal domain of N. clavipes FSP (Fig. S1). The C-terminal domain contains one small helical section, maintained by a disulphide bridge formed by two highly conserved cysteine residues. According to Heim et al.54 studies with recombinant FSP domains, this disulphide bridge apparently has no influence on secondary or tertiary structure.
MD simulations were performed in a virtual box in presence of water molecules and all model validation parameters revealed that the molecular model was reliable, despite of the predominance of random structures in the solid fibres of the silk. Apparently, the FSP seems to be an intrinsically disordered protein (IDP) or natively unfolded proteins; and this property is a computational challenge for MD simulations, because by definition there are no available 3D structures of the whole molecule that is IDP55,56,57. The structural properties of IDPs are more sensitive to protein-water interactions than those of folded proteins56. Protein intrinsic disorder is a state related to protein function58,59. Studies have demonstrated that disorder is a metastable state susceptible to the changes in the environment; and that IDPs behaviour is often controlled by PTMs, such as phosphorylation60. It is important to mention that our findings pointed a series of PTMs such as phosphorylation and hydroxylation of proline on the FSP; these PTMs could potentially act on mechanoelastic properties and on protein-protein interactions in the web fibres. The data shown in figure S6a indicate that the FSP conformations were stabilized from 6 ns, despite the high abundance of random structures; in the figure S6b,c we can note that FSP exhibited higher molecular radii and higher potential energy in the presence of water. The number of hydrogen bonds in a molecular structure may be considered as an indicator of its stability. FSP contains 600 intramolecular hydrogen bonds in presence of water (Figure S6d), indicating that FSP is structured and stable in presence of water.
The spider silk has a great potential as a biomaterial. To explore this potential, it is needing a better understanding about the structure-function relationship of the proteins present in N. clavipes silk fibres; thus, chemical information such as PTMs can provide a basis for the mechanoelastic properties comprehension of silk fibres for biomedical and biotechnology applications. In the present study a large amount of hydroxyproline was observed along the sequence of the FSP, which constitutes the silk of the capture spiral. This silk is known to be highly elastic and the observation of a large amount of hydroxylated proline residues could potentially characterize this elastic property presented by the fibres. Sequence data reported herein may be relevant for the development of novel approaches for the synthetic or recombinant production of novel silk-based spider polymers, forms the basis for understanding previous work and for designing future studies of silks.
Material and Methods
Web silk samples
Web silk (radii and spiral) from N. clavipes spider was collected at the São Paulo State University at Rio Claro, SP in southeast Brazil. Approximately 25 mg (dry weight) of web silk from 20 orb-web spiders were dissolved in 2 mL of 38 M lithium thiocyanate (LiSCN hydrate) at 25 °C for 2 h under continuous shaking. The web-silk samples were processed as mentionedpreviously by dos Santos-Pinto et al.15. The protein concentration was estimated by the Bradford assay61.
Scanning electron microscopy
To examine the structural characteristics and the solubilization of the silk fibre surface, a small piece (approximately 5 mm) was placed into the lithium thiocyanate solution. Next, solution aliquots were placed on microscopy stubs and dried on a heating plate at 70 °C for several hours. Thereafter, the stubs were coated with gold in a Cressington sputter coater and then analyzed in a Hitachi, TM-3000 scanning electron microscope at 5–15 kV with a working distance of approximately 8 mm.
Two-dimensional gel electrophoresis
Silk protein samples (200 μg) were subjected to 2-DE, which was performed without any modification as reported previously by dos Santos-Pinto et al.15. The gels were stained with Coomassie Brilliant Blue R-250 (CBB) and thereafter, were scanned for documentation.
In-gel digestion
The spots of interest were excised from 2-DE gels, de-stained and treated with the following eight proteolytic enzymes: 40 ng/mL of trypsin (Promega, Madison, USA); 50 ng/mL of chymotrypsin (Roche Diagnostics); 40 ng/mL of proteinase 10 (syn.: Thermolysin); 40 ng/µL of Glu-C/V8 protease (Sigma); and 40 ng/µL of subtilisin (Sigma) in different conditions as described previously by dos Santos-Pinto et al.15 without any modification. Peptide recovering was performed using 0.5% (v/v) formic acid and 0.5% (v/v) formic acid in 30% (v/v) acetonitrile. The extracted peptides were then pooled for nanoLC-ESI-CID/ETD-MSn analysis.
NanoLC-ESI-CID/ETD-MSn
The HPLC used was a Nano-Advance UHPLC system (Bruker, Daltonics, Bremen, Germany) equipped with a PepMap100 C-18 trap column (300 mm × 5 mm) and a PepMap100 C-18 analytical column (75 mm × 150 mm). An Amazon ETD (Bruker Daltonics, Bremen, Germany) equipped with a CaptiveSpray source (Bruker, Daltonics, Bremen, Germany) was used to record the MS2 spectra in information-dependent data acquisition (three data-dependent CID MS/MS spectra and three ETD MS/MS spectra) mode over the mass range of m/z 100–3500. At the end, the peak lists from MS/MS data were generated by DataAnalysis 4.1 (Bruker Daltonics), and the spectra were interpreted. The generated spectral data, combining CID MS/MS and ETD MS/MS, were analysed by a MASCOT protein engine search and Modiro®.
Database search proteomics
Database searches were performed by using the MASCOT 2.3.02 (Matrix Science, London, UK) against the latest available spider protein sequences deposited in the NCBInr database (http://blast.ncbi.nlm.nih.gov, on July 24, 2017).
Thus, it was selected all 150,235 entries contained in the taxa Araneae for protein identification as reported previously by dos Santos-Pinto et al.15. Afterwards the identified proteins were subjected to additional filtering by Scaffold 4.3.2 (Proteome Software Inc., Portland, OR) to validate peptide identification. The false discovery rate (FDR) of less than 1%was calculated by requiring significant matches to at least 2 different sequences. Considering the Scaffold Local FDR algorithm, the peptide probability identification was set to a minimum of 99%, while the protein probability identification was set at 95%. PTM searches were performed using ModiroTM - PTM Explorer 1.1 software (Protagen AG, Dortmund, Germany). Using the advanced PTM-explorer search strategies it was possible to perform searches for amino acid substitution and unknown mass shifts. The Modiro software is complementary to the MASCOT software, using already identified sequences and has the advantage that also unknown mass shifts can be handled62. A list of 172 common modifications was selected and applied to virtually cleaved and fragmented peptides that were compared with experimentally obtained MS/MS spectra.
Quantitative proteomics
For the quantification of proteolytic peptides, it was used the spectral counting of the data generated during the mass spectrometry analysis15,16,63,64,65 of the individual digests of N. clavipes FSP with trypsin, chymotrypsin, Glu-C/V8 protease, proteinase 10 and subtilisin. The extracted ion chromatograms of each peptide were manually inspected for spectral counting; and the results were normalized dividing the individual counting of each peptide by the lowest counting observed amongst all the peptides.
Western blotting
In order to check the phosphotyrosine and nitrotyrosine modifications of the FSP, 50 μg of web silk protein extract was loaded onto 1D-SDS-PAGE gels; thereafter, the proteins were transferred onto PVDF membranes (Millipore). The entire procedure was performed as described previously by dos Santos-Pinto et al.15.
Secondary structure analysis and molecular modelling
Flagelliform silk protein was subjected to molecular modelling using a restrained-based modelling approach as implemented in the program MODELLER 9v11. Templates for flagelliform silk protein were searched using THREADER v3.566, BLASTp67 and FASTA68. Template searching by sequential identity using the program BLASTp only found templates that exhibited more than 30% identity to the N-terminal and C-terminal regions. Thus, we decided to search for templates using the THREADER program, which searches according to structural similarity, such that two proteins are considered homologous if they share similar sequences and structures. The output of these tools was formatted and used as input for the MODELLER program, which implements an automated approach for comparative modelling based on the fulfilment of spatial restraints. One thousand models were generated, and the final model was selected based on stereochemical quality and a MODELLER objective function. Images of the three-dimensional structures of the models were generated using PyMOL34.
Molecular dynamics simulations
Molecular dynamics (MD) simulations were performed using the GROMACS 5.1.4 software package69, using the force field 43a370 combined with the flexible Simple Point Charge (SPC) water mode71. Flagelliform silk protein was subjected to molecular dynamics simulation in a cubic box containing water, and a 1.0-nm minimum distance to the box face was used in all directions. The overall charge on the protein was neutralized, and physiological salt concentrations were simulated using Na+ and Cl- counter-ions. During the simulations, the lengths of the bonds within the protein were constrained under the conditions set by LINCS72 and SETTLE73 for water geometry. In the initial MD simulations, all hydrogen atoms, ions, and water molecules were subjected to 500 steps of energy minimization to remove close van der Waals contacts. The water system was then subjected to a short MD simulation with position restraints for 1,000 pico-seconds (ps). The final MD simulations were performed under the same conditions, except that the position restraints were removed for 10,000 ps. Energy minimization and MD were performed under periodic boundary conditions. Simulation was accomplished in the isothermal-isobaric ensemble at 300 K using temperature coupling74 and a constant pressure of 1 atm with isotropic molecule-based scaling75. Temperature and pressure were modulated using coupling techniques74 with coupling and isothermal compressibility constants of 0.01 ps (solvent and protein) and 6.5 × 10−5 bar−1, respectively. Electrostatic interactions among non-ligand atoms were evaluated using the particle-mesh Ewald method76. Cut-off distances for the calculation of the Coulomb and van der Waals interactions were 1.0 and 1.4 nm, respectively. Molecular visualization was performed in the graphical environments VMD - Visual Molecular Dynamics77 and PyMOL34.
References
Römer, L. & Scheibel, T. The elaborate structure of spider silk: structure and function of a natural high performance fiber. Prion. 2(4), 154–61 (2008).
Lefèvre, T., Boudreault, S., Cloutier, C. & Pézolet, M. Diversity of molecular transformations involved in the formation of spider silks. J. Mol. Biol. 405, 238–253 (2011).
Becker, N. et al. Molecular nanosprings in spider capture-silk threads. Nature Materials 2(4), 278–283 (2003).
Adrianos, S. L. et al. Nephila clavipes flagelliform silk-like GGX motifs contribute to extensibility and spacer motifs contribute to strength in synthetic spider silk fibers. Biomacromolecules 14(6), 1751–1760 (2013).
Craig, C. L. & Riekel, C. Comparative architecture of silks, fibrous proteins and their encoding genes in insects and spiders. Comp. Biochem. Physiol. 133, 493–507 (2002).
Hu, X. et al. Molecular mechanisms of spider silk. Cell. Mol. Life Sci. 63, 1986–1999 (2006).
Ohgo, K., Kawase, T., Ashida, J. & Asakura, T. Solid-state NMR analysis of a peptide (Gly-Pro-Gly-Gly-Ala) 6-Gly derived from a flagelliform silk sequence of Nephila clavipes. Biomacromolecules 7, 1210–1214 (2006).
Rousseau, M. E., Lefèvre, T. & Pézolet, M. Conformation and orientation of proteins in various types of silk fibers produced by Nephila clavipes spiders. Biomacromolecules. 10(10), 2945–53 (2009).
Perea, G. B. et al. Identification and dynamics of polyglycine II nanocrystals in Argiope trifasciata flagelliform silk. Sci Rep. 3, 3061 (2013).
Hardy, J. G., Römer, L. M. & Scheibel, T. R. Polymeric Materials Based on Silk Proteins. Polymers 49, 4309–4327 (2008).
Rising, A., Widhe, M., Johansson, J. & Hedhammar, M. Spider silk proteins: recent advances in recombinant production, structure-function relationships and biomedical applications. Cell. Mol. Life Sci. 68, 169–184 (2011).
Cranford, S. W., Tarakanova, A., Pugno, N. M. & Buehler, M. J. Nonlinear Material Behavior of Spider Silk Yields Robust Webs. Nature 482, 72–76 (2012).
Michal, C. A., Simmons, A. H., Chew, B. G., Zax, D. B. & Jelinski, L. W. Presence of phosphorus in Nephila clavipes dragline silk. Biophys J 70, 489–493 (1996).
dos Santos-Pinto, J. R. A. et al. Structure and post-translational modifications of the web silk protein spidroin-1 from Nephila spiders. J. Proteomics 105, 174–185 (2014).
dos Santos-Pinto, J. R. A. et al. Structural Model for the Spider Silk Protein Spidroin-1. Journal of Proteome Research 14, 3859–3870 (2015).
dos Santos-Pinto, J. R. A., Arcuri, H. A., Lubec, G. & Palma, M. S. Structural characterization of the major ampullate silk spidroin-2 protein produced by the spider Nephila clavipes. Biochimica et Biophysica Acta. Proteins and Proteomics 1864, 1444–1454 (2016).
Weiskopf, A., Senecal, K., Vouros, P., Kaplan, D. & Mello, C. M. The carbohydrate composition of a spider silk: Nephila clavipes dragline. Glycobiology 6, 1703–1703 (1996).
Sponner, A. et al. Composition and hierarchical organisation of a spider silk. PLoS ONE 2(10), 998 (2007).
Chen, W. Q., Priewalder, H., John, J. P. P. & Lubec, G. Silk cocoon of Bombix mori: Proteins and posttranslational modifications - heavy phosphorylation and evidence for lysine-mediated cross links. Proteomics 10, 369–379 (2010).
Redeker, V. Mass spectrometry analysis of C-terminal posttranslational modifications of tubulins. Methods Cell Biol. 95, 77–103 (2010).
Bae, N., Li, L., Lödl, M. & Lubec, G. Peptide toxin glacontryphan-M is present in the wings of the butterfly Hebomoia glaucippe (Linnaeus, 1758) (Lepidoptera: Pieridae). Proc Natl Acad Sci USA 109, 17920–17924 (2012).
Heo, S., Jung, G., Beuk, T., Höger, H. & Lubec, G. Hippocampal glutamate transporter 1 (GLT-1) complex levels are paralleling memory training in the Multiple T-maze in C57BL/6J mice. Brain Struct Funct. 217(2), 363–378 (2012).
Resende, V. M., Vasilj, A., Santos, K. S., Palma, M. S. & Shevchenko, A. Proteome and phosphoproteome of aAfricanized and european honeybee venoms. Proteomics 13(17), 2638–48 (2013).
Mann, M. & Jensen, O. N. Proteomic analysis of post-translational modifications. Nat. Biotechnol. 21, 255–261 (2003).
Wiesner, J., Premsler, T. & Sickmann, A. Application of electron transfer dissociation (ETD) for the analysis of posttranslational modifications. Proteomics 8, 4466–4483 (2008).
Johnson, H. & Eyers, C. E. Analysis of post-translational modifications by LC-MS/MS. Methods Mol Biol. 658, 93–108 (2010).
Salles, H. C., Volsi, E. C. F. R., Marques, M. R., Santos, L. D. & Palma, M. S. The venomous secrets of the web droplets from the viscid spiral of the orb-weaver spider Nephila clavipes (Araneae, Tetragnatidae). Chemistry & Biodiversity 3, 727–741 (2006).
Blackledge, T. A. et al. Reconstructing web evolution and spider diversification in the molecular era. Proc. Natl. Acad. Sci. USA 106, 5229–5234 (2009).
Tso, I. M., Wu, H. C. & Hwang, I. R. Giant wood spider Nephila clavipes alters silk protein in response to prey variation. J. Exp. Biol. 208, 1053–1061 (2005).
Blamires, S. J., Chao, I. C. & Tso, I. M. Prey type, vibrations and handling interactively influence spider silk expression. J. Exp. Biol. 213, 3906–3910 (2010).
Jaudzems, K. et al. pH-Dependent Dimerization of Spider Silk N-Terminal Domain Requires Relocation of a Wedged Tryptophan Side Chain. J.Mol.Biol. 422, 477–487 (2012).
Orgel, J. P., Irving, T. C., Miller, A. & Wess, T. J. Microfibrillar structure of type I collagen in situ. Proc.Natl.Acad.Sci. 103, 9001–9005 (2006).
Andersson, M. et al. Carbonic anhydrase generates CO2 and H+ that drive spider silk formation via opposite effects on the terminal domains. Plos Biol. 12, e1001921–e1001921 (2014).
Delano, W.L. The PyMOL Molecular Graphics System DeLano Scientific, Palo Alto, CA, USA (2002).
Hayashi, C. Y. & Lewis, R. V. Evidence from flagelliform silk cDNA for the structural basis of elasticity and modular nature of spider silks. J. Mol. Biol. 275, 773–784 (1998).
Hayashi, C. Y. & Lewis, R. V. Spider flagelliform silk: lessons in protein design, gene structure, and molecular evolution. Bioessays 23, 750–756 (2001).
Rising, A., Hjalm, G., Engstrom, W. & Johansson, J. N-terminal non-repetitive domain common to dragline, flagelliform, and cylindriform spider silk proteins. Biomacromolecules 7, 3120–3124 (2006).
Ayoub, N. A., Garb, J. E., Tinghitella, R. M., Collin, M. A. & Hayashi, C. Y. Blueprint for a high-performance biomaterial: full-length spider dragline silk genes. PLoS ONE 2, e514 (2007).
Hagn, F. et al. A conserved spider silk domain acts as a molecular switch that controls fibre assembly. Nature 465, 239–242 (2010).
Zhang, P. et al. Proteomic studies of isoforms of the P25 component of Bombyx mori fibroin. Biosci. Biotechnol. Biochem. 69, 2086–2093 (2005).
Zhang, P. et al. Proteome analysis of silk gland proteins from the silkworm. Bombyx mori, Proteomics 6, 2586–2599 (2006).
Stadtman, E. R. & Levine, R. L. Free radical-mediated oxidation of free amino acids and amino acid residues in proteins. Amino Acids 25, 207–218 (2003).
Peluffo, G. & Radi, R. Biochemistry of protein tyrosine nitration in cardiovascular pathology. Cardiovasc Res 75(2), 291–302 (2007).
Kaelin, W. G. Proline hydroxylation and gene expression. Annu Rev Biochem. 74, 115–28 (2005).
Qin, X. X. & Waite, J. H. Exotic collagen gradients in the byssus of the mussel Mytilus edulis. J Exp Biol. 198, 633–44 (1995).
Qin, X. X., Coyne, K. J. & Waite, J. H. Tough tendons. Mussel byssus has collagen with silk-like domains. J Biol Chem. 272(51), 32623–7 (1997).
Hagenau, A., Papadopoulos, P., Kremer, F. & Scheibel, T. Mussel collagen molecules with silk-like domains as load-bearing elements in distal byssal threads. J Struct Biol. 175(3), 339–47 (2011).
Sutherland, T. D. et al. A new class of animal collagen masquerading as an insect silk. Sci Rep. 3, 2864 (2013).
Rauscher, S., Baud, S., Miao, M., Keeley, F. W. & Pomès, R. Proline and glycine control protein self-organization into elastomeric or amyloid fibrils. Structure 14(11), 1667–76 (2006).
Savage, K. N. & Gosline, J. M. The effect of proline on the network structure of major ampullate silks as inferred from their mechanical and optical properties. J Exp Biol. 211(Pt 12), 1937–47 (2008).
Guinea, G. V., Cerdeira, M., Plaza, G. R., Elices, M. & Pérez-Rigueiro, J. Recovery in viscid line fibers. Biomacromolecules 11(5), 1174–1179 (2010).
Askarieh, G. et al. Self-assembly of spider silk proteins is controlled by a pH-sensitive relay. Nature 465, 236–238 (2010).
Hagn, F., Thamm, C., Scheibel, T. & Kessler, H. pH-dependent dimerization and salt-dependent stabilization of the N-terminal domain of spider dragline silk - Implications for fiber formation. Angew. Chem. Int. Ed. 50(1), 310–3 (2011).
Heim, M., Ackerschott, C. B. & Scheibel, T. Characterization of recombinantly produced spider flagelliform silk domains. J Struct Biol. 170(2), 420–5 (2010).
Battisti, A. & Tenenbaum, A. Molecular dynamics simulation of intrinsically disordered proteins. Molecular Simulation. 1–5 (2011).
Kosciolek, T., Buchan, D. W. A. & Jones, D. T. Predictions of backbone dynamics in intrinsically disordered proteins using de novo fragment-based protein structure predictions. Scientific Reports 7(1), 6999, https://doi.org/10.1038/s41598-017-07156-1 (2017).
Chong, S. H., Chatterjee, P. & Ham, S. Computer Simulations of Intrinsically Disordered Proteins. Annu Rev Phys Chem. 68, 117–134 (2017).
Dyson, H. J. & Wright, P. E. Intrinsically unstructured proteins and their functions. Nat. Rev. Mol. Cell Biol. 6, 197–208 (2005).
Babu, M. M., Kriwacki, R. W. & Pappu, R. V. Versatility from Protein Disorder. Science 337, 1460–1461 (2012).
Bah, A. et al. Folding of an intrinsically disordered protein by phosphorylation as a regulatory switch. Nature 519, 106–109 (2015).
Bradford, M. M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye biding. Anal. Biochem. 72, 248–254 (1976).
Nuwal, T., Heo, S., Lubec, G. & Bunchner, E. Mass spectrometric analysis of synapsins in Drosophila melanogaster and identification of novel phosphorylation sites. J Proteome Research 10, 541–550 (2011).
Arike, L. & Peil, L. Spectral counting label-free proteomics. Methods Mol Biol. 1156, 213–222 (2014).
Zhang, S., Cao, X., He, Y., Hartson, S. & Jiang, H. Semi-quantitative analysis of changes in the plasma peptidome of Manduca sexta larvae and their correlation with the transcriptome variations upon immune challenge. Insect Biochem Mol Biol 47, 46–54 (2014).
Ramus, C. et al. Benchmarking quantitative label-free LC-MS data processing workflows using a complex spiked proteomic standard dataset. J Proteomics 132, 51–62 (2016).
Jones, D. T., Taylor, W. R. & Thornton, J. M. A new approach to protein fold recognition. Nature 358, 86–89 (1992).
Lipman, D. J. & Pearson, W. R. Rapid and sensitive protein similarity searches. Science 227, 1435–1441 (1985).
Pearson, W. R. & Lipman, D. J. Improved tools for biological sequence comparison. Proc Natl Acad Sci USA 85(8), 2444–2448 (1988).
Van Der Spoel, D., Lindahl, E., Hess, B., Groenhof, G., Mark, A. E. & Berendsen, H. J. GROMACS: fast, flexible, and free. J Comput Chem 26, 1701–1718 (2005).
Scott, W. R. P. The GROMOS biomolecular simulation program package. J. Phys. Chem. A103, 3596–3507 (1999).
Berendsen, H. J. C., Postma, J. P. M., van Gunsteren, W. F. & Hermans, J. Interaction models for water in relation to protein hydration. In (ed.). Reidel Publishing Co:, in Intermolecular Forces (ed. Pullman, B.) 331–342 (1981).
Hess, B., Bekker, H., Berendsen, H. J. C. & Fraaije, J. G. E. M. LINCS: a linear constraint solver for molecular simulations. J Comput Chem 18, 1463–1472 (1997).
Miyamoto, S. & Kollman, P. A. SETTLE: An analytical version of the SHAKE and RATTLE algorithm for rigid water models. J. Comput. Chem. 13, 952–962 (1992).
Berendsen, H. J. C., Postma, J. P. M., van Gunsteren, W. F., DiNola, A. & Haak, J. R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 81, 3684–3690 (1984).
Chowdhuri, S., Tan, M. L. & Ichiye, T. Dynamical properties of the soft sticky dipole-quadrupole-octupole water model: a molecular dynamics study. J Chem Phys 125, 144513 (2006).
Essmann, U. A smooth particle mesh Ewald method. J. Chem. Phys. 103, 8577–8593 (1995).
Humphrey, W., Dalke, A. & Schulten, K. VMD - Visual Molecular. Dynamics. J. Molec. Graphics 14, 33–38 (1996).
Acknowledgements
This work was supported by grants from FAPESP (Proc. 2010/19051-6, Proc. 2011/51684-1 Proc. 2013/26451-9, Proc. 2015/14220-8, Proc. 2016/16212-5), CNPq (Proc. 301656/2013-4, Proc. 150699/2017-4) and the Gert Lubec Proteomics Laboratory. M.S.P. is a researcher from the National Research Council of Brazil-CNPq; G.L. is a researcher from the Gert Lubec Proteomics Laboratory at the Paracelsus Medical University, Salzburg, Austria and J.R.A.S.P. is a Post-Doc Research fellow from FAPESP at São Paulo State University - UNESP, Rio Claro, Brazil.
Author information
Authors and Affiliations
Contributions
M.S.P. and G.L. obtained the major funding, conceived and designed the experiments, supervised the research and drafted the text. J.R.A.S.P. obtained samples for protein extraction, designed and performed all the proteomic-related experiments, prepared the figures, tables and drafted the text. H.A.A. developed the studies of molecular dynamics of flagelliform silk protein. F.G.E. carried out scanning microscopical analyses and prepared the figures and tables. All authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
dos Santos-Pinto, J.R.A., Arcuri, H.A., Esteves, F.G. et al. Spider silk proteome provides insight into the structural characterization of Nephila clavipes flagelliform spidroin. Sci Rep 8, 14674 (2018). https://doi.org/10.1038/s41598-018-33068-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-33068-9
Keywords
This article is cited by
-
Silk: A Promising Biomaterial Opening New Vistas Towards Affordable Healthcare Solutions
Journal of the Indian Institute of Science (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
