
Abstract
IgM antibodies (Abs) are thought to play a major role in humoral immunity but only at the early stage of the primary immune response. However, two subsets of IgM+ memory B cells (MBCs), one with high affinity gained by means of multiple somatic hypermutation (SHM) and the other with low affinity and no SHMs, are generated through the germinal center (GC)-dependent and GC-independent (non-GC) pathway, respectively, after immunization with (4-hydroxy-3-nitrophenyl)acetyl (NP)-chicken γ-globulin. Surprisingly, an analysis of antibody-secreting cells reveals that a large amount of anti-NP IgM Ab with few SHMs is secreted during the recall response, indicating that only non-GC MBCs have terminal differentiation potential. Since secondary IgM Abs are capable of binding to dinitrophenyl ligands, they likely provide broad cross-reactivity in defense against microbial infection.
Similar content being viewed by others


Introduction
The (4-Hydroxy-3-nitrophenyl)acetyl (NP)-hapten system of the C57BL/6 mouse is advantageous for studying the structural bases of Ab affinities since anti-NP Abs are largely encoded by the V186.2 gene, as well as by minor genes such as V23, J558.55, and V124, referred to as Vanalogs1. However, these immunoglobulin (Ig) heavy chains possess different CDR3 structures, particularly around the amino acid residue at position 95 that corresponds to the VH-DH junction, where Tyr95 and Gly95 pair with His100 as well as X95 (where X indicates amino acids other than Tyr95 and Gly95)2,3. The structure at position 95 has been shown to contribute to variations in initial affinities1,4 and to raising the maximum affinity attainable by somatic hypermutation (SHM), namely, the ceiling affinity5.
By leveraging the NP system with a strong understanding of Ab affinity maturation, hapten-chromophore proteins with different numbers of hapten molecules, such as NPhi- or NPmed-allophycocyanin (APC), have often been used to estimate the relative affinities of BCRs6,7. Using these NP-APCs, Hara et al. showed that there are two subsets of IgM+ memory B cells (MBCs) in the spleens of mice immunized with NP-chicken γ-globulin (NP-CGG): one expressing BCRs with increased affinities due to SHM, similar to those of IgG1+ MBCs acquired through a germinal center (GC) reaction (referred to as MBCGC), and the other expressing BCRs lacking SHMs that are supposedly generated through the GC-independent (non-GC) pathway (referred to as MBCnon-GC)7.
Classically, MBCs are defined as B cells that have undergone antigen-driven proliferation and become resting cells. Upon re-exposure to primed antigens, MBCs are considered to proliferate and differentiate into Ab-secreting cells (ASCs)8,9. The expectation is that plasma cells secreting high-affinity IgM Abs are generated by secondary immunization; however, Murakami et al. showed that IgM Abs secreted from hybridomas prepared by cell fusion technology are characterized by consistently low affinity and few SHMs10. The results are in agreement with previous findings that IgM+ MBCs mainly comprise SHM− cells11,12,13. To explain the discrepancy in maturation in terms of affinities between BCRs on MBCsGC and secreted Abs from ASCs, an analysis of MBCs and ASCs at the same time points of immunization with NP-CGG was considered necessary.
Here, we report that IgM+ SHM+ MBCsGC are unable to differentiate into IgM ASCs. On the other hand, IgM+ SHM− MBCsnon-GC are capable of differentiating into plasma cells that secrete IgM Abs as part of the recall response. We also show that primary and secondary IgM Abs have different CDR3 structures that bring about different cross-reactivities to antigens; the secondary IgM Abs generally have very low initial affinities and can cross-react with the dinitrophenyl (DNP) hapten. Since neither primary IgM Abs nor affinity-matured IgG1 Abs bind to DNP, the broad cross-reactivity observed is considered to be characteristic of secondary IgM Abs, and their immunological role is discussed.
Results
We analyzed the number of NP-specific B-lineage cells as well as Ab affinity with time after immunization with NP40-CGG by dividing the time course into three phases: Phases I, II, and III14. Phase I involves naïve B cell activation with antigens and the production of primary Abs by day 7. Phase II corresponds to the subsequent period up to the time of secondary immunization. GC structures are known to reside in follicles at this phase15. Phase III starts with secondary immunization, which induces MBC activation followed by MBC differentiation into ASCs that secrete secondary Abs.
Kinetic analysis of activated B cell differentiation into MBCs and plasmablasts in Phases I and II
First, we analyzed the cell fate of IgM+ B cells in Phase I. NP-specific IgM+ B cells were detected as NPbright B220+ cells with NPhi-APC (Fig. 1A), although they were below the detection limit in unimmunized- and CGG-immunized mice (data not shown). This indicated that NP-specific naïve B cells were stimulated with NP40-CGG/alum and proliferated rapidly soon after immunization (Fig. 1B), followed by differentiation into either GL7− B cells or GL7+ B cells by day 7 (Fig. 1B,C). Since IgG1+ B cells were also detected on day 3, class-switch recombination (CSR) was thought to occur at the early stage of immunization7,16. GL7+ B cells were also detected on day 3 as NPdull B220+ cells (Fig. 1C), which would be pre-GC B cells destined to enter into GC structures that were not observed in follicles at this time point15,17. Since SHM was shown to be induced in GL7+ B cells on day 7 but not on day 57 (Tashiro et al., unpublished observation), we distinguished pre-GC B cells from GC B cells on the basis of SHM expression. In other words, we used the SHM frequency as an index of B cells that had resided for a time in GCs.

Analysis of the cell number and SHM frequency of B cells after immunization with NP40-CGG in Phase I and Phase II. (A) Flow cytometry of NP-specific (NPhi+ Igκ−) B cells in the spleen 7 days postimmunization with NP40-CGG/alum. The B cells were separated into NPbright and NPdull fractions, followed by the gating of GL7+ CD38− cells or GL7− CD38+ cells. The numbers in outlined areas indicate the percentages of GL7+ CD38− cells (middle right) and GL7− CD38+ cells (top left). (B) Changes in the number of NP-specific B cells (NPhi-APC-binding B220+ cells) with time postimmunization. (C) Changes in the number of pre-GC B cells (NPhi-APC-binding B220+ GL7+ cells) with time postimmunization. (D) Comparison of the numbers of GL7− B cells on days 14 and 42. Right panel, IgM+ B cells; Left panel, IgG1+ B cells. (E) Comparison of the number of SHMs per V186.2 gene between IgM+ and IgG1+ MBCs on day 42. Data were pooled from four independent experiments with one mouse per experiment in (E). (F) Flow cytometry of antigen-specific (NPhi+ Igκ−) B cells in the spleen on day 42 postimmunization with NP40-CGG/alum. The B cells were separated into three fractions based on the expression of IgM and IgD BCRs. Comparison of the numbers of IgM+ IgD− cells (white bars) and IgM+ IgD+ cells (black bars) on days 14 and 42. Fractional ratios of B cells with different numbers of SHMs are compared between IgM+ IgD− and IgM+ IgD+ cells. Fractional ratios of SHM+ V186.2+ B cells are divided into three based on the amino acid residues at positions 33 and 95. The results are presented as pie graphs pooled from five independent experiments with one mouse per experiment. The number of VH sequences analyzed is indicated in the center. *p < 0.05. Data are from three independent experiments with one mouse per time point indicated in the figures for each experiment (A–C), from four independent experiments with one mouse per time point indicated in the figure for each experiment (D) or from 4–5 independent experiments with one mouse per time point indicated in the figures for each experiment (F).
We identified MBCs as NPhi-APC-binding GL7− B220+ cells, and their number remained almost constant throughout Phase II (14 to 42 days postimmunization) (Fig. 1D). VH sequence analysis of MBCs on day 42, when the GC reaction was almost complete, revealed two subsets: MBCs that had no SHMs (SHM−) and MBCs with multiple SHMs (SHM+) (Fig. 1E). Since pre-GC B cells had converted to GC B cells and since SHM was induced only in GC B cells, SHM− GL7− B cells were considered to be MBCsnon-GC. Although both of these subsets evidently resided among IgM+ MBCs, there were only a few SHM− cells among IgG1+ MBCs, suggesting that the IgG1+ fraction largely comprised MBCsGC (Fig. 1E).
Next, we sorted the NPhi-APC-binding IgM+ MBCs into IgM+ IgD+ and IgM+ IgD− cells according to a report by Dogan et al.11. These cells had increased in number on day 42 (Fig. 1F). The nucleotide sequences of the V186.2 genes showed that the IgM+ IgD+ fraction contained more SHM+ cells than the IgM+ IgD− fraction. In addition, since SHM+ cells in the IgM+ IgD+ fraction contained those with a Trp33Leu mutation (Leu33+), which was shown to increase affinity18,19, and since Leu33+ cells were rare in the IgM+ IgD− fraction, IgD expression seemed to depend on the affinity of IgM BCRs.
IgM+ GC B cells differentiate into MBCs but not into plasmablasts
We previously developed a method for discriminating between plasmablasts and plasma cells, enabling us to examine these ASCs separately14. Because most CD138+ cells were found to be mIgλ, they were largely plasmablasts on day 7 (Fig. 2A). In fact, the ratio of plasma cells in the total ASC population on day 7 was less than 0.02 (data not shown). Both IgM+ and IgG1+ plasmablasts were observed on day 5 and reached maximum cell numbers of ~104 for IgM+ cells and ~3 × 105 for IgG1+ on day 7 (Fig. 2B).

Comparison of the cell number, VH usage, and SHM frequency between IgM+ and IgG1+ plasmablasts residing in spleens during Phase I and Phase II. (A) Flow cytometry of B220− CD138+ cells in the spleen on day 7 postimmunization with NP40-CGG/alum. The cells were separated based on the expression of Igκ and Igλ. The numbers in the outlined areas indicate the percentages of Igκ+ Igλ− cells (bottom right), Igκ− Igλ+ cells (top left) and Igκ− Igλ− cells (bottom left). (B) Kinetic analysis of the number of IgM+ or IgG1+ plasmablasts with time. (C) Comparison of VH usage between IgM+ and IgG1+ plasmablasts on day 7. (D) Comparison of the SHM frequency per V186.2 gene between IgM+ and IgG1+ plasmablasts in the spleen on day 42 postimmunization. Data were pooled from four independent experiments with one mouse per experiment in Figure (B). Data are from 3–4 independent experiments with one mouse per time point indicated in the figures for each experiment (A–C).
During Phase II, the number of plasmablasts decreased (Fig. 2B). Judging from the rapid decrease in number by day 14, plasmablasts in Phase I were considered to be short-lived. VH sequence analysis of Phase I plasmablasts revealed that both IgM and IgG1 Abs were encoded by germline V186.2+ Tyr95+ genes (Fig. 2C). We sorted these day-42 plasmablasts and analyzed VH nucleotide sequences, which revealed that SHMs were essentially absent in IgM+ cells while multiple SHMs were observed in the counterpart IgG1+ cells (Fig. 2D), suggesting that IgM+ plasmablasts had not arisen from IgM+ GC B cells on day 42 because GC B cells at this time point had multiple SHMs7. The presence of only SHM+ IgM+ MBCs (Fig. 1E) but not SHM+ IgM+ plasmablasts (Fig. 2D) suggested that IgM+ GC B cells are capable of differentiating into MBCs but not into plasmablasts or plasma cells.
Comparison of transcription factor expression in IgM+ and IgG1+ GC B cells
We next compared the expression of transcription factors of IgM+ and IgG1+ GC B cells to determine whether the defect in IgM+ GC B cells in terms of their inability to differentiate into ASCs would relate to the expression of these factors. Blimp1 is the key regulator of ASC development, directly represses Pax5 and is negatively regulated by Bach220. We isolated IgG1+ and IgM+ GC B cells on the basis of Fas expression, separated centroblasts and centrocytes based on the expression of CXCR4 and GL7 (Fig. 3A) and examined the expression of each transcription factor by RT-PCR (Fig. 3B). The results showed that IgM+ centrocytes expressed Prdm1 mRNA at the same level as their IgG1+ counterparts and that there were no differences in Pax5 or Bach2 mRNA expression between these cells. Therefore, the defect in IgM+ GC B cells in differentiation into plasmablasts and plasma cells could not be explained in terms of transcription factor expression.

RT-PCR analysis of transcription factors expressed in IgM+ and IgG1+ centroblasts and centrocytes. (A) Isolation of centroblasts and centrocytes with IgM or IgG1 BCRs was performed on the basis of the expression of CXCR4 and GL7. (B) RT-PCR analysis of Pax5 (Pax5), Bach2 (Bach2), and Prdm1 (Blimp1) mRNA expression between IgM+ and IgG1+ centroblasts or centrocytes obtained from spleens at 42 days postimmunization with NP40-CGG/alum. Data are representative of three independent experiments with one mouse per experiment.
Self-renewal and differentiation potential of MBCs after secondary immunization
Although IgM+ plasmablasts and plasma cells were nearly below the detection limit in Phase II (Fig. 2B), they appeared in the spleen and bone marrow after secondary immunization (Fig. 4A). In Phase III, the increase in the number of plasma cells was remarkable, as shown in a previous report14, which is true for IgM+ cells. We first examined which cells, MBCs or plasmablasts, were responsible for the generation of plasma cells, since both MBCs and plasmablasts express BCRs capable of receiving antigenic stimulation and since plasmablasts are thought to be precursors of plasma cells21,22. The results clearly showed that only transferred MBCs, and not plasmablasts, provided plasma cells that secreted anti-NP Abs (Fig. 4C). Although these experiments were carried out using plasmablasts regardless of the BCR isotype since few IgM+ plasmablasts were available, the results indicated that plasmablasts were unable to differentiate into plasma cells by secondary immunization with NP40-CGG.

Comparative analysis of MBCs, plasmablasts, and plasma cells in Phase II. (A) Comparison of the numbers of IgM+ and IgG1+ plasmablasts and plasma cells in the spleen and bone marrow 6 weeks postimmunization. Closed circles (individual mice) and solid lines (averaged values) represent NPhi+ cells. Open circles (individual mice) and dotted lines (averaged values) pertain to NPmed+ cells. (B) SHM frequency in VH sequences of IgM+ and IgG1+ MBCs in the spleen and in plasmablasts and plasma cells in the spleen and bone marrow on day 42. Each dot represents an NP-specific clone: circle, V186.2; triangle, Vanalog. The number represents the frequency of SHMs per VH gene. Data were pooled from 3–4 independent experiments with one mouse per experiment in (A). Data are from 3–4 independent experiments with one mouse per experiment.
Nucleotide sequence analysis revealed again that most of the IgM Abs secreted from plasmablasts and plasma cells had few SHMs (Fig. S1). This was even more evident in the VH sequences of IgM+ plasma cells that secreted only V186.2− SHM− Abs. Taken together with the data showing that the number of plasmablasts was low compared with the number of plasma cells, these results suggested that IgM+ MBCsnon-GC had differentiated into plasma cells that secrete low-affinity IgM Abs after secondary immunization. In other words, IgM+ MBCsGC were hardly differentiated into ASCs. This inability to differentiate into ASCs was not due to inefficient signaling through BCR since the VH sequence analysis revealed that both IgM+ primary MBCsnon-GC and MBCsGC were able to proliferate and self-renew by antigen stimulation, similar to IgG1+ MBCs (Fig. 4B).
Isotype-dependent variation in Ab production and affinity with time during immunization
Ab affinities are often estimated by ELISA using hapten-protein conjugates with different numbers of hapten molecules, such as NP2-BSA and NP26-BSA. In the case of germline IgG Abs (V186.2+ Tyr95+ Trp33+), Abs such as N1G9 cannot bind to NP2-BSA, but affinity-matured IgG Abs such as B2 (V186.2+ Tyr95+ Leu33+) or E11 (V186.2+ Gly95+ SHM+) can (Fig. 5A). On the other hand, IgM Ab has a pentameric structure and thus possesses ten antigen-combining sites per molecule, which allows binding to multivalent antigens with higher avidity than that of IgG Abs, as shown with control IgM Abs encoded by the same VH and VL genes as IgG counterparts, N1G9 and B2 (Fig. 6A). From the viewpoint of the VH-D-JH gene nucleotide sequence, N1G9-IgM is identical to B1-818,23. On the other hand, some IgM Abs, such as 8A38 and 1A810, which are V186.2+ X95+, are unable to bind to NP2-BSA, and their NP2/NP26 ratios are lower than that of N1G9-IgM (Fig. 5A), suggesting that they have very low affinities. In fact, they are unable to bind to NP26-BSA when converted into the IgG form (Murakami et al., unpublished observation). Since these IgM Abs are encoded by V186.2 genes with no SHMs10, their very low affinities are considered to arise from differences in CDR3H sequences (Fig. 5B).

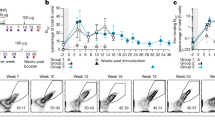
Changes in the amount of Ab bound to NP2-BSA or NP26-BSA and the NP2/NP26 ratio after immunization of C57BL/6 mice with NP40-CGG/alum. (A) Binding of IgG and IgM Abs to NP2-BSA (open bars) and NP26-BSA (closed bars). NP2/NP26 ratios are represented by hatched circles. Left panel, IgG mAbs with different Ka values to NPs, namely, N1G9 (Ka = 5 × 105 M−1), B2 (Ka = 3.4 × 106 M−1), and E11 (Ka = 108 M−1), were used. Right panel, IgM mAbs secreted from hybridomas (8As38, 1A8, and 1A86, respectively) as well as recombinant IgM mAbs whose V-D-J genes were from N1G9 and B2 (N1G9-IgM and B2-IgM, respectively) were used. (B) Binding of IgM mAbs to NP2-BSA (open bars) and NP26-BSA (closed bars). NP2/NP26 ratios are represented by hatched circles. The CDR3 sequences of the IgM mAbs are shown. (C) Changes in the binding of IgM and IgG2b Abs in the immune sera. The serum from an immunized mouse (n = 3) was diluted (200-fold for IgM, 800-fold for IgG2b), and binding was measured by ELISA. Open circles (individual mice) and open bars (averaged values) represent binding to NP2-BSA. Closed circles (individual mice) and closed bars (averaged values) represent binding to NP26-BSA. The time course of Ab production was divided into four periods: Phase I, Phase II, Phase III and Phase IV. Phase IV corresponds to the period post tertiary immunization. Data are representative of three independent experiments per experiment (A,B) or three independent experiments with one mouse per experiment (C).

NP-binding IgM Abs possess cross-reactivity to analogs of the original antigen. (A) Binding of IgG mAbs to NP26-BSA (open bars) and DNP26-BSA (closed bars). IgG mAbs with different Ka values to NPs, namely, N1G9 (Ka = 5 × 105 M−1), B2 (Ka = 3.4 × 106 M−1), and E11 (Ka = 108 M−1), were used. (B) Binding of IgM mAbs to NP26-BSA (open bars) and DNP26-BSA (closed bars). Recombinant IgM mAbs whose V-D-J genes were from N1G9, B2 and E11 (N1G9-, B2- and E11-IgM, respectively) were used. (C) Binding of IgM mAbs secreted by hybridomas (1A350, 1A8, and 1A86, respectively) to NP26-BSA (open bars) and DNP26-BSA (closed bars). (D) Binding of IgM mAbs performed as in (B) in the presence of the indicated concentrations of NP-Cap. (E) Binding of IgM Abs to NP2-, NP26-, and DNP26-BSA in serum collected from an individual mouse (n = 3) at 1 week after primary immunization (primary 1w) and secondary immunization (secondary 1w). The serum was diluted 200-fold, and binding was measured by ELISA. *p < 0.05, **p < 0.01, and ***p < 0.001. Error bars indicates the s.d. Data are representative of three independent experiments per experiment (A–D) or four independent experiments with one mouse per experiment (C).
Next, we examined the binding of IgM Abs to NP2- and NP26-BSA in immune sera obtained from mice immunized with NP40-CGG in Phase II (Fig. 5C). The total amount of IgM Ab bound to NP26-BSA was estimated with the OD490 value, which decreased steadily with time until day 49, indicating that there was a short supply of IgM Abs in Phase II. On the other hand, a large amount of Ab production was observed on day 54 or 56 after secondary immunization (Phase III), indicating that recall Ab production was induced. However, IgM Abs on day 56 showed inefficient binding to NP2-BSA and provided a low NP2/NP26 ratio of ~0.2–0.3, which was smaller than that of day 7 Abs (~0.7), suggesting that the secondary Abs have lower affinity (avidity) than the primary Abs. The lower NP2/NP26 ratio for day 56 IgM Abs was not due to inhibition by the binding of high-affinity IgG Abs to NP2-BSA since similar results were obtained using antisera from which IgG Abs had been removed with protein G-beads (Fig. S2A).
An Ab production profile that was different from that of IgG1 Abs was observed for other isotypes. The peak of primary IgG2b Ab production was detected at approximately 7–14 days, followed by a gradual decrease with time. The ratio of NP2/NP26 increased slightly (0.2–0.4), but the decrease was not as evident as that of IgG1 Abs. Secondary immunization, however, resulted in an obvious increase in the NP2/NP26 ratio (0.9–1.0), indicating that the recall response of IgG2b Abs was accompanied by the production of affinity-matured Abs (Fig. 5D). We also examined the production of IgG3 and IgA Abs (Fig. S3). The peak production of IgG3 Abs was observed at approximately 7–14 days, followed by a gradual decrease during Phase II, with little change in the NP2/NP26 ratio, which was as low as <0.2. In the case of IgA Abs, neither a production peak at approximately day 7 nor subsequent Ab production in Phase II was observed, indicating that the primary response of these Abs was different from those of the others. In conclusion, these results show that each isotype has a unique Ab production profile and may not necessarily be accompanied by an increase in Ab affinity (Figs 5C and S3).
Cross-reactive binding of secondary IgM Abs to DNP-hapten
To understand the immunological role of secondary IgM Abs, we examined their cross-reactivity to various chemical compounds, since cross-reactions sometimes play an important role in viral infection24. Anti-NP Abs are known to react with NP analogs, such as (4-hydroxy-3-iodo-5-nitrophenyl)acetyl (NIP) or (4-hydroxy-3,5-dinitrophenyl)acetyl (NNP), which are haptens with a higher affinity than that of NP alone; this phenomenon is referred to as heterocliticity25,26. We also examined whether anti-NP IgG1 Abs can bind to DNP26-BSA and found that they are unable to bind to DNP regardless of their affinity (Fig. 6A), even though the structures of the IgG Abs were converted to the pentameric IgM form (Fig. 6B). On the other hand, the IgM Abs 1A350, 1A8, and 1A86, which are encoded by germline V186.2 genes10, could bind to DNP26-BSA (Fig. 6C). Such cross-reactive binding to DNP would not be caused by a nonspecific hydrophobic interaction since the Abs did not bind to (4-hydroxyphenyl)acetyl (HP)26-BSA, FITC-BSA or acetyl (Ac)-BSA (Table 1). In addition, this binding was inhibited with NP-Cap in a dose-dependent manner, suggesting that the same binding sites were recognized by both NP and DNP (Fig. 6D).
We also compared the binding of IgM Abs to NP2, NP26, and DNP26-BSA using NP-CGG immune sera (Fig. 6E). Day 7 IgM Abs showed efficient binding to NP2-BSA, while day 56 IgM Abs showed weak binding to NP2-BSA but significant binding to DNP26-BSA and to NP26-BSA (Fig. 6E). In terms of NP/DNP cross-reactivity at the molecular level, the appearance of Igλ+ NP+ DNP+ MBCs supported these observations (Fig. S4).
Discussion
In this study, we analyzed IgM+ MBCs phenotypically and functionally based on our previous reports7,10, focusing on IgM+ ASCs and Abs secreted from these cells. Subcategorizing IgM+ MBCs on the basis of their expression of IgD BCR, we found that the SHM+ and SHM− fractions corresponded to IgM+ IgD− and IgM+ IgD+ cells, respectively. Although these two types of IgM+ MBCs possessed the same memory characteristic of self-renewal potential, there was a difference in the ability to differentiate into ASCs; IgM+ IgD+ MBCs, but not IgM+ IgD− MBCs, showed poor potential for ASC formation, in contrast to classical views on memory definition8,9. Notably, our study revealed that secondary IgM Abs exhibited cross-reactivity, while their affinities were lower than those of canonical IgM Abs with strict antigen specificity. In contrast, Krishnamurty et al. reported that IgM+ IgD−, but not IgM+ IgD+ MBCs, were highly mutated in the malaria immunization system27. However, since Pape et al.28 demonstrated that IgM+ IgD+ MBCs in the PE/CFA immunization system dominantly comprised those possessing fewer SHMs, IgM+ IgD+ MBCs likely expressed few SHMs, and the discrepancy may be explained in terms of antigens employed, namely, NP, PE or malaria. Obviously, malaria-antigen epitopes would be more complex than NP and PE and activate more diverse B cell repertoires with a broad range of affinities to be involved in Ab production. This brought about the enrichment of heterogeneous MBCs due to wider gating of antigen-specific populations and a broader range for Ig gene sequence analysis.
Our results showing that IgM+ MBCsnon-GC with low-affinity BCRs are able to differentiate into ASCs responsible for recall IgM Ab production are not consistent with the notion that high-affinity B cells are selected to differentiate into plasma cells14,29. We hypothesized that high-affinity IgG1 Abs may prevent the secretion of IgM Abs, as reported for TI antigens30; however, no such effect of IgG1 Ab on the recall response of IgM Abs was observed (Fig. S4). Therefore, it is likely that a lack of differentiation of IgM+ MBCGC into ASCs might be caused by isotype-specific and affinity-dependent regulation of BCR signaling31,32,33,34. Our finding that the two IgM+ MBC subsets seemed to be distinguishable on the basis of IgD BCR expression (Fig. 1E) will be useful to elucidate the underlying mechanisms.
In addition to MBCs, as described above, a rapid decline in the number of IgM+ plasmablasts indicated poor potential to differentiate into ASCs among IgM+ GC B cells (Fig. 2B). Furthermore, most of the IgM Abs secreted from plasmablasts on day 42 had no SHMs (Fig. 2D). These results indicated the preference for skewed MBC differentiation in IgM+ GC B cells in contrast to IgG1+ GC B cells. Surprisingly, the inability of IgM+ GC B cells to differentiate into plasmablasts was not due to insufficient expression of transcription factors, since no significant differences in the levels of transcription factors were observed between IgM+ and IgG1+ GC B cells (Fig. 3). Considering the small difference in BCR affinity between IgM+ and IgG1+ GC B cells7, we supposed that IgM+ GC B cells were prohibited from differentiating into plasmablasts capable of secreting high-affinity IgM Abs that are inferior to low-affinity Abs in such functions as complement-dependent hemolysis10, although these cells were allowed to differentiate into MBCs, whose regulatory mechanism has not yet been revealed.
During Phase III, a large amount of IgM Ab capable of binding to NP26-BSA, but not to NP2-BSA, was secreted. Although both Phase I- and Phase III-IgM Abs lack SHMs, they are different in terms of their initial affinities (Fig. 5C). Despite their low affinities, secondary IgM Abs have a unique antigen specificity that makes them capable of binding to DNP and TNP; no other anti-NP Abs, including primary IgM Abs and affinity-matured secondary IgG1 Abs, showed any ability to bind to NP analogs (Fig. 6 and data not shown). We suppose that the immune system prepares two different types of IgM Abs in terms of antigen recognition in Phases I and III, which conclude on days 7 and 54, respectively (Fig. 5). Although the intrinsic affinities of the combining sites are similar, IgM Abs have the advantage over IgG1 Abs of being able to bind to multivalent antigens through their higher avidity because the IgM Abs have 10 combining sites per molecule, suggesting that primary IgM Abs play a role in compensating for the low affinities of IgG1 Abs. Secondary immunization also provides both IgM and IgG1 Abs, whose affinities are significantly differ (Fig. 5C). Since affinity maturation has been shown to accompany specificity maturation35, affinity-matured IgG1 Abs recognize antigens more efficiently and more precisely than their precursors. However, microbes that invade our bodies sometimes change their surface structures to escape attack from affinity-matured IgG1 Abs. Immune systems seem to prepare IgM Abs with a broad specificity, which is expected to extend beyond the narrow specificity of IgG1 Abs and function as a first-line barrier against the invasion of microbes whose epitopes are altered by mutations through an Ab cross-reaction mechanism. Krishnamurty et al. provided new knowledge on IgM+ MBCs and ASCs by utilizing the malaria immunization system27. Tracing the changes in the SHM frequencies, affinities and specificities of IgM Abs through comparative analysis of the changes in those of IgG Abs in a clinically relevant setting is informative.
Classically, MBCs are defined as cells that have undergone antigen-driven proliferation, expressed isotype-switched BCRs, and then become resting cells. They are activated upon re-exposure to primed antigens and then proliferate and subsequently differentiate into ASCs capable of secreting high-affinity Abs8,9. These types of MBCs are sometimes referred to as effector MBCs, and IgG1+ MBCsGC meet this definition. Judging from the profiles of Ab production and affinity maturation, IgG2b+ MBCs would be characterized as the effector type (Fig. 6), although their frequency is quite low during late Phase II at both the cellular level (IgM+, 63.3 ± 7.0%; IgG1+, 28.5 ± 6.3%; IgG2b+, 2.2 ± 0.8% in NPhi+ B cells) and sequence level7. On the other hand, IgM+ MBCs are referred to as central MBCs because they lack the ability to differentiate into ASCs but are able to re-enter into GCs and differentiate into IgG+ MBCs12,36. However, since we found that IgM+ MBCsnon-GC have the potential to differentiate into plasma cells that secrete secondary IgM Abs, they can be referred to as the effector type. Zuccarino-Catania et al. recently demonstrated remarkable data that effector MBCs and central MBCs were distinguished by the expression of CD80 and PD-L237. Further characterization of IgM+ MBCsnon-GC with the expression patterns of surface markers12,37 and transcriptional factors20 is expected. Although little information is available regarding their function of IgM+ MBCsGC, we speculate that IgM+ MBCsGC may be a new type of MBC possessing affinity-matured BCRs capable of switching to other isotypes when necessary. The immunological role of IgM+ MBCsGC needs to be clarified as well as that of MBCs with isotypes other than IgG1.
In a series of reports6,7,14, we have described the basis of the cellular dynamism of Ab production and affinity change and showed divergent and unique patterns that are specific to such isotypes as IgG1, IgG2b, and IgM. Vaccines have been used to activate naïve B cells to differentiate into MBCs and ASCs and are expected to prevent the invasion of microbes, which are known to stimulate the production of various Ab isotypes. For efficient and safe vaccination, further knowledge on the regulation of the MBCs and ASCs of each isotype is required.
Methods
Mice and Immunizations
C57BL/6 mice were purchased from Japan SLC. AID−/− mice were provided by Prof. T. Honjo (Kyoto University, Kyoto, Japan). All mice were maintained in Tokyo University of Science (TUS) mouse facility under specific pathogen-free conditions. Mouse procedures were performed under protocols approved by the TUS Animal Care and Use Committee and are in accordance with the current national regulation. Mice were immunized i.p. with 100 μg of NP40-CGG precipitated in alum and boosted with the same Ag in PBS.
NP-Protein Conjugates
NP-protein conjugates were prepared as described previously38. Briefly, the hydroxysuccinimide ester of NP (NP-OSu) was coupled with proteins in 0.1 M sodium bicarbonate buffer (pH 8.5) containing 0.15 M NaCl for 1 h at room temperature. Any excess of reagents or reaction byproducts was removed via passage through a Sephadex G-25 column (GE Healthcare) or by dialysis against PBS. In the case of NP-allophycocyanin (NP-APC) and streptavidin-NP-APC (NP-APCavi), the number of NPs per APC was assumed on the basis of the molar ratio of NP-Osu to APC or APCavi in the reaction mixture6. DNP- and TNP-BSA were prepared by reacting BSA with 2,4-dinitrobenzenesulfonic acid or 2,4,6-trinitrobenzenesulfonic acid39. HP-BSA was prepared by reacting BSA with the hydroxysuccinimide ester of HP. The number of HPs introduced was estimated by titration of amino groups with 2,4,6-trinitrobenzenesulfonic acid40.
Anti-NP Abs
Hybridomas producing the anti-NP Abs 8A38, 1A8, 1A86, 9T13, B2 and E11 were prepared as described previously5,10,38. N1G9 was provided by Prof. K. Rajewsky (Max-Delbrück-Centrum für Molekulare Medizin, Berlin, Germany). The VH-DH-JH genes of N1G9 or B2 were fused with the secreted form of Cμ-genes and transfected into J558L myeloma cells by electroporation. N1G9-IgM and B2-IgM were purified from culture supernatants6.
Flow Cytometry and Detection Reagents
Cells were treated with 5 mM EDTA in PBS before staining, which reduced the binding of cytophilic nonendogenous Abs to B cells (Supplemental information). Cells were treated with unlabeled anti-CD16/CD32 (2.4G2, eBioscience) in FACS buffer (1% BSA/PBS) to block FcγRs. After washing, cells were stained with the relevant antibodies: anti-Igκ-FITC (187.1, BD Pharmingen), anti-IgM-PerCP-Cy5.5 (II/41, eBioscience), anti-IgD-FITC (11–26 c.2a, BD Pharmingen), anti-IgG1-PE (A85-1, eBioscience), anti-CD45R/B220-PE-Cy7 (RA3-6B2, eBioscience), anti-CD138-Biotin (281.2, BD Pharmingen), anti-GL7-FITC (GL7, BD Pharmingen), anti-CD38-PE (90, eBioscience), anti-CD95/Fas-PE (Jo2, BD Pharmingen), anti-CXCR4-APC (2B11, eBioscience), and PE–streptavidin (eBioscience). Cells were analyzed on FACSCanto II and FACSAria II (BD Biosciences). Cell sorting was performed on FACSAria II (BD Biosciences). The data were analyzed using FlowJo software (Tree Star).
Detection of Cells Secreting Antibodies by Flow Cytometry
We employed the method as described previously14. Cells were treated with unlabeled rat anti-mouse CD16/CD32 (2.4G2, eBioscience) in medium (10% FCS/IMDM) to block FcγRs. After washing with medium, cells were stained with rat anti-Igλ-PE (JC5-1, Southern Biotech) and biotinylated rat anti-mouse CD138 (281-2, BD Pharmingen) in medium. FACS buffer (1% BSA/PBS) containing no azide was used for washing cells and subsequent staining with streptavidin-APC (BioLegend) coupled with NP. After cultivation, the cells were washed and stained in FACS buffer containing sodium azide. Finally, cells were stained with rat anti-Igλ-FITC (JC5-1, Southern Biotech) and rat anti-mouse B220 (RA3-6B2, eBioscience). Cell sorting was carried out using a FACSAriaII (BD Biosciences). Files were analyzed using FlowJo software (Tree Star).
VH Sequence Analysis
mRNAs were purified from sorted cells of each B cell subset using Trizol (Invitrogen). cDNAs were synthesized using PrimeScript Reverse Transcriptase (Takara) and Oligo-dT primers, followed by amplification using PrimeSTAR DNA polymerase (Takara) and forward primer: 5′-TCTAGAATTCAGGTCCAACTGCAGCAGCC-3′, paired with the following reverse primers: mCμ5′-CTTGAACAGGGTGACGG-3′ for IgM+ MBCs, sCμ5′-GTGTCAGACATGATCAGG-3′ for IgM+ plasmablasts/plasma cells; mCγ5′-TCTGCTTCAGCTCCACCAC-3′ for IgG+ MBCs; sCγ1 5′-TCTGCTTCAGCTCCACCAC-3′ and sCγ2b/2c 5′-GTGCTGAGCTCATTTACC-3′ for IgG+ plasmablasts/plasma cells.
Transcriptional Factor Expression Analysis
mRNAs were purified from 1,000 sorted cells of each B cell subset using Trizol (Invitrogen). cDNAs were synthesized using PrimeScript Reverse Transcriptase (Takara) with random primers. PCR was performed using Takara Ex Taq HS (Takara) and the following primers41,42,43: Pax5 5′-CCTACCCTATTGTCACAGGCC-3′, 5′-CCTCTGTCTGTCTCAGGGGGTT-3′; Bach2 5′-GCTGCTCCACACTGTGACAT-3′, 5′-ACCTCCCGGATGTTCTCTCT-3′; and Blimp1 5′-AACACGTGGTACAACCCAAAG-3′, 5′-AGGCTGCAGAGATGGATGTAG- 3′.
ELISA
To measure the binding of Abs to hapten, plates (Nunc) were coated with 50 μl per well of hapten-BSA in PBS and left at 4 °C overnight, followed by blocking with FACS buffer (1% BSA/0.05% sodium azide/PBS). After washing with PBS-Tween 20 (PBST), Abs of known concentrations or antisera at various dilutions were added, and the plates were then incubated for 2 h at room temperature. After washing with PBST, the bound Abs were detected with HRP-conjugated goat antibodies specific to each mouse isotype (Southern Biotech).
Statistics
Statistical differences were analyzed with the 2-tailed Student’s t-test for 2-group comparisons and one-way ANOVA with Tukey’s test for multiple comparisons, with Prism 7 software (GraphPad Software, La Jolla, CA, USA). Data were considered significant at *p < 0.05, **p < 0.01, and ***p < 0.001.
Data Availability
The datasets generated and/or analyzed during this study are available from the corresponding author on reasonable request.
References
Dal Porto, J. M., Haberman, A. M., Kelsoe, G. & Shlomchik, M. J. Very low affinity B cells form germinal centers, become memory B cells, and participate in secondary immune responses when higher affinity competition is reduced. J. Exp. Med. 195, 1215–1221 (2002).
Kabat, E. A., Reed-Miller, M., Perry, H. M. & Gottesman, K. S. Sequences of Proteins of Immunological Interest. 5th ed. Department of Health and Human Services, Public Health Service, National Institutes of Health, Bethesda, MD. (1991).
Hofle, M., Linthicum, D. S. & Ioerger, T. Analysis of diversity of nucleotide and amino acid distributions in the VD and DJ joining regions in Ig heavy chains. Mol. Immunol. 37, 827–835 (2000).
Murakami, A. et al. The amino acid residue at position 95 and the third CDR region in the H chain determine the ceiling affinity and the maturation pathway of an anti-(4-hydroxy-3-nitrophenyl)acetyl antibody. Mol. Immunol. 48, 48–58 (2010).
Furukawa, K. et al. T. Junctional amino acids determine the maturation pathway of an antibody. Immunity 11, 329–338 (1999).
Nishimura, M., Murakami, A., Hara, Y. & Azuma, T. Characterization of memory B cells responsible for affinity maturation of anti- (4-hydroxy-3-nitrophenyl)acetyl (NP) antibodies. Int. Immunol. 23, 271–285 (2011).
Hara, Y. et al. High affinity IgM+ memory B cells are generated through a germinal center-dependent pathway. Mol. Immunol. 68, 617–627 (2015).
Tarlinton, D. M. B-cell memory:are subsets necessary? Nat. Rev. Immunol. 6, 785–790 (2006).
Kurosaki, T., Kometani, K. & Ise, W. Memory B cells. Nat. Rev. Immunol. 15, 149–159 (2015).
Murakami, A. et al. Low-affinity IgM antibodies lacking somatic hypermutations are produced in the secondary response of C57BL/6 mice to (4-hydroxy-3-nitrophenyl)acetyl hapten. Int. Immunol. 26, 195–208 (2014).
Dogan, I. et al. Multiple layers of B cell memory with different effector functions. Nat. Immunol. 10, 1292–1299 (2009).
Pape, K. A. et al. Different B cell populations mediate early and late memory during an endogenous immune response. Science 331, 1203–1207 (2011).
Taylor, J. J., Pape, K. A. & Jenkins, M. K. A germinal center-independent pathway generates unswitched memory B cells early in the primary response. J. Exp. Med. 209, 597–606 (2012).
Tashiro, Y. et al. An asymmetric antibody repertoire is shaped between plasmablasts and plasma cells after secondary immunization with (4-hydroxy-3-nitrophenyl)acetyl chicken γ-globulin. Int. Immunol. 27, 609–620 (2015).
Jacob, J., Kassir, R. & Kelsoe, G. In situ studies of the primary immune response to (4-hydroxy-3-nitrophenyl)acetyl. I. The architecture and dynamics of responding cell populations. J. Exp. Med. 173, 1165–1175 (1991).
Chan, T. D. et al. Antigen affinity controls rapid T-dependent antibody production by driving the expansion rather than the differentiation or extrafollicular migration of early plasmablasts. J. Immunol. 183, 3139–3149 (2009).
Coffey, F., Alabyev, B. & Manser, T. Initial clonal expansion of germinal center B cells takes place at the perimeter of follicles. Immunity 30, 599–609 (2009).
Cumano, A. & Rajewsky, K. Clonal recruitment and somatic mutation in the generation of immunological memory to the hapten NP. EMBO J. 5, 2459–2468 (1986).
Torigoe, H. et al. The affinity maturation of anti-4-hydroxy-3-nitrophenylacetyl mouse monoclonal antibody. A calorimetric study of the antigen-antibody interaction. J. Biol. Chem. 270, 22218–22222 (1995).
Nutt, S. L. et al. The genetic network controlling plasma cell differentiation. Semin. Immunol. 23, 341–349 (2011).
Radbruch, A. et al. Competence and competition: the challenge of becoming a long-lived plasma cell. Nat. Rev. Immunol. 6, 741–750 (2006).
Oracki, S. A. et al. Plasma cell development and survival. Immunol. Rev. 237, 140–159 (2010).
Bothwell, A. L. M. et al. Heavy chain variable region contribution to the NPb family of antibodies: somatic mutation evident in a gamma 2a variable region. Cell 24, 625–637 (1981).
Eisen, H. N. Affinity enhancement of antibodies: How low-affinity antibodies produced early in immune responses are followed by high-affinity antibodies later and in memory B-cell responses. Cancer Immunol. Res. 2, 381–392 (2015).
Imanishi, T. & Makela, O. Strain differences in the fine specificity of mouse anti-hapten antibodies. Eur. J. Immunol. 3, 323–330 (1973).
Reth, M., Hammerling, G. J. & Rajewsky, K. Analysis of the repertoire of anti-NP antibodies in C57BL/6 mice by cell fusion. Eur. J. Immunol. 8, 393–400 (1978).
Krishnamurty, A. T. et al. Somatically Hypermutated Plasmodium-Specific IgM+ Memory B Cells Are Rapid, Plastic, Early Responders upon Malaria Rechallenge. Immunity 45, 402–414 (2016).
Pape, K. A. et al. Naive B Cells with High-Avidity Germline-Encoded Antigen Receptors Produce Persistent IgM+ and Transient IgG+ Memory B Cells. Immunity 48, 1135–1143 (2018).
Smith, K. G. C., Light, A., Nossal, G. J. V. & Tarlinton, D. M. The extent of affinity maturation differs between the memory and antibody-forming cell compartments in the primary immune response. EMBO J. 16, 2996–3006 (1997).
Obukhanych, T. V. & Nussenzweig, M. C. T-independenttype II immune responses generate memory B cells. J. Exp. Med. 203, 305–310 (2006).
Kaisho, T., Schwenk, F. & Rajewsky, K. The roles of gamma 1 heavy chain membrane expression and cytoplasmic tail in IgG1 responses. Science 276, 412–415 (1997).
Weiser, P., Muller, R., Braun, U. & Reth, M. Endosomal targeting by the cytoplasmic tail of membrane immunoglobulin. Science 276, 407–409 (1997).
Wakabayashi, C., Adachi, T., Wienands, R. & Tsubata, T. A distinct signaling pathway used by the IgG-containing B cell antigen receptor. Science 298, 2392–2395 (2002).
Kometani, K. et al. Repression of the transcription factor Bach2 contributes to predisposition of IgG1 memory B cells toward plasma cell differentiation. Immunity 39, 136–147 (2013).
Oda, M. & Azuma, T. Affinity maturation of anti-(4-hydroxy-3-nitrophenyl)acetyl antibodies accompanies a modulation of antigen specificity. Mol. Immunol. 70, 8–12 (2016).
Burton, B. R. et al. Variant proteins stimulate more IgM+ GC B-cells revealing a mechanism of cross-reactive recognition by antibody memory. elife 7, e26832, https://doi.org/10.7554/eLife.26832 (2018).
Zuccarino-Catania, G. V. et al. CD80 and PD-L2 define functionally distinct memory B cell subsets that are independent of antibody isotype. Nat. Immunol. 15, 631–637 (2014).
Azuma, T., Sakato, N. & Fujio, H. Maturation of the immune response to (4-hydroxy-3-nitrophenyl)acetyl (NP) haptens in C57BL/6 mice. Mol. Immunol. 24, 287–296 (1987).
Little, J. R. & Eisen, H. N. Preparation and characterization of antibodies specific for 2,4,6-trinitrophenyl group. Biochemistry 5, 3385–3395 (1966).
Okuyama, T. & Satake, K. On the preparation and properties of 2,4,6-trinitrophenyl-amino acids and -peptides. J. Biochem. 47, 454–466 (1960).
Corcoran, L. M. et al. Differential requirement for OBF-1 during antibody-secreting cell differentiation. J. Exp. Med. 201, 1385–1396 (2005).
Schebesta, A. et al. Transcription factor Pax5 activates the chromatin of key genes involved in B cell signaling, adhesion, migration, and immune function. Immunity 27, 49–63 (2007).
Kallies, A. et al. Initiation of plasma-cell differentiation is independent of the transcription factor Blimp-1. Immunity 26, 555–566 (2007).
Acknowledgements
We thank Dr. T. Honjo for providing AID-deficient mice and Dr. K. Rajewsky for providing N1G9. This work was supported in part by research funds from Kyokuto Pharmaceutical Industrial Co., Ltd. and Antibody Engineering Research Center Co., Ltd.
Author information
Authors and Affiliations
Contributions
Y.T. conceived the project, designed and performed all of the experiments, interpreted the data, and wrote the manuscript. A.M., T.S., M.K., R.G. and H.K. provided critical advice. Y.H. provided technical support. T.A. supervised the project, designed the experiments and wrote the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Tashiro, Y., Murakami, A., Hara, Y. et al. High-affinity IgM+ memory B cells are defective in differentiation into IgM antibody-secreting cells by re-stimulation with a T cell-dependent antigen. Sci Rep 8, 14559 (2018). https://doi.org/10.1038/s41598-018-32926-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-32926-w
Keywords
This article is cited by
-
B cell subsets were associated with prognosis in elderly patients with community acquired pneumonia
BMC Pulmonary Medicine (2022)
-
A Trade-off Between Thermostability and Binding Affinity of Anti-(4-hydroxy-3-nitrophenyl)Acetyl Antibodies During the Course of Affinity Maturation
The Protein Journal (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
