
Abstract
The current application of genome editing to crop plants is limited to cultivars that are amenable to in vitro culture and regeneration. Here, we report an in planta genome-editing which does not require callus culture and regeneration. Shoot apical meristems (SAMs) contain a subepidermal cell layer, L2, from which germ cells later develop during floral organogenesis. The biolistic delivery of gold particles coated with plasmids expressing CRISPR/Cas9 components designed to target TaGASR7 were bombarded into SAM-exposed embryos of imbibed seeds. Bombarded embryos showing transient GFP expression within SAM were selected and grown into adult plants. Mutations in the target gene were assessed in fifth-leaf tissue by cleaved amplified polymorphic sequence analysis. Eleven (5.2%) of the 210 bombarded plants carried mutant alleles, and the mutations of three (1.4%) of these were inherited in the next generation. Genotype analysis of T1 plants identified plants homozygous for the three homeologous genes, which were all derived from one T0 plant. These plants showed no detectable integration of the Cas9 and guide RNA genes, indicating that transient expression of CRISPR/Cas9 introduced the mutations. Together, our current method can be used to achieve in planta genome editing in wheat using CRISPR/Cas9 and suggests possible applications to other recalcitrant plant species and variations.
Similar content being viewed by others

Introduction
Genome editing has been successfully applied in major crops, such as rice (Oryza sativa L.), maize (Zea mays L.) and wheat (Triticum aestivum L.), using clustered regularly interspaced short palindromic repeats (CRISPR) and CRISPR-associated protein9 (Cas9) nuclease, which is a simple and versatile tool for inducing DNA double-stranded breaks at target DNA sites1,2,3. In these cases, CRISPR/Cas9 expression cassettes as well as a selectable marker gene were introduced into plant genomes using Agrobacterium tumefaciens-mediated or biolistic delivery. Recently, DNA-free genome editing has been achieved in several crops, including lettuce (Lactuca sativa L.), maize and wheat, with a mixture of Cas9 mRNA and guide RNA or pre-assembled ribonucleoproteins (RNPs) directly introduced into the cells4,5,6. However, current applications of the CRISPR/Cas9 system in plants rely on the conventional plant transformation procedure that includes callus culture and regeneration processes. This limits application of this technology to cultivars that are amenable to tissue culture. Many elite commercial cultivars lack this property and thus are recalcitrant to transformation. In addition, the callus culture process is generally time-consuming and can suffer from somatic variation.
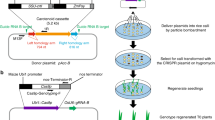
To avoid the problems associated with the callus culture and regeneration processes during plant transformation, an in planta method, in which transgenes are introduced directly into intact immature florets through Agrobacterium, has been developed in Arabidopsis thaliana. In planta transformation procedures have also been reported in several crops, such as rice, maize, tomato and wheat7,8,9,10. However, these methods seem to suffer from problems with reproducibility and efficiency and are not established as standard protocols. Recently, an alternative method using biolistic delivery, in planta particle bombardment (iPB), has been developed in wheat11. The iPB method utilizes shoot apical meristem (SAM) of imbibed seeds as the target cells for transformation. With this method, it is now possible to transform wheat cultivars that are recalcitrant to conventional transformation procedures.
Here, we report an in planta genome-editing procedure in wheat using iPB. We show that transient expression of CRISPR/Cas9 genes within the SAM of imbibed seed embryos induces genome editing and the plants grown from the embryos inherit the edited sequence to the next generation. The method can be used to introduce DNA-free genome-editing in wheat.
Results
Detection of CRISPR/Cas9-mediated genome editing in SAM
To achieve in planta genome-editing in wheat, we selected TaGASR7, which is involved in the control of grain length and weight and has been shown to be amenable to genome editing with CRISPR/Cas9, as the target gene12,13. We designed a single guide RNA (sgRNA) targeting all three homeologous TaGASR7 genes (TaGASR7-A1, -B1 and -D1) according to the previous report12. We mixed three plasmid constructs carrying expression cassettes for Staphylococcus pyogenes Cas9, the sgRNA and a GFP reporter gene, respectively, coated gold particles with the mixture, and then bombarded shoot apical meristems (SAMs) of mature embryos with the coated gold particles according to the standard iPB delivery protocol11. We observed the bombarded SAMs under a fluorescence microscope to check for transient GFP expression (Supplementary Fig. S1a,b). We defined embryos showing one or more GFP spots within the SAM (19 of the 30 bombarded) as GFP positive and selected these for further study (Supplementary Fig. S1c,d). No wound-induced auto-fluorescence was observed in the bombarded SAMs11. Three days after the bombardment, we excised the SAMs of the embryos to test for targeted mutagenesis of the TaGASR7 genes. Cleaved amplified polymorphic sequences (CAPS) analysis revealed that five embryos (nos. 3, 5, 6, 7 and 12) showed undigested bands after BcnI digestion, suggesting that mutations had occurred at the BcnI target site (Fig. 1a). Subsequent small-scale sequence analysis of the undigested bands revealed several patterns of mutations in TaGASR7 genes (Fig. 1b). These results suggested that Cas9 and sgRNA expression cassettes were successfully delivered into the meristematic region and caused targeted mutagenesis within 3 days.

CAPS analysis of TaGASR7 locus in meristematic tissue. (a) Genomic DNA was isolated from the meristematic tissue of wild-type (Wt) and GFP-positive embryos (nos. 1–19) 3 days after plant bombardment and then subjected to a cleaved amplified polymorphic sequences (CAPS) assay. M, marker; −, undigested PCR products; + , BcnI-digested PCR products. Solid and dashed arrows indicate the positions of uncut and cut PCR products, respectively. The full-length gel image is shown in Supplementary Fig. S2. (b) DNA extracted from the undigested bands (nos. 3, 5, 6, 7 and 12 in (a)) indicated by white arrowheads were randomly sequenced. The guide sequence is indicated by a green arrow. The protospacer-adjacent motif (PAM) sequence and BcnI restriction site are indicated by yellow and blue bars, respectively.
Confirmation of targeted mutagenesis in T0 progeny and T1 progeny
Next, we conducted a larger-scale screening of genome-edited plants in T0 progeny. Of the 210 bombarded embryos, we selected 176 (83.8 ± 4.3%, n = 7) that transiently expressed GFP in their meristematic regions and grew these into mature plants (Table 1). We tested the targeted mutagenesis of the TaGASR7 genes in the fifth leaf of each plant using a CAPS assay. We detected undigested bands in 11 plants, accounting for 5.2% of the bombarded embryos (Fig. 2, Table 1), which implies that targeted mutations in the meristematic region can be reflected in the genotype of the young leaf of T0 plants. We grew the 11 selected plants to obtain seeds and then, because of the possible chimeric nature of the T0 plants, subjected all of these T1 seeds to genotype analysis. The CAPS assay detected mutant alleles of TaGASR7 genes in one or more T1 plants (2-7-1 to 2-7-8, 2-21-1, and 7-2-1 to 7-2-10) derived from 3 of the 11 putative T0 mutant plants (2-7, 2-21 and 7-2) (Fig. 3a, Tables 1, 2). T1 plants derived from the 2–7 T0 plant contained mutations in the B and D genomes but not the A genome. The 2–21 T0 plant set five T1 seeds, of which one had heterozygous mutations in the A, B and D genomes (Table 2). Three T1 plants (7-2-3, 7-2-8 and 7-2-10) from the 7–2 T0 plant carried homozygous mutations in all three genomes (Fig. 3b, Table 2). Sequencing analysis of the BcnI-resistant PCR amplicons revealed either a T, A or GT insertion or a 35-bp deletion in one or more of the TaGASR7 genes in each of these T1 plants (Fig. 3c, Supplementary Table 1). The 7–2–8 plant showed insertions of T, GT and A into the TaGASR7 genes of the A, B and D genomes, respectively, and thus was considered to carry a complete knockout of TaGASR7 (Tagasr7). The inheritance of the mutations in T1 plants suggested that the Cas9 and sgRNA cassettes are efficiently introduced into meristematic L2 cells and cause mutations in germline cell(s).

CAPS analysis of TaGASR7 locus in T0 plants. Genomic DNA was isolated from each fifth leaf of 11 bombarded plants and one wild-type (Wt) plant and then subjected to PCR and subsequent BcnI restriction enzyme digestion. M, marker; −, undigested PCR products; + , BcnI-digested PCR products. Black and dashed arrowheads indicate the positions of uncut and cut PCR products, respectively. The full-length gel image is shown in Supplementary Fig. S3.

CAPS analysis of the TaGASR7-A1, -B1 and -D1 loci in T1 plants. Genomic DNA isolated from each first leaf of independent T1 plants derived from three T0 mutants (2-7, 2-21, and 7-2) and one wild-type (Wt) plant. The DNA was subjected to PCR with TaGASR7-A1, -B1 and -D1 conserved (a) and specific (b) primer sets. PCR products were digested with BcnI restriction enzyme. −, undigested PCR products; + , BcnI-digested PCR products. Solid and dashed arrows indicate the positions of uncut and cut PCR products, respectively. The full-length gel image of Fig. 3(a) is shown in Supplementary Fig. S4. (c) Mutant alleles identified in T1 mutant plants. The guide sequence is highlighted by a green arrow. The PAM sequence and BcnI restriction site are indicated by yellow and blue bars, respectively.
Detection of CRISPR/Cas9 vector DNA sequences
Genes introduced into plant cells via particle bombardment can be expressed transiently without genome integration. This suggested that genome editing using iPB can be achieved by transient expression of the CRISPR/Cas9 system without stable chromosomal integration of the genes required for CRISPR/Cas9. To test this possibility, we carried out genomic PCR to detect the possible integration of the introduced plasmids. We designed primer sets to detect eight distinct DNA regions within the expression plasmids pE(R4-R3)ZmUbi_OsCas9_ver3, pTAKN-sg-GR7 and pUba-GFP (Fig. 4a, Supplementary Table 2) and analysed genomic DNA from all 19 T1 plants (2-7-1 to -8, 2-21-1, and 7-2-1 to -10). None of the T1 mutants, including the three homozygous mutants, showed an amplification signal for any of the eight DNA regions that was comparable in level to that of TaLOX2, which is a single-copy gene in wheat genomes (Fig. 4b, Supplementary Table 1). These results indicated that none of the genome-edited plants contained a functional copy of the CRISPR/Cas9 genes, suggesting that the mutations were created through transient expression of CRISPR/Cas9 in SAM.

Detection of foreign DNA integration in T1 mutant lines. (a) Schematic structure of the bombarded plasmids and primer sets used to detect DNA integration. ZmPubi, maize ubiquitin promoter; T35S, cauliflower mosaic virus 35S terminator; kanR, kanamycin-resistance gene; TaU6, wheat U6 promoter; ampR, ampicillin-resistance gene. (b) Genomic PCR analysis of T1 mutants (2-7-1 to 2-7-8, 2-21-1, and 7-2-1 to 7-2-10) and wild-type (Wt) plants. Genomic DNA was extracted from the first leaf of each T1 plant. The corresponding vector DNA was used as a positive control (P). The full-length gel image is shown in Supplementary Fig. S5.
Discussion
Genome editing has been applied to major crops such as rice, maize, wheat and potato1,2,3,12,14. The current strategy for editing genes in such crops depends on tissue culture, and this limits application of genome editing to the specific cultivars amenable to callus culture and regeneration. In this study, we report an in planta method for genome editing in wheat that does not require callus culture and regeneration.
We utilized meristematic tissue of the mature embryo as the target tissue for bombardment. As indicated by GFP expression analysis (Supplementary Fig. S1, Table 1), gold particles can be efficiently delivered to the SAM region of the embryos. Analysis of genomic DNA isolated from SAM of the bombarded embryos indicated that genome editing occurred in a high proportion of the embryos within 3 days after bombardment (Fig. 1). These data suggested that genome editing might occur without integration and stable expression of CRISPR/Cas9 genes. This was confirmed by PCR analysis of T1 plants (Fig. 4). It is interesting to note that the pattern of mutations detected in the embryos was similar to that detected in T1 plants (Figs 1b and 3c), even though these mutations were created independently. Clearly, there is a preference for the base modification during DNA repair. The preference may be varied by cultivar and experimental system utilized12.
The iPB method is designed to target particle delivery of CRISPR/Cas9 expression cassettes to SAM. In imbibed seeds, the first three leaves have already developed. The fourth and later leaves appear after this point and therefore can show phenotypic evidence of mutations at this stage. We therefore performed our first selection of the genome-edited plants with fifth-leaf tissue, and we found that about 5% of T0 plants showed detectable mutations (Fig. 2, Table 1). Within the SAM, L2 cells are the only source of germ cells. Therefore, heritable mutations should be introduced into the L2 cells. In total, 1.4% of the T0 plants produced T1 seeds with inherited mutations (Table 1). This suggested that a fairly high proportion of mutations can be induced in L2 cells under the conditions used in this study.
Besides its potential application to commercial cultivars, the iPB method has several merits over conventional genome-editing methods. i) Experiments can be started quickly and easily with dry mature seeds; it is not necessary to prepare immature embryos. ii) No antibiotic selection is necessary. iii) In addition to DNA, RNA and protein can also be introduced into plant cells with gold particles; CRISPR/Cas9 RNP has been successfully delivered into maize and wheat cells by this method to induce genome editing5,6.
DNA-free genome editing is favoured for commercial applications of genome-edited crops, because it may reduce the chance of off-target changes and it alleviates concerns about genetically modified organisms12. The iPB method with CRISPR/Cas9 DNA successfully created genome-edited wheat plants through transient expression of the CRISPR-Cas9 genes. However, it is still possible that a small fragment of the plasmids might be integrated into the chromosomes of the mutants. To avoid this possibility, the next avenue of exploration will be the use of CRISPR/Cas9 RNP instead of DNA. In any event, we believe the iPB method will be a useful tool for genome editing in a wide range of wheat cultivars.
Materials and Methods
Preparation of mature embryos
Preparation of mature embryos was performed as previously reported11. In brief, mature seeds of wheat (Triticum aestivum L. cv. Bobwhite) were sterilised and rinsed and then germinated overnight at 22 °C. The parts of the coleoptile and leaf primordia covering the SAM were excised. The embryos were separated from endosperms and placed upright on Murashige and Skoog (MS) basal medium supplemented with plant preservative mixture (3%; Nacalai Tesque, Japan) in culture plates. Thirty embryos per plate were placed in a circle (diameter 0.8 cm).
Expression vector
The wheat U6 promoter and gRNA scaffold15 were synthesised de novo and cloned into the plasmid pTAKN-2 by TA cloning. The guide sequence12 for the TaGASR7-A1, -B1, and -D1 genes were inserted between the two BbsI sites of the plasmid. The resulting vector (pTAKN-sg-GR7) was used for expression of sgRNA. Wheat SAMs were bombarded with pE(R4-R3)ZmUbi_OsCas9_ver3, pTAKN-sg-GR7 and pUba-GFP11.
Preparation of microprojectiles and biolistic delivery
Microprojectiles were prepared as previously reported with slight modifications11. Briefly, pE(R4-R3)ZmUbi_OsCas9_ver3 (5 μg), pTAKN-sg-GR7 (3 μg) and pUba-GFP (2 μg) were mixed with gold particles (InBio Gold, Australia) of 0.6 μm. Bombardment was conducted using a PDS-1000/He™ device (Bio-Rad, USA) with a target distance of 6.0 cm from the stopping plate. The vacuum in the chamber was 27 inches of Hg and the helium pressure was 1350 psi. Bombardment was repeated four times per plate.
Microscopic analyses and plant growth conditions
About 12 h after bombardment, SAMs in the bombarded mature embryo were observed with an MZFLIII microscope equipped with a GFP filter (excitation wavelength, 470/40 nm; emission wavelength, 525/50 nm). Mature embryos expressing GFP in the SAMs were transferred into a Phytatray™ II (Sigma-Aldrich, USA) with basal MS medium and cultivated for 2–3 weeks in a growth chamber under long day conditions (16 h light/8 h darkness) at 22 °C. Seedlings were planted in pots (3 seedlings/pot, φ 10.5 cm) and grown in a phytotron under long day conditions at 22 °C.
Detection of vector DNA sequences
To detect vector DNA sequences in T1 mutants, polymerase chain reaction (PCR) analysis was conducted. DNA was isolated from the first leaf of the T1 progeny, as described previously16. Each round of PCR was conducted in a reaction mixture (10 µL) containing dNTP (0.2 mM of each), 1 × PrimeSTAR GXL Buffer, primer (300 nM of each), PrimeSTAR GXL DNA Polymerase (0.25 U; TaKaRa, Japan) and genomic DNA (15 ng). The mixture was denatured (for 2 min at 98 °C) in a thermocycler and then subjected to 32 cycles of amplification (98 °C for 10 sec, 60 °C for 15 sec, and 68 °C for 30 sec). PCR amplification to select DNA-integrated plants was performed with the primers described in Supplementary Table 2. A lipoxygenase gene (LOX2) was analysed as a quantitative control. Half of each PCR product was resolved by agarose gel electrophoresis and visualised by staining with ethidium bromide under UV light.
Cleaved amplified polymorphic sequences (CAPS)
Meristematic tissue was dissected from an embryo 3 days after bombardment. DNA was isolated from the meristematic tissue using 2 µL DNAzol Direct (Cosmo Bio, Japan). DNA from the fifth leaf (of the T0 progeny) and the first leaf (of the T1 progeny) was isolated as described previously16. Each round of PCR was conducted in a reaction mixture (10 µL) containing dNTP (0.2 mM of each), 1 × PCR Buffer for KOD FX Neo, primers (300 nM of each), KOD FX Neo (0.20 U; TOYOBO, Japan) and genomic DNA (0.3 µL for meristematic tissue or 15 ng for leaf tissue). The mixture was denatured for 2 min at 94 °C in a thermocycler and then subjected to 32 cycles for leaf tissue or 35 cycles for meristematic tissue of amplification (98 °C for 10 sec, 68 °C for 30 sec). TaGASR7 genes conserved or specific primer sets were described in Supplementary Table 2. The specific primer sets were designed according to the previous report12. PCR products were digested with the restriction enzyme BcnI and then analysed by agarose gel electrophoresis. The purified PCR products from the enzyme-durable bands were subjected to further sequencing analysis.
Sequencing analysis
PCR products used in the CAPS analysis were cloned into pCR-BluntII-TOPO (Thermo Fisher Scientific, USA) and sequenced with a 3130xL genetic analyser (Applied Biosystems, USA).
Data Availability
All data generated or analysed during this study are included in this published article (and its Supplementary Information files).
References
Miao, J. et al. Targeted mutagenesis in rice using CRISPR-Cas system. Cell Res. 23, 1233–1236 (2013).
Svitashev, S. et al. Targeted Mutagenesis, Precise Gene Editing, and Site-Specific Gene Insertion in Maize Using Cas9 and Guide RNA. Plant Physio. 169, 931–945 (2015).
Shan, Q., Wang, Y., Li, J. & Gao, C. Genome editing in rice and wheat using the CRISPR/Cas system. Nat. Protoc. 9, 2395–2410 (2014).
Woo, J. W. et al. DNA-free genome editing in plants with preassembled CRISPR-Cas9 ribonucleoproteins. Nat. Biotechnol. 33, 1162–1164 (2015).
Svitashev, S., Schwartz, C., Lenderts, B., Young, J. K. & Cigan, A. M. Genome editing in maize directed by CRISPR–Cas9 ribonucleoprotein complexes. Nat. Commun. 7, 13274 (2016).
Liang, Z. et al. Efficient DNA-free genome editing of bread wheat using CRISPR/Cas9 ribonucleoprotein complexes. Nat. Commun. 8, 14261 (2017).
Yasmeen, A. et al. In Planta Transformation of Tomato. Plant Mol. Biol. Rep. 27, 20–28 (2009).
Mu, G. et al. Genetic Transformation of Maize Female Inflorescence Following Floral Dip Method Mediated by Agrobacterium. Biotechnology 11, 178–183 (2009).
Rod-in, W., Sujipuli., K. & Ratanasut, K. The floral-dip method for rice (Oryza sativa)transformation. J. Agric. Technol. 10, 467–474 (2014).
Zale, J. M., Agarwal, S., Loar, S. & Steber, C. M. Evidence for stable transformation of wheat by floral dip in Agrobacterium tumefaciens. Plant Cell Rep. 28, 903–913 (2009).
Hamada, H. et al. An in planta biolistic method for stable wheat. Sci. Rep. 282, 34013 (2017).
Zhang, Y. et al. Efficient and transgene-free genome editing in wheat through transient expression of CRISPR/Cas9 DNA or RNA. Nat. Commun. 7, 12617 (2016).
Ling, H. et al. Draft genome of the wheat A-genome progenitor Triticum urartu. Nature 496, 87–90 (2013).
Clasen, B. M. et al. Improving cold storage and processing traits in potato through targeted gene knockout. Plant Biotechnol. J. 14, 169–76 (2016).
Shan, Q. et al. Targeted genome modification of crop plants using a CRISPR-Cas system. Nat Commun. 31, 686–688 (2013).
Zhu, H., Qu, F. & Zhu, L. H. Isolation of genomic DNAs from plants, fungi and bacteria using benzyl chloride. Nucleic Acids Res. 21, 5279–5280 (1993).
Acknowledgements
We would like to thank Drs. M. Endo, and S. Toki for providing the pE(R4-R3)ZmUbi_OsCas9_ver3 and also thank M. Fukunaga for technical support. This work was supported in part by Cabinet Office, Government of Japan, Cross-ministerial Strategic Innovation Promotion Program (SIP), “Technologies for creating next-generation agriculture, forestry and fisheries” (funding agency: Bio-oriented Technology Research Advancement Institution, NARO).
Author information
Authors and Affiliations
Contributions
H.H., Y.N., R.M., N.T., and R.I. designed the experiments. H.H. and Y.L. performed most of the experiments. H.H. Y.L. and R.I. wrote the manuscript and generated the figures. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hamada, H., Liu, Y., Nagira, Y. et al. Biolistic-delivery-based transient CRISPR/Cas9 expression enables in planta genome editing in wheat. Sci Rep 8, 14422 (2018). https://doi.org/10.1038/s41598-018-32714-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-32714-6
Keywords
This article is cited by
-
A comprehensive review of in planta stable transformation strategies
Plant Methods (2024)
-
Plant regeneration in the new era: from molecular mechanisms to biotechnology applications
Science China Life Sciences (2024)
-
Current approaches and future potential for delivering CRISPR/Cas components in oilseeds and millets
The Nucleus (2024)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
