
Abstract
MiRNAs are part of the epigenetic machinery, and are also epigenetically modified by DNA methylation. MiRNAs regulate expression of different genes, so any alteration in their methylation status may affect their expression. We aimed to identify methylation differences in miRNA encoding genes in breast cancer affecting women under 35 years old (BCVY), in order to identify potential biomarkers in these patients. In Illumina Infinium MethylationEPIC BeadChip samples (metEPICVal), we analysed the methylation of 9,961 CpG site regulators of miRNA-encoding genes present in the array. We identified 193 differentially methylated CpG sites in BCVY (p-value < 0.05 and methylation differences ±0.1) that regulated 83 unique miRNA encoding genes. We validated 10 CpG sites using two independent datasets based on Infinium Human Methylation 450k array. We tested gene expression of miRNAs with differential methylation in BCVY in a meta-analysis using The Cancer Genome Atlas (TCGA), Clariom D and Affymetrix datasets. Five miRNAs (miR-9, miR-124-2, miR-184, miR-551b and miR-196a-1) were differently expressed (FDR p-value < 0.01). Finally, only miR-124-2 shows a significantly different gene expression by quantitative real-time PCR. MiR-124-hypomethylation presents significantly better survival rates for older patients as opposed to the worse prognosis observed in BCVY, identifying it as a potential specific survival biomarker in BCVY.
Similar content being viewed by others

Introduction
Breast cancer has the highest incidence rate of all cancers in women worldwide1. Although early breast cancer generally has an excellent prognosis, breast cancer in young women is associated with a high risk of systemic disease at long-term follow-up2,3,4,5,6,7. Young women tend to be diagnosed at a later stage with highly proliferative, high-grade tumours with the presence of lymphovascular invasion3,4,5,8,9,10,11,12.
Hypomethylation may result in aberrant or inappropriate gene expression that contributes to neoplastic transformation, tumorigenesis or cancer progression (oncogenes)13. In addition, genome-wide loss of methylation contributes to chromosomal instability by destabilizing pericentromeric regions of certain chromosomes14,15,16. Gene-specific hypermethylation typically reflects hypermethylation of CpG-rich regions within gene promoter sequences that lead to gene silencing events17.
MicroRNAs (miRNAs) are short non-coding RNAs that act as important regulators of gene expression as part of the epigenetic machinery. Epigenetic modifications were reported to play an important role in many disease onsets and progressions and can be used to explain several features of complex diseases, such as late onset and fluctuation of symptoms. In addition, miRNAs not only function as part of the epigenetic machinery but can also be epigenetically modified by DNA methylation and histone modifications themselves like any other gene. Methylation regulates the CpG islands on miRNA promoters altering their expression, the histone modifications affecting chromatin structure or changing the affinities for chromatin-associated proteins, thereby modulating gene expression, and therefore, miRNA gene expression. On the other hand, miRNAs can directly target epigenetic factors, such as DNA methyltransferases or histone deacetylases, thus regulating chromatin structure. Moreover, several studies have reported coordinated actions between miRNAs and other epigenetic mechanisms to reinforce the regulation of gene expression18. There is a strong connection between epigenome and miRNome, and any dysregulation of this complex system can result in various physiological and pathological conditions19.
The aim of the current study is to analyse the methylation alterations of CpG associated with miRNA encoding genes in breast cancer tumours in very young women (≤35 years old) and older ones (>50 years old). DNA methylation was analysed using Illumina Infinium HumanMethylation EPIC array20 (EPICarray); this array measures DNA methylation at approximately 850 000 CpG sites across the genome and replace the previous Infinium HumanMethylation450 BeadChip, which analyses 450,000 CpG sites. The EPICarray incorporates CpG sites located in enhancer regions identified by the ENCODE21 and FANTOM522 projects. We hypothesized that methylation differences in miRNA-encoding genes could also represent gene expression differences. This could be a contributing factor in the poorer outcome of tumours in young women.
Results
Methylation differences in CpG probes regulating gene-encoding miRNAs
We analysed data from 6,567 CpG sites regulators of miRNA encoding genes present in the Illumina Infinium MethylationEPIC BeadChip in BC samples from Hospital Clínico Universitario of Valencia (metEPICVal). Wilcoxon rank sum test shown 193 CpG probes that were significantly differentially methylated in BCVY (p-value < 0.05 and methylation differences ±0.1) and that regulated 83 unique miRNA encoding genes. Among them, 90 were hypomethylated and 103 hypermethylated in BCVY (Fig. 1A,B). Significant differentially methylated CpG sites obtained are included in Supplementary Table 1. However, hierarchical clustering showed two principal sample groups: one consisting of BCVY samples and other including BCO, four BCO samples clustered with BCVY patients. Generalized linear model (GLM) showed that methylation differences distinguishing BCVY from BCO were not related to ER status (p-value = 0.06) or molecular subtypes (p-values = 0.65) and no molecular subtype clusters were identified in heatmap representation (Fig. 1A). All data has been analysed again with the addition of five samples from healthy female donors, showing highly similar results (data not shown) proving that differences found are those related to breast cancer affecting young women and not due just produced by age differences.

Differential miRNA methylation study in BCVY vs. BCO from metEPICVal samples. (A) Heatmap representing a supervised cluster centred on the median of the methylation levels at the 193 CpG sites that regulated miRNA genes distinctive in BCVY. Hypermethylated CpG probes in BCVY (red) and hypomethylated probes (green). Samples represented as BCVY (light pink) and BCO samples (purple). Molecular subtypes are indicated as: Her2 (yellow), triple negative (red), luminal A (dark blue) and luminal B (light blue). (B) Volcano-plot representation of methylation for significant CpG regulators of miRNA genes. Hypomethylated probes in BCVY are represented in green colour and hypermethylated probes in BCVY are represented in red. Red lines delimit ±0.1 methylation differences between BCVY vs. BCO and the dotted line represents a p-value threshold of 0.05.
Genomic and functional context of significant CpG sites
In terms of CpG context, significant methylation differences for miRNA CpG probes were localized in islands (p-value = 5.29 × 10−7) and regions away from them called open sea (p-value = 4.73 × 10−5). Specifically, significant CpG probes localized in island regions were mainly hypomethylated in BCVY and those localized in open sea sites were hypermethylated in this age group (Fig. 2A). We analysed the functional localization of differentially methylated miRNA probes and the most important differences were identified in DNasa I hypersensitive sites (DHSs) (p-value = 4 × 10−3), which were generally hypermethylated in BCVY. Although not significant, transcription factor binding sites, promoters and gene body regions presented methylation differences of more than 4% between BCVY and BCO (Fig. 2B).

Genomic and functional context of significant CpG site regulators of genes encoding miRNAs which are differentially methylated in BCVY-metEPICVal samples. Percentage of methylation differences for statistically significant CpG sites from BCVY-BCO comparison according to location of the CpG relative to the island (A) and to the UCSC gene region feature category and regulatory elements (B). Red bars represent hypermethylation in BCVY and green bars represents hypomethylation. *Statistically significant p-values (p < 0.001). N/S: north/south; upstream or downstream to the CpG island.
Methylation Validation in two populations
Both data validation sample sets were analysed using Infinium HumanMethylation450 BeadChip (HM450K), which includes around 50% of probes included in the EPICarray. Of 193 significant differentially methylated miRNA probes identified in the metEPICVal sample set, only 85 were shared with the HM450K array and could be analysed in the validation study, whereas the 108 remaining probes were exclusively for the EPICarray and could not be included in the validation study. We verified the methylation differences for 17 CpG sites between BCVY and BCO in TCGA data (p-value < 0.05 and methylation differences ±0.1). Combined data showed 30 differentially methylated miRNA CpG probes among the 85 analysed in the validation study. Finally, we were able to validate a total of 10 methylation probes regulating miRNAs that were significant in both validation data sets, and all of them were hypomethylated in BCVY compared to older patients (Table 1).
Meta-analysis expression of miRNAs regulated by significant CpG probes
To elucidate whether differentially methylated CpG probes affected miRNA gene expression, we analysed expression of affected miRNA in a meta-analysis using three gene expression data sets (TCGA, Clariom D and Peña-Chilet et al.23). We analysed expression in miRNAs regulated by the 10 validated CpG sites. In addition, we included miRNAs regulated by those 108 CpG probes that were differently methylated in BCVY-BCO comparison and exclusive of the EPICarray, which were therefore excluded from the methylation validation study in the HM450K data sets. Finally, we had a total of 118 CpG probes that regulated 62 unique miRNAs. Gene expression for unique miRNAs was evaluated by t-test in the three data sets separately. Only 10 miRNAs were included in the three studies and could therefore be evaluated in the meta-analysis (Table 2), and 4 of them were significantly de-regulated with adjusted p-value < 0.05 (miR-9-1, miR-184, miR-551b and miR-196a-1).
We also analysed whether we observed different expression in miRNAs regulated by differentially methylated regions. For this purpose, we selected unique miRNAs for each significant region obtained (CpG Island, open sea and DHSs) and their expression was analysed in a meta-analysis using the three previously mentioned data sets. We identified 14 differentially methylated miRNAs regulated by CpG islands regions and 3 of them were also significantly differently expressed between BCVY and BCO (miR-181c, miR-196a-1 y miR-212). Significant open sea regions regulated 55 miRNA genes and miR-184, miR-211 and miR-383 were significantly deregulated in the meta-analysis. Finally, DHSs significant in methylation analysis regulated 38 miRNAs and 5 of them presented different expression (miR-196a-1, miR-184, miR-345, miR-212 and miR-211). Results are shown in Table 3. Interestingly, we found some miRNAs that were regulated by more than one significant CpG categories and were deregulated in BCVY compared with older patients.
Pathway enrichment analysis of significant miRNA
MiRNA genes obtained in the meta-analysis that were regulated by different methylated regions, were related to different pathways reported in Fig. 3. The miRNAs miR-9-1 and miR-196a-1 were involved in pathways related to adherent junctions, proteoglycans involved in cancer or transcriptional misregulation in cancer, among others. MiR-196a-1 took part in multiple pathways, the vast majority of them implicated in other cancer types.

Pathway enrichment analysis results obtained by DIANA mirpath. Plot represents the main pathways in which miRNA regulated by significant CpG probes in BCVY were involved. Dot colour indicates p-values and miRNA count indicates the number of miRNAs involved in the represented pathways.
MiRNAs regulated by CpG probes localized in island regions were involved in multiple pathways related to cancer (transcriptional misregulation in cancer, prostate, lung and endometrial cancer pathway, among others), adherent junctions, oestrogen signalling pathways, etc. For deregulated miRNAs in BCVY whose methylation was regulated by open sea regions, we identified pathways related to cancer as previously observed for island miRNAs and, moreover, they were implicated in tumour-necrosis signalling pathways, extracellular matrix-receptor interaction and prolactin signalling pathways. Finally, for DHSs we obtained, again, most of the pathways previously observed in island regions. Pathway enrichment results by category region are reported in Supplementary Fig. 1.
qRT-PCR Validation of miRNA expression
Validation by quantitative real-time PCR (qRT-PCR) was performed on an independent set of samples with similar characteristics to those used in the metEPICVal study. We analysed the expression of the four significant miRNAs from the meta-analysis (miR-9-1, miR-184, miR-551b and miR-196a-1) and additionally, we analysed the expression of the precursor miR-124-2 that was hypomethylated in both methylation validation data sets but could not be evaluated in the expression study because it was not included in all data sets used in the meta-analysis expression. MiR-124-2 was significantly overexpressed in BCVY samples (p-value < 0.05) compared with older patients and this result correlated with the significant hypomethylation detected in BCVY (Fig. 4A). MiR-9-1 (p-value = 0.07), miR-196a-1 (p-value = 0.36) and miR-184 (p-value = 0.7) were overexpressed in BCVY, which are in agreement with the hypomethylation detected and miR-551b was repressed in BCVY (p-value = 0.07) that was consistent with the hypermethylation found. However, the differences did not reach statistical significance (Supplementary Fig. 2).

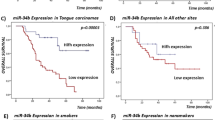
MiR124-2 expression validation by qRT-PCR and overall survival studies. First track represents the chromosome position of the different methylated region. Gene Region track represents genome position of the significant CpG regions obtained. Methylation track shows the β-values for the significant CpG probes regulating miR-124-2. Validation results for miR-124-2 expression using qRT-PCR are plotted in the qRT-PCR Validation track. Boxplots represent the relative mean expression for BCO and BCVY samples, p-values were obtained by Wilcoxon rank sum test (A); Representation of overall survival curves for miR-124-2 according to their methylation status in BCO (B) and BCVY (C). Green curves represent miRNA hypomethylation and red colour line hypermethylation. P-values were obtained by multivariate Cox analysis adjusting for oestrogen receptor status and molecular subtype.
Clinical importance of miRNA methylation as an independent prognostic factor for relapse-free survival and overall survival in BCVY
To test the hypothesis of miRNA-methylation association to relapse-free survival (RFS) and overall survival (OS), we investigated the metEPICVal samples and methylation samples from TCGA that present follow-up data. We next performed a univariate cox regression study to determine whether hypo- or hypermethylation of significant miRNAs were correlated with underlying clinical conditions in BCVY and BCO. Whereas no significance was reached in the RFS analysis, miRNA hypomethylation was related with higher relapse risk in both BC groups (BCO and BCVY) for all miRNAs analysed with the exception of miR-184, in which hypomethylation reduces the risk of relapse in BCVY, contrary to the results for BCO (Supplementary Fig. 3). For OS studies, univariate cox analysis showed that miR-124-2 hypermethylation was significantly related with reducing survival in BCO (p-value = 9 × 10−4) (Fig. 4B). In order to evaluate whether other clinical factors were impacting in the survival in BCO patients apart from miR-124-2 methylation, we performed a multivariable Cox regression analysis including data from oestrogen receptor status and BC molecular subtypes. Multivariate analysis shown a significant relation with miR-124-2-hypermethylation in BCO and poor survival (p-value 1.02 × 10−3, hazard ratio HR = 3.23). However, oestrogen receptor status and molecular subtype were not contributing to survival in BCO patients. Although no significant results were obtained in univariate cox study among BCVY survival and miR-124-2 methylation, we observed a contrary effect on them, in which poorer survival was related with miR-124-2 hypomethylation (Fig. 4C). BCVY sample size is limited compared with older patients and further studies with larger dataset should be addressed to evaluate mir-124-2-hypomethylation and prognosis in this group. Additionally, OS analysis demonstrated a good prognostic factor for miR-184 hypomethylation in both BCVY and BCO groups, and a reduced survival rate for miR-196a-1-hypomethylation in BCVY: however, these results did not reach statistical significance (Supplementary Fig. 4).
Discussion
Several miRNAs have been involved in cancer pathogenesis24. Accordingly, altered miRNA expression profiles have been found in every type of human cancer including colon, brain, lung and breast cancer25,26 suggesting miRNAs as possible biomarkers for early cancer detection. Furthermore, miRNAs not only function as part of the epigenetic machinery, but are also epigenetically modified by DNA methylation themselves. DNA methylation plays a key role in silencing numerous cancer-related genes affecting several processes, and there is considerable evidence supporting the idea that DNA methylation is actively involved in the dysregulation of miRNAs in cancer27.
Breast cancer affecting young women has been previously associated with a more aggressive type of tumours than those diagnose in older women which could be responsible for the poor prognosis that characterizes these tumours. Previous group studies have shown differing methylation signatures for breast cancer in young women than in older patients28. Although the mechanism underlying miRNA dysregulation in cancer is not yet fully understood, recent studies have shown that epigenetic mechanisms play an important role in regulating miRNA expression19.
In this study, we analysed the differences in DNA methylation of miRNA encoding genes between breast cancer samples in young and old women. Results from the methylation study in metEPICVal samples performed in the Illumina Infinium MethylationEPIC BeadChip showed 193 CpG sites that were significantly differentially methylated between groups. Most highlighted methylation differences were localized in open sea and island CpG regions. Specifically, open sea sites were hypermethylated and islands and regions near them were hypomethylated in BCVY. These results are in agreement with our previous global methylation study28 that found hypomethylation localized in CpG island regions and hypermethylation in open sea and regions away from the islands. Relative to that, it has been seen that several gene-encoding miRNAs are more frequently hypermethylated and consequently repressed in regions away from the islands than the CpG island itself27, agreeing with our observations.
In terms of regulatory localization, most significantly hypomethylated probes in BCVY were located on DHSs. These regions of chromatin are sensitive to cleavage by DNase I enzyme, and chromatin has lost its condensed structure, making the DNA more accessible. This remodelled state is essential to increased transcriptional activity. DHSs therefore tended to be enriched on highly expressed genes throughout whole gene regions while not showing significant changes for low and silently expressed genes. Also, DHSs are enriched in regions away from CpG islands, suggesting preference to act within active chromatin domains that present low density CpG islands29. In contrast to the previously mentioned hypermethylation observed in regions away from islands, DHS regions present hypomethylation in BCVY, contributing to the more accessible DNA and consequently upregulating their expression.
In order to validate the results found in the metEPICVal sample, we used data from two previous methylation analysis (TCGA methylation and combined data) performed with HM450K array, with the limitation that we could only analyse 85 CpG probes that were shared between EPICarray and HM450K. The methylation validation study identified 10 CpG significant probes, all of them hypomethylated in young women with breast cancer. These results are in agreement with HM450K array content, given that the number of CpG islands probes is higher than open sea probes, the latter poorly presented in the HM450K array. Based on this we were able to validate some of the hypomethylation probes for BCVY localized in islands regions or near them, using the HM450K validation data sets. However, hypomethylation was concentrated mainly in open sea regions, which were underrepresented in HM450K data sets and could not be validated.
Advances in microarray and sequencing technologies have enabled comprehensive analysis of the epigenome and miRNA expression in cancer cells, which has led to the identification of miRNA which are frequent targets of aberrant DNA methylation in cancer30. In the present study we were able to validate significant methylation differences in a set of differentially methylated miRNAs in cancer such as: miR-181c, miR-129-2, miR-196a-1, miR-137, miR-9-1 and miR-124, of which methylation differences were observed in miR-124-2 and miR-124-3. Unfortunately, some differentially methylated miRNA could not be analysed in the expression meta-analysis because they were not included in the three data sets analysed.
We explored the expression of miRNAs regulated by differently methylated probes in BCVY taking into account their genomic/functional category. Some significant miRNAs were regulated by probes situated in more than one genomic/functional category. Differentially deregulated MiR-196a-1 and miR-212 in BCVY were in turn regulated by differently methylated regions located in islands and DHS sites. Another similar case is miR-211 and miR-184, which were regulated by probes situated in open sea and DHS sites and were deregulated in BCVY. These results suggest an important regulatory role for DNA methylation in the miRNA-encoding genes which are significantly deregulated in BCVY vs. BCO in CpG from different regions.
Next, meta-analysis of expression in miRNAs regulated by validated regulatory CpG probes and new significant probes included in the EPICarray, revealed 4 miRNAs (miR-9-1, miR-184, miR-551b and miR-196a-1) that were significantly differently expressed and methylated in BCVY. Although miR-124-2 expression could not be evaluated in the meta-analysis expression study, its hypomethylation was validated in both methylation data sets and qRT-PCR revealed significant miR-124-2 upregulation in BCVY. However, none of the four miRNAs identified (miR-9-1, miR-184, miR-551b and miR-196a-1) could be validated by qRT-PCR.
Enrichment analysis showed that the significant differentially methylated miRNAs have functions linked to cancer. Most of them were implicated in pathways related with adherent junctions, important in cancer initiation and progression. Furthermore, some miRNAs were involved in extracellular matrix organization through proteoglycans and extracellular matrix-receptor interaction.
MiRNA-methylation has been extensively investigated as a prognostic factor, and survival analysis performed with our data and TCGA revealed that hypermethylation of miR-124-2 was significantly associated with poorer OS in BCO, in contrast to BCVY, in which miR-124-2-hypomethylation presented the lowest survival rates. These results suggest that hypomethylation of miR-124-2 may be a potential prognostic risk factor specific to BCVY. Although no significant results were reached for the rest miRNAs, our study suggested that miR-184-hypomethylation could be a good prognostic factor in breast cancer patients, in which methylation loss increases survival and reduces relapse rates. Conversely, miR-9-1 and miR-196a-1 hypomethylation were associated with reduced survival and higher relapse rates.
Within the human genome, three independent loci (miR-124-1, miR-124-2 and miR-124-3) encode the identical mature miR-124, and all are associated with CpG islands, which have been described as targets of hypermethylation in colon, stomach, liver, leukaemia and cervix cancers31. MiR-124 exerts a tumour suppressor effect by targeting cyclin-dependent kinase 6, and epigenetic silencing of miR-124 leads to CDK6 activation and Rb phosphorylation31,32. Interestingly, our results show a hypomethylation of miR-124-2 gene in BCVY compared with older patients and a higher miRNA expression. Additionally, the miR-124-2 has been found to be the most abundant miRNA expressed in neuronal cells and their differentiation. Neuronal elements have been described that are related to cancer microenvironment promoting cell growth, although the mechanisms mediating neuronal influences on cancer growth and progression are likely incompletely understood33. Previous studies in BCVY have shown that most of the pathways deregulated in this group were related to neuronal-system processes (Oltra SS et al., unpublished data). However, more work is needed to gain insight into the miRNA-124-2-hypomethylation role in breast cancer affecting young women and its potential role as a biomarker of poor survival in BCVY.
Several limitations of the present study need to be acknowledged. Methylation validation analysis has been possible for only 85 of the total 193 significant CpG probes due to the lack of methylation studies performed with the EPICarray. The TCGA dataset only provides data on methylation studies performed with HM450K or previous arrays. In the case of miRNA expression meta-analysis, the three datasets employed for the study do not include all significantly differentially methylated miRNAs. Thus, the miRNAs that were significantly deregulated in some but not all of the three studies were not included in the meta-analysis and information about expression status could not be evaluated; despite obtaining the methylation status of significant CpG regions for BCVY, we could not translate them into differences in gene expression.
The present work is the first to analyse methylation of miRNA-encoding genes using the EPICarray in breast cancer samples from young women, and the association with miRNA expression. Our main finding is the relationship of miR-124-hypomethylation with significantly better survival rates for BCO as opposed to the worse prognosis observed in BCVY, identifying it as a potential specific survival biomarker in this group.
Material and Methods
Methylation and gene expression samples
Illumina Infinium MethylationEPIC BeadChip samples (metEPICVal samples)
Samples included in the Illumina Infinium MethylationEPIC BeadChip experiment were archived in formalin fixed paraffin-embedded and all were stored at the Pathology Department at the Hospital Clínico Universitario, Valencia, Spain. We used 26 samples of BCVY and 15 samples from older women with breast cancer (BCO). Additionally, we have 5 samples from healthy female donors (3 from young and 2 from old women). The clinical characteristics of the patients whose samples were included in the study are shown in Supplementary Table 2. Informed consent was obtained from all subjects and the study was approved by the Hospital Clinico Ethical Committee and all research was performed in accordance with relevant guidelines.
DNA methylation quantification and normalization
Samples were extracted using a commercial kit (QIAamp DNA FFPE Tissue Kit, Qiagen, Hilden, Germany) following the manufacturer’s instructions. DNA samples were quantified (Quant-iT PicoGreen dsDNA Assay, Life Technologies, CA, USA), and assessed for purity by NanoDrop (Thermo Scientific, MA, USA). Samples were checked for suitability for FFPE restoration, as indicated in the Infinium HD FFPE QC Assay (Illumina Inc.); 500 ng of FFPE DNA were bisulphite converted using the EZ-96 DNA Methylation-Gold™ kit (Zymo Research Corp., CA, USA). Bisulfite-converted DNA from FFPE samples was restored following instructions from Infinium assay. Next, DNA was hybridised to the Illumina Infinium MethylationEPIC BeadChip array following the Illumina protocol. BeadChips were washed and scanned using the Illumina HiScan SQ scanner, and the intensities were extracted from GenomeStudio (v.2011.1) and Methylation module (1.9.0) software which normalises within-sample data. Raw microarray data and processed normalized data are available from Gene Expression Omnibus (GEO) (GSE100850). Background subtraction and colour correction for the dye bias seen in Infinium II probes, as well as removal of bad quality samples were performed using minfi package implemented in R Bioconductor34. The β-values (indicating methylation level at each CpG) were calculated using minfi package. A total of 30,271 single nucleotide polymorphism loci were excluded from subsequent analysis. Probes that were not detected in more than 20% of the samples were excluded from the analyses. We selected samples with >98% of probes detected. Additionally, we removed cross reactive probes for the methylation EPICarray described by McCartney et al.35. After pre-processing, we analysed 793,483 CpG sites in 21 samples from BCVY and 13 from BCO.
Analysis of miRNA-associated probes
Our study focused on probes associated with gene encoding for miRNAs. According to the information available in the Illumina annotation file, EPICarray platform includes 9,961 probes that are linked to miRNAs. After the pre-processing described in previous section, we obtained a set of 6,567 CpG probes associated with 1,264 unique miRNAs which were used in the present study.
Analysis of probe distribution by gene-coding miRNAs revealed probes associated with one miRNA and probes associated with multiple miRNAs; the maximum number was for probes regulating 7 miRNAs. Information about correspondence between CpG probes and miRNAs regulated by them are included in Supplementary Table 3. Additionally, miRNAs may be regulated by unique or multiple probes (1–293). Probe count by miRNA is summarized in Supplementary Table 4.
Statistical analysis
We analysed methylation differences using the Wilcoxon Rank Sum test. CpG sites with both p-values < 0.05 and a minimum change of ±0.1 in β-values between BCVY and BCO were considered significant. Additionally, we performed a GLM analysis to assess whether specific methylation differences observed between BCVY and BCO were independent of molecular subtype and ER status. The study procedure diagram is included in Fig. 5.

Workflow diagram of the procedure. The diagram shows the sample set analysed and the significant probes obtained in each step. The table includes the sample and probe size for each sample set. *diff: methylation differences between BCVY minus BCO; sig: significant.
MiRNA probes were classified into different categories according to their description in the Illumina manifest file, depending on function (promoter, gene body, TSS or UTRs), relation to the CpG context (N/S shore, N/S shelf and island), enhancer regions provided by ENCODE and FANTOM5 projects, and ENCODE regulatory elements (CpG sites in transcription factor binding sites [TFBS], open chromatin regions and DHSs). Methylation differences by categories in the significant probe set were assessed by Welch’s t-test and p-values < 0.001 were considered to be statistically significant.
Univariate Cox analysis was used to analyse the effect of clinical variables and miRNA methylation on patients’ relapse-free survival and overall survival. Multivariate Cox method was performed to adjust for clinical variables such as oestrogen receptor status and molecular subtype that could be influencing in relapse and/or survival.
Methylation Validation in two populations
We performed a combined study using methylation data from Flower et al.36 analysed by Infinium HumanMethylation450 BeadChip and data from the metEPICVal study previously described. First, we analysed HM450K array samples according to the methods described in the data pre-processing section. We retained methylation levels for 405,068 probes present in both metEPICVal and Flower et al.’s36 HM450K study in a total of 64 samples (32 BCO samples, 32 from BCVY) that were used in the validation study.
Additionally, we used methylation data from independent breast tissue samples available from TCGA, that includes data for 485,577 probes in 720 BCO and 27 BCVY samples. Gene expression study for TCGA data was done with 50 permutations, and 50 samples were randomly selected and balanced by subtype.
Both studies were performed using the Illumina HM450K array, which includes only half of the probes present in the EPICarray. To validate our results from the BCVY-BCO study we selected differentially methylated miRNA probes that were included in HM450K and a Wilcoxon Rank Sum test was performed between BCVY and BCO tumour samples.
miRNA expression meta-analysis
We used our own gene expression data from 42 breast cancer patients (13 from BCO and 27 from BCVY) analysed by Clariom D array (Applied Biosystems by Life Technologies, Carlsbad, California, USA)37. Total RNA was isolated using RecoverAll Total Nucleic Acid Isolation Kit (Applied Biosystems by Life Technologies, Carlsbad, California, USA) following the manufacturer’s protocol. RNA concentration was measured by Qubit® 3.0 (InvitrogenTM by Termo Fisher Scientific, Waltham, MA, USA) using the Qubit® RNA HS Assay Kit (Molecular Probes® by Life Technologies Carlsbad, California, USA). RNA quality was assessed by qRT-PCR; 25 ng of RNA were hybridized in the Clariom D array. Raw files (*.CEL) were normalized by Robust Multichip Average method using from the R Bioconductor affy package.
Expression of miRNAs regulated by differentially methylated probes were analysed using data from Clariom D samples. This study includes expression for 1,217 miRNA precursors. Additionally, in the meta-analysis we included previous miRNA expression published data from Peña-Chilet et al.23, which were analysed by GeneChip® miRNA 2.0 Array (Affymetrix, Santa Clara,CA, USA) with GEO accession number GSE48088. This data set includes 21 BCVY and 12 BCO samples. Additionally, we downloaded gene expression from TCGA and we used data from 39 BCVY and 50 BCO samples, randomly selected and balanced by subtype.
We performed a meta-analysis study to combine p-values from three studies by Fisher method using the metap R package; p-values were adjusted by Benjamini-Hochberg FDR procedure38; genes with FDR <0.01 and gene expression differences ±0.1 were considered statistically significant.
Pathway enrichment analysis
DIANA miRPath pathway enrichment analysis was used to gain insight into global molecular networks and canonical pathways related to differentially expressed miRNAs (http://diana.imis.athena-innovation.gr/DianaTools/index.php?r=mirpath/index). The software performs an enrichment analysis of multiple miRNA target genes comparing each set of miRNA targets to all known KEGG pathways. Pathways showing a FDR p-value < 0.05 were considered significantly enriched between classes under comparison.
miRNA expression validation by quantitative real-time PCR
Validation of significant miRNAs was performed in a different sample set composed of 27 samples from BCVY and 13 from BCO using qRT-PCR (clinical characteristics in Supplementary Table 2) using Advance TaqMan Gene Expression Assays and TaqMan microRNA Assays (Applied Biosystems by Life Technologies, Carlsbad, California, USA) and normalising to hsa-mir-21 or RNU43 and RNU6B expression. The data were managed using the QuantiStudio Design & Analysis software (v1.4). Relative expression was calculated by using the comparative Ct method and obtaining the fold-change value (ΔΔCt). The Wilcoxon rank sum test was used for non-parametric samples, p-value < 0.05 were considered statistically significant.
References
Ferlay, J. et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. International journal of cancer 136, E359–386, https://doi.org/10.1002/ijc.29210 (2015).
Azim, H. A. et al. Elucidating prognosis and biology of breast cancer arising in young women using gene expression profiling. Clinical cancer research: an official journal of the American Association for Cancer Research 18, 1341–1351, https://doi.org/10.1158/1078-0432.CCR-11-2599 (2012).
Fredholm, H. et al. Breast cancer in young women: poor survival despite intensive treatment. PloS one 4, e7695, https://doi.org/10.1371/journal.pone.0007695 (2009).
Fredholm, H. et al. Long-term outcome in young women with breast cancer: a population-based study. Breast cancer research and treatment 160, 131–143, https://doi.org/10.1007/s10549-016-3983-9 (2016).
Gnerlich, J. L. et al. Elevated breast cancer mortality in women younger than age 40 years compared with older women is attributed to poorer survival in early-stage disease. Journal of the American College of Surgeons 208, 341–347, https://doi.org/10.1016/j.jamcollsurg.2008.12.001 (2009).
Kroman, N. et al. Factors influencing the effect of age on prognosis in breast cancer: population based study. Bmj 320, 474–478 (2000).
Partridge, A. H. et al. Subtype-Dependent Relationship Between Young Age at Diagnosis and Breast Cancer Survival. Journal of clinical oncology: official journal of the American Society of Clinical Oncology 34, 3308–3314, https://doi.org/10.1200/JCO.2015.65.8013 (2016).
Anders, C. K. et al. Young age at diagnosis correlates with worse prognosis and defines a subset of breast cancers with shared patterns of gene expression. Journal of clinical oncology: official journal of the American Society of Clinical Oncology 26, 3324–3330, https://doi.org/10.1200/JCO.2007.14.2471 (2008).
Bharat, A., Aft, R. L., Gao, F. & Margenthaler, J. A. Patient and tumor characteristics associated with increased mortality in young women (<or = 40 years) with breast cancer. Journal of surgical oncology 100, 248–251, https://doi.org/10.1002/jso.21268 (2009).
Colleoni, M. et al. Very young women (<35 years) with operable breast cancer: features of disease at presentation. Annals of oncology: official journal of the European Society for. Medical Oncology 13, 273–279 (2002).
Sabiani, L. et al. Breast cancer in young women: Pathologic features and molecular phenotype. Breast 29, 109–116, https://doi.org/10.1016/j.breast.2016.07.007 (2016).
Swain, S. M., Nunes, R., Yoshizawa, C., Rothney, M. & Sing, A. P. Quantitative Gene Expression by Recurrence Score in ER-Positive Breast Cancer, by Age. Advances in therapy 32, 1222–1236, https://doi.org/10.1007/s12325-015-0268-3 (2015).
Feinberg, A. P. & Vogelstein, B. Hypomethylation of ras oncogenes in primary human cancers. Biochemical and biophysical research communications 111, 47–54 (1983).
Eden, A., Gaudet, F., Waghmare, A. & Jaenisch, R. Chromosomal instability and tumors promoted by DNA hypomethylation. Science 300, 455, https://doi.org/10.1126/science.1083557 (2003).
Gaudet, F. et al. Induction of tumors in mice by genomic hypomethylation. Science 300, 489–492, https://doi.org/10.1126/science.1083558 (2003).
Narayan, A. et al. Hypomethylation of pericentromeric DNA in breast adenocarcinomas. International journal of cancer 77, 833–838 (1998).
Herman, J. G. & Baylin, S. B. Gene silencing in cancer in association with promoter hypermethylation. The New England journal of medicine 349, 2042–2054, https://doi.org/10.1056/NEJMra023075 (2003).
Bannister, A. J. & Kouzarides, T. Regulation of chromatin by histone modifications. Cell research 21, 381–395, https://doi.org/10.1038/cr.2011.22 (2011).
Piletic, K. & Kunej, T. MicroRNA epigenetic signatures in human disease. Archives of toxicology 90, 2405–2419, https://doi.org/10.1007/s00204-016-1815-7 (2016).
http://support.illumina.com. Infinium MethylationEPIC BeadChip Kit Support.
Consortium, E. P. An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74, https://doi.org/10.1038/nature11247 (2012).
Lizio, M. et al. Gateways to the FANTOM5 promoter level mammalian expression atlas. Genome biology 16, 22, https://doi.org/10.1186/s13059-014-0560-6 (2015).
Pena-Chilet, M. et al. MicroRNA profile in very young women with breast cancer. BMC cancer 14, 529, https://doi.org/10.1186/1471-2407-14-529 (2014).
Calin, G. A. et al. Frequent deletions and down-regulation of micro- RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proceedings of the National Academy of Sciences of the United States of America 99, 15524–15529, https://doi.org/10.1073/pnas.242606799 (2002).
Baffa, R. et al. MicroRNA expression profiling of human metastatic cancers identifies cancer gene targets. The Journal of pathology 219, 214–221, https://doi.org/10.1002/path.2586 (2009).
Di Leva, G., Briskin, D. & Croce, C. M. MicroRNA in cancer: new hopes for antineoplastic chemotherapy. Upsala journal of medical sciences 117, 202–216, https://doi.org/10.3109/03009734.2012.660551 (2012).
Suzuki, H., Maruyama, R., Yamamoto, E. & Kai, M. DNA methylation and microRNA dysregulation in cancer. Molecular oncology 6, 567–578, https://doi.org/10.1016/j.molonc.2012.07.007 (2012).
Oltra, S. S. et al. Increased DNA methylation age acceleration in breast cancer tumours from very young women. Breast cancer research and treatment In revision (2018).
Cockerill, P. N. Structure and function of active chromatin and DNase I hypersensitive sites. The FEBS journal 278, 2182–2210, https://doi.org/10.1111/j.1742-4658.2011.08128.x (2011).
Kaur, S., Lotsari-Salomaa, J. E., Seppanen-Kaijansinkko, R. & Peltomaki, P. MicroRNA Methylation in Colorectal Cancer. Advances in experimental medicine and biology 937, 109–122, https://doi.org/10.1007/978-3-319-42059-2_6 (2016).
Lujambio, A. et al. Genetic unmasking of an epigenetically silenced microRNA in human cancer cells. Cancer research 67, 1424–1429, https://doi.org/10.1158/0008-5472.CAN-06-4218 (2007).
Agirre, X. et al. Epigenetic silencing of the tumor suppressor microRNA Hsa-miR-124a regulates CDK6 expression and confers a poor prognosis in acute lymphoblastic leukemia. Cancer research 69, 4443–4453, https://doi.org/10.1158/0008-5472.CAN-08-4025 (2009).
Venkatesh, H. & Monje, M. Neuronal Activity in Ontogeny and Oncology. Trends in Cancer 3, 89–112, https://doi.org/10.1016/j.trecan.2016.12.008.
Aryee, M. J. et al. Minfi: a flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics 30, 1363–1369, https://doi.org/10.1093/bioinformatics/btu049 (2014).
McCartney, D. L. et al. Identification of polymorphic and off-target probe binding sites on the Illumina Infinium MethylationEPIC BeadChip. Genomics data 9, 22–24, https://doi.org/10.1016/j.gdata.2016.05.012 (2016).
Flower, K. J. et al. DNA methylation profiling to assess pathogenicity of BRCA1 unclassified variants in breast cancer. Epigenetics 10, 1121–1132, https://doi.org/10.1080/15592294.2015.1111504 (2015).
Vidal-Tomas, V. et al. Global transcriptome deregulation of Breast Cancer in Very Young Women samples. Annals of Oncology 28, 7, https://doi.org/10.1093/annonc/mdx361 (2017).
Benjamini, Y. & Y., H. Controlling the false discovery rate: a practical and powerful approach to multiple testing. Journal of the Royal Statistical Society 57, 289–300 (1995).
Acknowledgements
We would like to thank all the patients and volunteers who gave their consent for inclusion in this study, the medical staff in the Medical Oncology Unit at the INCLIVA Biomedical Research Institute in Valencia (Spain) for collecting the samples and providing the material for all the analyses, and the expert personnel in the Genotyping and Epigenetics Laboratory at the Central Biomedical Research Unit (UCIM) in the University of Valencia. We would like to thank the thorough English revision by an expert colleague. This study was supported by grants from The Ministry of Economy and Competitiveness and the Carlos III Health Institute (PI13/00606) and FEDER. SSO is funded on a FPU pre-doctoral fellowship (FPU13/04976) from MECD, (Spanish Government); G.R. is funded on a Miguel Servet II contract (CPII14-00013) from the Carlos III Health Institute; M.P.C. is funded by the private patients Foundation LeCado. CIBERONC (CB16/12/00481-CB16/12/00473) is an initiative of the Carlos III Health Institute. J.M.F. and K.J.F. acknowledge funding support from Breast Cancer Now, the Cancer Research UK (CRUK) Centre, the Experimental Cancer Medicine Centre (ECMC), and the National Institute for Health Research (NIHR), Biomedical Research Centre (BRC) based at Imperial College Healthcare NHS Trust and Imperial College London.
Author information
Authors and Affiliations
Contributions
G.R. and A.L. conceived the study. G.R. and M.P. supervised the study. G.R. and J.F. coordinate the work. J.F. and K.F. contributed in the statistical analysis of Illumina methylation data. G.R. and S.O. designed all aspects of the research. S.O. collected the data and performed computational analysis. E.A. and O.B. contribute in the pathology aspects. A.L. and M.M. contributed in the clinical patient data collection. V.V.T. contributed in the gene expression study by Clariom D array. S.O. and G.R. wrote the manuscript. All authors revised, read, and approved the final manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Oltra, S.S., Peña-Chilet, M., Vidal-Tomas, V. et al. Methylation deregulation of miRNA promoters identifies miR124-2 as a survival biomarker in Breast Cancer in very young women. Sci Rep 8, 14373 (2018). https://doi.org/10.1038/s41598-018-32393-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-32393-3
Keywords
This article is cited by
-
A semi-supervised approach for the integration of multi-omics data based on transformer multi-head self-attention mechanism and graph convolutional networks
BMC Genomics (2024)
-
Genome-wide methylation analyses identifies Non-coding RNA genes dysregulated in breast tumours that metastasise to the brain
Scientific Reports (2022)
-
Review article epithelial to mesenchymal transition‑associated microRNAs in breast cancer
Molecular Biology Reports (2022)
-
Exosomal microRNA-551b-3p from bone marrow-derived mesenchymal stromal cells inhibits breast cancer progression via regulating TRIM31/Akt signaling
Human Cell (2022)
-
MiR-214 inhibits apoptosis in thyroid epithelial follicular cells induced by amiodarone through the FASL/MAPK pathway
Molecular & Cellular Toxicology (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
