
Abstract
This study investigated the effect of supplementing the diet of calves with two direct fed microbials (DFMs) (Saccharomyces cerevisiae boulardii CNCM I-1079 (SCB) and Lactobacillus acidophilus BT1386 (LA)), and an antibiotic growth promoter (ATB). Thirty-two dairy calves were fed a control diet (CTL) supplemented with SCB or LA or ATB for 96 days. On day 33 (pre-weaning, n = 16) and day 96 (post-weaning, n = 16), digesta from the rumen, ileum, and colon, and mucosa from the ileum and colon were collected. The bacterial diversity and composition of the gastrointestinal tract (GIT) of pre- and post-weaned calves were characterized by sequencing the V3-V4 region of the bacterial 16S rRNA gene. The DFMs had significant impact on bacteria community structure with most changes associated with treatment occurring in the pre-weaning period and mostly in the ileum but less impact on bacteria diversity. Both SCB and LA significantly reduced the potential pathogenic bacteria genera, Streptococcus and Tyzzerella_4 (FDR ≤ 8.49E-06) and increased the beneficial bacteria, Fibrobacter (FDR ≤ 5.55E-04) compared to control. Other potential beneficial bacteria, including Rumminococcaceae UCG 005, Roseburia and Olsenella, were only increased (FDR ≤ 1.30E-02) by SCB treatment compared to control. Furthermore, the pathogenic bacterium, Peptoclostridium, was reduced (FDR = 1.58E-02) by SCB only while LA reduced (FDR = 1.74E-05) Ruminococcus_2. Functional prediction analysis suggested that both DFMs impacted (p < 0.05) pathways such as cell cycle, bile secretion, proteasome, cAMP signaling pathway, thyroid hormone synthesis pathway and dopaminergic synapse pathway. Compared to the DFMs, ATB had similar impact on bacterial diversity in all GIT sites but greater impact on the bacterial composition of the ileum. Overall, this study provides an insight on the bacteria genera impacted by DFMs and the potential mechanisms by which DFMs affect the GIT microbiota and may therefore facilitate development of DFMs as alternatives to ATB use in dairy calf management.
Similar content being viewed by others


Introduction
The microbiota composition of the gastrointestinal tract (GIT) influences the health outcome of animals as well as their productivity1,2. The diversity and composition of the GIT microbiota can be influenced by many factors including age, diet, feeding method (management), and feed additives3,4. In particular, diet plays pivotal roles on the composition of the GIT microbiota5,6,7. Furthermore, diet and the weaning process affect the development of the GIT and microbial colonization in calves during the early period of growth8,9. Calf GIT is rapidly colonized by the maternal and environmental microorganisms during and after birth4,10. Consequently, exposure to beneficial microbes in the early period of growth will have relevant roles in health outcome11. It has been shown that diet and feeding management can be used to manipulate the rumen microbiota in ruminants with long lasting effects12. Likewise, microbial colonization and subsequent fermentation processes in the rumen during the early period of growth was influenced by feeding (natural or artificial) practice13.
Direct fed microbials (DFMs) have been shown to provide health benefits to the host mainly by modulating the GIT microbiota in cattle or other ruminants, and humans2,14,15. By modifying the composition of the GIT microbiota, DFMs may contribute to optimize beneficial functions of GIT microbial communities such as digestion, production of vitamin K, promotion and development of the immune system, and detoxification of harmful chemicals resulting in improvement of GIT health16. While the diversity, composition, and complexity of calves GIT microbiota has been mostly derived from the analyses of fecal17,18,19 and rumen microbiota20,21, few studies have characterized the diversity and community composition in the different sections of the GIT of 5 years old cows and 10 months old sheep22,23.
Manipulating the microbiota of the GIT through supplementation with DFMs is an attractive approach to improve and maintain animal health24,25. DFMs including Saccharomyces cerevisiae and Lactobacillus acidophilus are naturally occurring microorganisms in the GIT26,27. Introducing Saccharomyces cerevisiae boulardii CNCM I-1079 (SCB) and Lactobacillus acidophilus BT1386 (LA) soon after birth could provide beneficial impact in the establishment of the GIT microbiota. An increase in the potentially beneficial phylum, Actinobacteria, and genera, Bifidobacterium and Collinsella, in the cecum and colon of yeast supplemented piglets28 has been observed. Also, Lactobacillus spp. and Bifidobacteruim spp. were increased following treatment with several Lactobacillus species in a simulator of human intestinal microbial ecosystem29. Furthermore, SCB significantly improved the growth of total lactobacilli in the GIT especially around the weaning period and improved colon morphology30. Our hypothesis was that supplementation of calf’s diet with SCB and LA will increase the colonisation and establishment of beneficial bacteria in the different GIT sites.
Therefore, the present study investigated the effect of feeding SCB and LA on the colonisation and development of the GIT microbiota, their effects on the composition of bacterial populations in different GIT sites and their potential mechanisms of action during the early period of calf’s growth.
Results
Data acquisition
A total of 8,824,437 sequences of the 16S rRNA genes were generated from amplicon sequencing of 159 samples representing rumen (RuD), ileum (IlD) and colon (CoD) digesta and ileum (IlM) and colon (CoM) mucosa of 16 calves on day 33 (pre-weaning) and another 16 on day 96 (post-weaning) for a total of 4 calves per treatment (Control (CTL), SCB, LA, and an antibiotic growth promoter (ATB)). The mean number of sequences was 55,494.00 ± 1,969.00 per sample. A random sub-sample of sequences per sample were utilised for the normalisation of sequence numbers for other analyses. The sequencing depth was sufficient to cover each microbial community as shown on the rarefaction curves for each sample (Fig. S1). Overall, a total of 23 different phyla with 428 genera, 131 families, 81 order and 41 classes were detected (Fig. 1, Table S1a–e).

Distribution of 159 samples with complete 16S rRNA gene sequences of bacteria phylum and genera.
Bacterial diversity across treatments in GIT sites at pre-weaning (day 33) and post-weaning (day 96)
A pairwise comparison of treatments was done within each GIT site on day 33 (pre-weaning) and day 96 (post-weaning) separately. The results of alpha diversity indices are shown in Table 1. In the pre-weaning period, ATB had bacterial communities with a tendency for a greater Shannon diversity index (p = 0.06) compared to CTL in IlM (Table 1). On the contrary, animals supplemented with ATB had bacterial communities with lower (p < 0.01) Shannon diversity index compared to that of CTL in RuD (Table 1). Moreover, SCB treatment had greater (p < 0.05) Simpson diversity index compared to ATB in CoM and greater bacterial richness (Chao1, p < 0.05) compared to ATB in CoD (Table 1). Meanwhile, LA had greater (p < 0.01) Shannon and Simpson diversity indices compared to ATB in CoM.
In the post-weaning period, LA treatment had bacterial communities with greater (p < 0.01) Shannon, Simpson and InvSimpson diversity indices compared to ATB in CoD. SCB had bacterial communities with greater Simpson (p < 0.05) diversity index compared to CTL in RuD (Table 1).
For beta diversity, dissimilarities were mostly observed between periods, i.e. pre-weaning vs. post-weaning, as shown by the clustering pattern of the principal coordinate analysis (PCoA) plots at the different GIT sites (Fig. 2a–e). There was no dissimilarity (p = 0.512) in bacterial communities between treatments in RuD but a tendency (p = 0.09) was observed in IlM (Fig. 2a and c). However, there was a clear difference (p < 0.01) between all treatments in the pre-weaning period compared to the post-weaning period in IlD (Fig. 2b), CoD (Fig. 2d) and CoM (Fig. 2e).

Principal coordinate analysis (PCoA) plots for treatment effect on each site at pre- and post-weaning periods. (a) Principal coordinate analysis (PCoA) plots for treatment effect on Rumen digesta at pre-weaning and post-weaning. (b) Principal coordinate analysis (PCoA) plots for treatment effect on ileum mucosa at pre-weaning and post-weaning. (c) Principal coordinate analysis (PCoA) plots for treatment effect on ileum digesta at pre-weaning and post-weaning. (d) Principal coordinate analysis (PCoA) plots for treatment effect on colon digesta at pre-weaning and post-weaning. (e) Principal coordinate analysis (PCoA) plots for treatment effect on colon mucosa at pre-weaning and post-weaning. Distances between the samples are based on similarity in OTU composition (OTU similarity 97%). A greater distance implies lower similarity, whereas similar OTUs will cluster together. The clustering pattern of the bacterial communities were tested using PERMANOVA and (P < 0.05) were considered significant.
Bacterial composition and differential abundance across treatments in GIT sites at pre-weaning and post-weaning periods
The most abundant phyla in all treatments (SCB, LA, ATB and CTL) at all GIT sites were either Firmicutes or Bacteriodetes at both pre- and post-weaning periods. However, Proteobacteria was the most abundant (33.31%) phylum in IlM for calves fed LA in the pre-weaning period (Fig. 3).

Stack bar charts of phylum level bacterial composition for the treatment effect on each site at pre- and post-weaning periods. CoM = colon mucosa, CoD = colon digesta, IM = ileum mucosa, IlD = ileum digesta, RuD = rumen digesta.
At the pre-weaning period, the most abundant genera for all treatments were Blautia, Lactobacillus and Prevotella_1 in CoD (17.1–21.9%), IlD (43.1–66.7%) and RuD (19.5–40.7%), respectively (Table S2). While the most abundant genera were Bacteriodetes for ATB (22.5%) and LA (14.3%), Streptococcus for CTL (16.7%) and Faecalibacteria for SCB (13.2%) in CoM (Fig. 4). The most abundant genera were Megamonas for CTL (30%) and ATB (31%), Escherichia Shigella for LA (30.7%) and Chlamydophilia for SCB (32.7%) in IlM (Fig. 4).

Stack bar charts of genus level bacterial composition for the treatment effect on each site at pre- and post-weaning periods. CoM = colon mucosa, CoD = colon digesta, IlM = ileum mucosa, IlD = ileum digesta, RuD = rumen digesta.
At the post-weaning period, Ruminococcaceae_UCG-005 was the most abundant genus in all treatments (13.2–47.5%) in CoD and CoM while Atopobium was the most abundant genus for both LA (28.8%) and CTL (17.5%) treatments and Intestinibacter for both ATB (20.9%) and SCB (13.6%) treatments in IlD. Candidatus_Arthromitus was the most dominant genus for both LA (28.60%) and SCB (19.9%) treatments while Bifidobacterium was the most abundant genus for CTL (14%) and Ruminococcus_gauvreauii_group for ATB (12.9%) in IlM (Fig. 4). Prevotella_1 was the most abundant genus for all treatments (24.9–38.1%) in RuD.
Significant differential abundant (DA) genera between treatments (SCB, LA and ATB) and CTL in the pre- and post-weaning periods are shown in Tables 2, 3 and 4, respectively. The numbers of DA genera and common genera between the three pairwise comparisons are also shown in Fig. 5 for pre- and post-weaning periods. At the pre-weaning period, SCB significantly reduced the abundance of Streptococcus (FDR = 8.49E-06) and Prevotella_7 (FDR = 1.49E-02) in CoM but increased (FDR = 1.30E-02) the abundance of Ruminococcaceae_UCG-005 in CoD compared to CTL (Table 2). SCB treatment also significantly changed the relative abundance of 42 and two genera in IlM and IlD, respectively, but had no impact on the relative abundance of genera in RuD at the pre-weaning period. In IlM, the genera Tyzzerella_4 (FDR = 4.27E-09) and Ruminococcaceae_UCG-008 (FDR = 2.38E-04) had the highest log fold change reduction, while Fibrobacter (FDR = 5.5E-04) and Roseburia (FDR = 7.01E-04) had the highest log fold change increase by SCB compared to CTL. In IlD, Ruminiclostridium_5 and Christensenellaceae_R-7 genera were two genera significantly reduced (FDR = 2.52E-02) by SCB compared to CTL in the pre-weaning period.

The common and specific genera in the (a) pre-weaning and (b) post-weaning periods for the different treatments.
In the post weaning period, SCB significantly reduced the abundance of Ruminococcaceae_UCG-008 in RuD (FDR = 1.32E-02) but increased (FDR = 2.24E-02) the relative abundance of four genera (Prevotella_1, Actinomycetes, Streptococcus and Rothia) in IlM compared to CTL. Genera relative abundance in other sites was not affected by SCB in the post-weaning period (Table 2).
In the pre-weaning period, no genus was significantly affected by LA treatment in the RuD, llD and CoD compared to CTL, but three and 18 genera were significantly affected in CoM and IlM respectively. In llM, Tyzzerella_4, Ruminococcaceae_UCG-008 and Lachnoclostridium were the top three genera significantly reduced (FDR ≤ 1.67E-06) while Fibrobacter was significantly increased (FDR = 3.09E-02) by LA treatment compared to CTL (Table 3). In the post-weaning period, LA treatment impacted only the IlD, by reducing (FDR ≤ 2.46E-02) the relative abundance of six genera (Ruminococcus_2, Lactobacillus, Ruminiclostridium_9, Prevotella_1, Acetitomaculum and Ruminococcaceae_NKA214_group (Table 3).
The ATB treatment had greater impact on genera relative abundance in llD and RuD at the pre-weaning period and in IlM at the post-weaning period (Table 4). ATB changed (FDR ≤ 9.08E-03) the relative abundance of 34 and 24 genera in IlD and RuD in the pre-weaning period and 16 genera in IlM. Streptococcus was significantly reduced (FDR = 5.97E-03) by ATB treatment in CoM at the pre-weaning period. In the post-weaning period, Dorea (FDR = 2.74E-03) and Anaerovibrio (FDR = 5.15E-03) were significantly increased by ATB (Table 4).
Comparisons between LA vs. ATB, SCB vs. ATB and SCB vs. LA are shown in Tables 5, 6 and S3. A total of 43 and 135 genera were significantly DA between LA vs. ATB (Table 5) and SCB vs. ATB (Table 6), respectively. Most DA genera for both pairwise comparisons were found in the pre-weaning period (40/43 for LA vs. ATB and 113/135 for SCB vs. ATB) as well as in the ileum (mucosa and digesta) (Tables 5 and 6). Tyzzerella 4 (FDR = 4.42E-11) and Ruminococcaceae_UCG-005 (FDR = 8.45E-07) were the most significant DA genera between SCB vs. ATB in the pre- and post-weaning period, respectively (Table 5). Tyzzerella 4 was also the most significant DA genus in the pre-weaning period when comparing LA vs. ATB (FDR = 7.91E-10) (Table 6).
Several genera were also found to be significantly DA between the two DFMs, and among them Ruminobacter (FDR = 1.72E-03) and Lachnospiraceae_UCG-008 (FDR = 3.71E-02) were the most significantly DA in pre- and post-weaning periods, respectively. Ruminobacter, Moryella, Acetitomaculum and Prevotellaceae UCG-001 were significantly reduced (FDR ≤ 7.96E-03) by SCB compared to LA (Table S3a).
Predicted pathways of the relative changes due to treatments
To investigate the potential molecular pathways by which the microbiota adapted to treatments, we performed metagenomics contribution of the communities observed and differential analyses of predicted pathways between control and treatments for each site in pre- and post-weaning periods using the Kyoto Encyclopedia of Genes and Genomes (KEGG) database. A total of 6,205 KEGG orthologies (Table S4a) were predicted for all samples and assigned into 261 KEGG pathways (Table S4b). Metabolic pathway, biosynthesis of amino acids, ribosome, carbon metabolism and purine metabolism were the top 5 predicted pathways by relative abundance values for all GIT sites in both pre- and post-weaning periods (Table S4c). ECM-receptor interaction and AGE-RAGE signaling pathway in diabetic complications were only predicted for RuD, while Fc epsilon RI signaling pathway was uniquely predicted for IlD (Table S4c). Several pathways such as endocrine resistance, spliceosome, rap1 signaling, gap junction, and cytosolic DNA-sensing pathway were also uniquely predicted for CoM (Table S4c). The changes in abundance values for predicted pathways varied between treatments, site and day.
At the pre-weaning period, the SCB treatment significantly (p < 0.05) influenced 6 pathways (cell cycle, EGFR tyrosine kinase inhibitor resistance, bile secretion, Fanconi anemia pathway, mRNA surveillance pathway and oxytocin signaling pathway) in IlM and 5 pathways (caffeine metabolism, cAMP signaling pathway, steroid biosynthesis, proteasome and dopaminergic synapse) in RuD but had no impact on other GIT sites (Table 7) compared to CTL treatment. The LA treatment significantly (p < 0.05) impacted 4 pathways (caffeine metabolism, cAMP signaling pathway, steroid biosynthesis, proteasome and dopaminergic synapse) in RuD only, compared to CTL. The ATB treatment had diverse effects including significant (p < 0.05) changes to steroid hormone biosynthesis pathway in CoM, bile secretion and caffeine metabolism in IlM and cAMP signaling pathway, steroid biosynthesis and proteasome pathways in RuD compared to CTL (Table 7).
At the post-weaning period, 5, 7 and 9 pathways were significantly (p < 0.05) changed by SCB compared to control in CoM, IlM and RuD, respectively (Table 7). The most significantly changed pathways by SCB during this period were caffeine metabolism (p < 1.72E-05), RIG-I-like receptor signaling pathway (p < 5.57E-05) and thyroid hormone signaling pathway (p < 7.57E-07) in CoM, IlM and RuD, respectively. Meanwhile, LA impacted the mucosa (IlM and CoM) only as it changed the abundance levels of caffeine metabolism (p < 7.13E-04) in CoM and of cell cycle (p < 2.64E-04) in IlM, EGFR tyrosine kinase inhibitor resistance, oxytocin signaling pathway, mRNA surveillance pathway and Fanconi anemia pathway (p ≤ 1.64E-03) in IlM. The ATB treatment significantly changed (p ≤ 5.63E-04) the abundance of thyroid hormone signaling pathway and ether lipid metabolism in IlD, cAMP signaling pathway in CoM and RIG-I-like receptor signaling pathway, D-arginine and D-ornithine metabolism and Butanoate metabolism in IlM.
Discussion
Overall, the phylum Firmicutes was the most abundant in all GIT sites except the RuD where Bacteroidetes was the most dominant. Our results are supported by earlier reports of high relative abundance of Firmicutes in the GIT of pre-weaned Holstein calves3 or of Brazilian Nelore steer31. It is well documented that the bacterial community diversity pattern and composition differ across GIT sites31,32. In the current study, each GIT site was host to different bacteria community structures. In fact, we observed that CoM harboured a greater bacterial community diversity compared to other GIT sites. The colon is considered a fermentation tank for microbial fermentation of indigestible dietary substrates and the digesta is retained in the colon (large intestine) for a longer time compared to the small intestine (ileum), the colon being the hub of a more complex bacterial community33. In the colon, dietary fiber that escaped digestion in the upper digestive tract are broken down into short chain fatty acids and, the increased availability of short chain fatty acids promotes the growth of some bacterial in the lower GIT sites. Therefore, the increased bacteria growth is expected to account for the richness of bacteria in the colon34. The IlD had the lowest diversity compared to all other GIT sites. Peristaltic movements ensure a relatively short passage time through the ileum (3–5 h) by pushing the microbiota migration towards the large intestine, hence limited time for microorganisms to replicate and increase in numbers35 in IlD compared with other GIT sites investigated. Mucosa-associated microorganisms live in close contact with host cells; hence they execute different functions within the GIT compared to digesta microorganisms. This might account for the differences in diversity and composition of the ileum mucosa and digesta as seen in the current study.
As expected, alpha diversity measures were higher for post-weaning compared to pre-weaning. Likewise bacterial community composition was different in the post-weaning period as compared to the pre-weaning period in this study. In the early period of growth, the bacterial populations undergo dynamic changes in diversity and abundance as calf age20. Also, the bacterial communities in the GIT sites are significantly influenced by weaning36. The increased consumption of large amounts of solid feed and dietary shift from milk replacer with age has been given as the reason for age dependent increase in bacterial diversity37. The fermentation processes in the rumen is activated by the introduction of solid feed but there is a dramatic shift when milk is completely removed (weaning), greatly altering the composition of the ruminal and intestinal microbiomes8. The ruminal bacterial community is established before intake of solid food, but solid food arrival in turn shapes this community38. Dias et al.39 indicated that diet and age concurrently drive changes in the structure and abundance of bacterial communities in the developing rumen in calves. The PCoA plots in this study clustered according to period (pre-weaning and post-weaning) which is in line with Wang et al.23 who also indicated that bacteria communities clustered based on different age groups.
Previously, we recovered viable SCB and LA (total lactobacilli) throughout the GIT (rumen, ileum and colon) and feces of calves at the pre- and post-weaning periods30,40. Although growth performance (weight gain, feed intake and efficiency) was not affected by treatments30, calves were generally healthy and the treatments (LA and SCB) improved innate immune response (oxidative burst and phagocytosis) and markers of the acute phase reaction (CRP and SAA2), especially during weaning40.
The current study indicated that DFMs had less impact on bacterial diversity but more impact on bacterial composition in the GIT sites in calves. The greater diversity of SCB or LA compared to ATB (Table 1) might be linked to the differences in the mechanisms of pathogen clearance by ATB in the GIT. ATB eliminates pathogen growth by direct killing including neighbouring commensals, and therefore completely changing the ecological niche41. The diversity of the GIT has been shown to decrease both by short-term and long-term usage of antibiotics42,43. Decreased diversity by the use of ATB resulted in dysbiosis of the GIT microbiota leading to undesired effects, such as antibiotic-associated diarrhea44. The effects of DFMs on bacterial composition of GIT microbiota was site specific. Interestingly, major changes associated with DFMs were mostly found in the ileum and rumen compared to the colon (Tables 2 and 3), while a higher impact was observed at the pre-weaning period compared to the post-weaning period. The DA communities were composed of bacteria genera with beneficial effects to the host. The genera were phylogenetically related, suggesting a high level of functional redundancy, which is often associated with stable microbial assemblages resistant to pathogens45. Changes in microbial community compositions have been attributed to diet46. Since, LA and SCB treatments had different impacts, we will discuss the specific potential mechanisms for each DFM separately. For specific mechanisms, we will also focus our discussion on results reported at the genus level.
Perhaps, the most interesting results for SCB treatment was the significant reduction in the presence of Tyzzerella_4 genus compared to control in IlM (Table 2). This genus belongs to Lachnospiraceae family and Clostridia class. Bacterial species of Clostridia class have the ability to form spores and some genera including Tyzzerella_4 are linked to human diseases47. For instance, Tyzzerella and Tyzzerella_4 were associated to increased cardiovascular disease risk47. SCB treatment reduced the presence of Streptococcus compared to control in CoM. The pathogenic Streptococcus genus is widely distributed on the mucosal surfaces of the animal GIT48. Therefore, it suggests that SCB was able to eliminate numerous pathogens in the colonic mucosa compared to CTL or the other treatments. The microbiota influences the immune system by obstructing invading pathogens and can also support the growth and production of immune cells49,50. SCB also reduced the abundance of Peptoclostridium (Clostridium difficile) in IlM a major pathogen linked with infectious diarrhea51 (Table 2). In general, Ruminococcaceae are common digestive tract microbes that break down complex carbohydrates. SCB consumption positively influenced the establishment of Ruminococcaceae genera in the ileum of calves in this study. Brousseau et al.52 also found Ruminococcaceae bacterial family in the colon of pigs fed SCB and suggested that SCB had the potential as feed additives to modulate bacterial populations associated with GIT health52. Ruminococcaceae, actively degrades plants; it has carbohydrate-active enzymes, sugar transport mechanisms, and metabolic pathways for the degradation of complex plant materials41,53. As a member of the Ruminococcaceae family, Ruminococcus is a mucin-degrader and this probably enhanced mucus production which could be the reason for improved inflammatory responses in calves54. In a previous study, we also observed an increase in the concentration of markers associated with inflammatory response (acute phase proteins: CRP and SAA2) in calves fed LA or SCB39. Additionally, SCB also significantly increased the abundance of Olsenella (Lactobacillus reclassified as Olsenella) in IlM, a lactic acid bacterium that ferments carbohydrates to lactic acid55. This genus is bile-resistant and has the ability to utilise mucin56. Since Olsenella is a re-classification of lactobacillus species, its higher abundance supports our recent data in which we observed that SCB promoted the growth of total lactobacilli in the GIT of calves33. Surprisingly, the relative abundance of lactobacillus in LA treatment was similar to control in IlM at pre-weaning but decreased significantly (p = 8.93E-03) in IlD at post-weaning as compared to control. One possible explanation for this observation is that LA was probably a substrate for some other beneficial bacteria which disallowed its increase in some GIT sites even after supplemental feeding of LA. It is known that the product of one microbe is usually the substrate for another57.The genus Roseburia was also significantly (p = 7.01E-04) increased by SCB in IlM pre-weaning as compared to control. This is a commensal related genus producing short-chain fatty acids, particularly butyrate, which provides energy for cells in the GIT58, affects motility, maintains immunity, and has anti-inflammatory properties59,60. Roseburia may affect various metabolic pathways and could also serve as biomarkers for beneficial flora in GIT health60. This genus metabolizes dietary components that stimulate their proliferation and metabolic activities60. In mice, it has been shown that an increase in the abundance of Roseburia is linked to reduction of glucose intolerance61.
Many mechanisms of action of SCB have been directed against pathogenic microorganisms which include regulation of intestinal microbial homeostasis, interference with pathogens ability to colonize and infect the mucosa, modulation of local and systemic immune responses, and induction of enzymatic activity favoring absorption and nutrition. Consistent with the DA analyses, the major pathways changed by SCB treatment were in the IlM at the pre-weaning period. During this period, SCB significantly changed cell cycle, EGFR tyrosine kinase inhibitor resistance, bile secretion, Fanconi anemia pathway, mRNA surveillance pathway and oxytocin signaling pathway in IlM (Table 7). Since cell cycle and EGFR pathways are important for the regulation of cell proliferation, differentiation, growth, survival and motility, the SCB treatment might alter the bacterial abundance by influencing the genes or enzymes controlling these processes. Bile secretion pathway was also increased by SCB. This is a vital secretion essential for digestion and absorption of fats and fat-soluble vitamins in the small intestine62. In addition, bile is also an important route for elimination of excess cholesterol and many waste products, bilirubin, drugs and toxic compounds63. Bile acids appear to be a major regulator of the gut microbiota; and significant reduction in Ruminococcaceae64 has been related to low bile acid levels in the intestine65. Bile acids have been shown to have direct and indirect (through FXR-induced antimicrobial peptides) antimicrobial effects on gut microbes66.
Moreover, SCB treatment also altered the abundance of caffeine metabolism, cAMP signaling pathway, steroid biosynthesis, proteasome and dopaminergic synapse in the RuD. Steroid biosynthesis and proteasome are crucial pathways for lipid and protein metabolism while cAMP signaling pathway is important for second messengers signaling and have wide ranges of impact on cellular processes; therefore, it is not surprising that these pathways were impacted by the SCB treatment. However, it is not clear how caffeine metabolism pathway is related to SCB treatment in RuD.
Overall, health benefits of DFMs interaction can be classified into three categories67 as they can act directly within the GIT (level 1), they can also interact directly with the gastrointestinal mucus layer and epithelium (level 2) or they can have effects outside the GIT (level 3). The third level might reflect the effects of SCB on the dopaminergic synapse pathway. SCB might have impact on dopamine, an important and prototypical slow neurotransmitter in the mammalian brain, where it controls a variety of functions including locomotor activity, motivation and reward, learning and memory, and endocrine regulation68. However, the exact mechanisms are not clear.
At the post-weaning period, SCB also had an effect on five different pathways (caffeine metabolism, dopaminergic synapse, cAMP signalling, serotonergic synapse and steroid biosynthesis) and among them serotonergic synapse was the only pathway not affected by SCB in the pre-weaning period. Notably, serotonin (5-Hydroxytryptamine, 5-HT) is a monoamine neurotransmitter that plays important roles in physiological functions such as learning and memory, emotion, sleep, pain, motor function and endocrine secretion, as well as in pathological states including abnormal mood and cognition (http://www.genome.jp/kegg-bin/show_pathway?map=hsa04726&show_description=show). Interestingly, beside the effects on steroid metabolism, SCB increased the thyroid hormone signaling pathway (p < 0.0001) in RUD during the post-weaning period. Thyroid hormones are important regulators of growth, development and metabolism69; therefore it could be an important pathway involved in the SCB mechanism of action.
Generally, the LA treatment had less impact on the bacterial diversity (Table 1) but similar impact with SCB treatment on bacterial composition. At the pre-weaning period, LA also had greater impact on bacterial diversity in llM compared to other GITs sites. Similar to SCB treatment, Tyzzerella_4 was the most significant genus decreased (FDR = 6.02E-08) and Fibrobacter was the most significant genus increased (FDR = 3.09E-02) by LA treatment in IlM (Table 3). However, some genera were significantly (FDR ≤ 2.33E-03) changed only by LA treatment including Phascolarctobacterium, Prevotella_9 and Candidatus_Soleaferrea. Little is known about the functions of Phascolarctobacterium, and Candidatus_Soleaferrea genera in calf’s GIT but in human, Phascolarctobacterium faecium demonstrated a high colonization rate in the GIT70. In CoM, LA treatment also reduced Turicibacter which has been shown to possess putative immunomodulatory71 and invasive properties and may cause subclinical infections in piglets72.
In the post-weaning period, Ruminococcus_2, most significantly reduced by LA, has been shown to potentially associate with hyperinsulinaemia, intestinal permeability and hepatic inflammation in rats73. However, there is no information about the detrimental effects of this genus in calves.
In the pre-weaning period, LA treatment had significant impact on KEGG pathways only in the RuD which is similar to the impact of SCB during this period. However, at the post-weaning period, LA did not have significant impact on these pathways in RuD, but significantly changed caffeine metabolism pathway in the CoM and five pathways (cell cylce, EGFR tyrosine kinase inhibitor resistance, oxytocin signaling, mRNA surveillance and Fanconi anemia pathway) in IlM. Since these pathways were also significantly changed by SCB, we might assume similar potential mechanisms for SCB and LA in IlM.
The effects of antibiotics growth promoter on the bacteria community in the GIT system have been well documented. Several studies have shown that treatment with ATB altered the bacteria diversity74,75 as well as the bacteria composition75,76 in the GIT. The genera Lactobacilli and C. perfringens decreased in the ileum in broiler chickens fed low dose avilamycin and salinomycin77. Meanwhile the abundance of lactobacilli particularly L. gasseri, was increased by tylosin in the ileum of pigs78. However, we observed less impact of ATB on the GIT bacteria community at the pre- and post-weaning periods in this study. ATB significantly changed the bacteria composition in IlD and RuD only, at the pre-weaning period. Unlike SCB or LA, ATB had greatest impact on genera composition in the IlD and RuD, since it significantly changed the abundance of 34 and 24 genera in these sites, respectively, at the pre-weaning period. Desulfovibrio and Ruminiclostridium_9 were the most significantly decreased or increased genera, respectively, by ATB treatment in IlD. Little is known about the roles of Ruminiclostridium_9 in the GIT sites. Interestingly, no pathway was significantly changed by ATB treatment in IlD and RuD at the pre-weaning period. Notably, ATB also reduced streptococcus in the CoM and also significantly changed the abundance of steroid hormone biosynthesis pathways in the CoM at the pre-weaning period. In fact, streptococcus was the top most DA general in all three treatments (LA, SCB and ATB) in the CoM. Some species of the Streptococcus genera are pathogenic such as Streptococcus pyogenes and Streptococcus pneumoniae. However, Streptococcus was reduced in the treated samples with the largest reduction by SCB, followed by LA and ATB.
At the post-weaning period, Sutterella was DA by ATB. The genus Sutterella are commensals in the GIT with mild pro-inflammatory capacity in the human GIT79.
At post-weaning, ATB impacted steroid biosynthesis pathway in the CoM but targeted three different pathways including RIG-I-like receptor signaling pathway, D-Arginine and D-ornithine metabolism and butanoate metabolism pathways in IlM. RIG-I-like receptor proteins including RIG-I, MDA5, and LGP2 are expressed in both immune and non-immune cells. Upon recognition of viral nucleic acids, RIG-I-like receptor proteins recruit specific intracellular adaptor proteins to initiate signaling pathways that lead to the synthesis of type I interferon and other inflammatory cytokines, which are important for eliminating viruses.
The results from direct comparison of DA genera between treatments confirmed that the GIT microbiota was more sensitive to treatments in the pre-weaning period compared to the post-weaning period since most genera were significantly DA in the pre-weaning period (Tables 5, 6 and S3). Moreover, fewer genera and sites were affected when comparing LA vs. ATB than the comparison between SCB vs. ATB. This suggests that there were more diverse impacts of SCB compared to other treatments. Notably, Tyzzerella_4 (potential pathogenic genera) was the most significant DA genera in the pre-weaning period in both comparisons (SCB vs. ATB and LA vs. ATB) (Tables 5 and 6) suggesting differences in mechanisms by which the antibiotics (ATB) and DFMs (SCB or LA) can modulate pathogenic bacterial populations. Nevertheless, more studies are required to examine the distinct mechanisms by which DFMs impact the GIT of calves to enable development of effective DFMs.
The functional prediction analysis revealed more effects in the RuD contrary to data on diversity and abundance, which mostly influenced the ileum and colon. However, it is known that the level of abundance might not reflect the function of the bacteria and that roles played by the bacteria might be more important than abundance80, thus our data should be interpreted with caution.
In summary, the current data showed that site and day had an effect on bacteria diversity. However, the effect of treatment on bacteria diversity was not significant for most sites even though an increase in diversity was observed in the colon. The bacterial composition of the GIT microbiota was altered due to supplementation with the two DFMs with most DA genera found in the ileum. Both DFM treatments reduced some pathogenic bacteria genera such as Streptococcus or Tyzzerella_4 and increased the potential beneficial bacteria, Fibrobacter. Other potential beneficial bacteria including Rumminococcaceae UCG 005, Roseburia and Olsenella were increased by SCB treatment only. The functional prediction via pathways enrichment analyses indicated that besides affecting the local pathways such as cell cycle, bile secretion, proteasome or cAMP signaling pathway both DFMs also impacted other pathways such as thyroid hormone synthesis or dopaminergic synapse in the brain pathway. Moreover, these DFMs also shared some common mechanisms with ATB; however they had more diverse target sites compared to the ATB which mainly targeted the colon microbiome. Although, this study indicates that DFM have site specific and age dependent effects on the calf gut microbiome, further system-omics related studies (meta-genomics, meta-transcriptomics, proteomics and metabolomics) are needed to better define the mechanisms related to these effects. Therefore, regional effects and age need to be taken into consideration when investigating the biological mechanisms by which DFMs affect the growth and development of calves at the early period of growth. Furthermore, the pre- and post-weaning samples were collected from different calves implying that some individual variation was expected to influence our results, thus our data should be cautiously interpreted.
Materials and Methods
Animal treatments and samplings
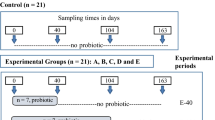
Animal management and use procedures were according to the Canadian Council on Animal Care81 and were approved by the animal care and ethics committee of Agriculture and Agri-Food Canada. Animal management procedures have been described in details previously30. Briefly, thirty two calves (2–7 days old) were randomly allocated to four treatments as follows: (1) Control (CTRL)- calves bucket fed with milk replacer (Goliath XLR 27–16, La Coop, Montreal, QC, Canada) at 6 L/day (2 L three times a day) for the first 4 days, and at 9 L/day (4.5 L twice a day) from day 5 to the end of weaning (day 53))and starter feed (Shur-Gain—Meunerie Sawyerville Inc., Cookshire-Eaton QC, Canada) fed ad libitum from day 8 of the experiment; (2) CTRL supplemented with Saccharomyces cerevisiae boulardii CNCMI-1079 (SCB; 7.5 × 108 colony forming units (CFU)/L milk replacer + 3 × 109 CFU/kg starter feed) (Levucell SB 20, Lallemand Animal Nutrition, Montreal, QC, Canada); (3) CTRL supplemented with Lactobacillus acidophilus BT1386 (LA; 2.5 × 108 CFU/L milk replacer + 1 × 109 CFU/kg starter feed) (Micro-Cell FS, Lallemand Animal Nutrition) and (4) CTRL supplemented with antibiotics (ATB) chlortetracycline and neomycin (528 and 357 mg/L milk replacer, respectively) pre-weaning, and chlortetracycline (55 mg/kg starter feed) (Vetoquinol Inc., Lavaltrie, QC, Canada) post-weaning. Calves were housed in individual pens, fed individually and had ad libitum access to water. The animal trial lasted for 14 weeks (experiment day 1 to 96). Weaning was initiated on day 43 by reducing the quantity of milk replacer offered by half every day and it was completed on day 53 when animals were able to eat 1 kg of starter feed per day. Four calves per treatment were euthanized on day 33 (pre-weaning) and another set of four calves per treatment on day 96 (post-weaning) to collect digesta samples from the rumen, ileum and colon, and mucosal samples from the ileum and colon. The pre- and post-weaning samples were collected from different calves. Digesta samples were aseptically collected placed in sterile tubes followed by storage at −20 °C until DNA isolation. Mucosal scrapings from intestinal tissues (colon and ileum) were collected using the inoculum method as described previously82 and stored at −80 °C until DNA isolation.
DNA isolation and quantification
Samples were thawed and kept on ice during the extraction process. The digesta were disrupted using a high speed blender and mucosa samples as described above. DNA was isolated from the homogenate using the bead beating method with the ZR fecal DNA kit (Zymo Research Corp., Irvine, CA, USA) following manufacturer’s instructions. The quantity and purity of isolated DNA was measured using spectrophotometry (Nano Drop Technologies, Wilmington, DE, USA) and diluted to a final concentration of 30 ng/µl.
Amplification of bacterial ribosomal DNA and sequencing
PCR primers targeting the 16S rRNA gene (V3–V4 region) were used to prepare amplicon libraries. Amplification of the 16S V3-V4 region was performed using sequence specific regions described previously83 in a dual indexed PCR approach. Briefly, the following generic oligonucleotide sequences were used for amplification: Bakt_341F-long AATGATACGGCGACCACCGAGATCTACAC[index1] TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGCCTACGGGNGGCWGCAG and Bakt_805R-longCAAGCAGAAGACGGCATACGAGAT[index2] GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAGGACTACHVGGGTATCTAATCC. The PCR was carried out in a total volume of 50 µL that contains 1X Q5 buffer (NEB), 0.25 µM of each primer, 200 µM of each dNTPs, 1 U of Q5 High-Fidelity DNA polymerase and 1 µL of template cDNA. The PCR started with an initial denaturation at 98 °C for 30 s followed by 10 cycles of denaturation at 98 °C for 10 s, annealing at 55 °C for 10 s and extension at 72 °C for 30 s, and 25 cycles of denaturation at 98 °C for 10 s, annealing at 65 °C for 10 s, extension at 72 °C for 30 s and a final extension step at 72 °C for 2 min. The PCR reactions were purified using the Axygen PCR cleanup kit (Axygen). Quality of the purified PCR product was checked on a DNA7500 BioAnalyzer chip (Agilent) and quantified using a Nanodrop 1000 Spectrophotometer (Thermo Fisher Scientific). Barcoded Amplicons were pooled in equimolar concentrations and sequenced on the Illumina MiSeq (paired–end 300 bases with two index reads). Library preparation and sequencing was performed by L’Institut de Biologie Intégrative et des Systèmes (IBIS), de Université Laval, Quebec City, Canada.
Bioinformatics analysis
The downstream analysis of output fastq files was done using the pipeline of the open source software package QIIME84. Paired end reads were merged using FLASh85. Chimera detection was applied to the merged reads using Uchime86. The GOLD87 database was used for reference based detection. Taxomomic affiliation of the 16S data was studied using QIIME84. Demultiplexed and quality filtered sequences from pre-processing step were clustered into OTUs using VSEARCH88. An OTU (Operational Taxonomic Unit) was formed based on sequence identity with threshold defined at 0.97.After the clustering step, a representative sequence was picked for each OTU and a taxonomic identity was assigned to each representative sequence. The 16S database used was Greengenes while Uclust86 was used for taxonomic assignment. Multiple alignments of the representative OTU sequences were generated with PyNAST89, which aligns the sequences to 16S reference sequences. The relationship between sequences was studied by generating a phylogenetic tree with FastTree90 followed by computing UniFrac distances. A rarefaction curve for each sample was plotted (observed OTUs metric) in order to estimate the depth of sequencing for each sample and to choose the rarefaction threshold for all samples. Results were generated after the cumulative sum scaling (CSS)91 normalization method. The Amplicon-Seq pipeline provides taxonomic affiliation of data at different levels (Kingdom, Phylum, Class, Order, Family and Genus).
Assessment of diversity and statistical analysis
Samples were rarefied for alpha-diversity calculations and rarefaction curves generated (Fig. S1) in order to eliminate the bias caused by the different sample sizes92. The OTU table was rarefied across samples to the lowest sample depth using QIIME based on the Messene Twister pseudorandom number generator. Alpha diversity estimators including Chao1, observed OTUs, Shannon, Simpson and Inverted Simpson (Invsimpson) were calculated for the overall bacterial community using Phyloseq93. Mean alpha diversity estimates for each site, day, treatment and treatment by site by day were compared using the two-sided t-test in R program94.
The dataset was also subsampled to the minimum95 to compare microbial composition between samples (β-diversity). Beta-diversity was measured by calculating the weighted and unweighted UniFrac distances96 using Phyloseq default scripts. Principal coordinate analysis (PCoA) was applied on the resulting distance matrices to generate two-dimensional plots. Permutational multivariate analysis of variance (PERMONOVA97) was used to calculate P-values and to test differences of β-diversity among treatment groups for significance.
Bacterial Community Composition and differential relative abundance analyses
To investigate the relative abundance of the different genera, The MicrobiomeAnalyst98 was used to obtain the most prevalent bacteria genera within each site.
To investigate the effect of treatment on the different genera, we did a pair wise comparison between each treatment and control, GIT site and day (33 and 96). Different abundance at genus level was compared between treatments and control as well as among treatments using the Wald Test method of DESeq299. The samples with OTU total count <10,000 were removed. The normalization step was done for each pair of comparison separately100 and taxa were considered significantly differentially abundant if p-corrected for false discovery rate (FDR) was <0.05. The FDR procedure is performed to reduce the type I error. In brief, this procedure includes the following steps: (1) uncorrected p-values are sorted in ascending order, (2) ranks to the p-values are assigned, (3) individual Benjamini-Hochberg critical p-values were calculated using the formula (i/m)q (i = the individual’s p-value rank, m = total number of tests, q = the false discovery rate). In this analysis, a q-value (FDR) of ≤0.05 was considered significant.
Functional prediction and differential analysis of predicted pathways
The phylogenetic investigation of communities by reconstruction of unobserved states (PICRUSt)101 software was used for the prediction of functional genes of the classified members of the GIT microbiota resulting from reference-based OTU picking against Greengenes database. Predicted genes were then hierarchically clustered and categorized under Kyoto Encyclopedia of Genes and Genomes102 orthologs (KOs). Predicted KOs was then converted into their associated pathways. The differential analyses of predicted pathways were done in DeSeq. 2 and only pathways predicted for at least 5 samples were used as input data. The pathways were considered significantly differentially predicted if p was <0.05. Since the enrichment relied on human data, we used a relaxed threshold (uncorrected p-values) to get a better overview of the impact of treatments on pathways.
References
Celi, P. et al. Gastrointestinal functionality in animal nutrition and health: New opportunities for sustainable animal production. Animal Feed Science and Technology 234, 88–100, https://doi.org/10.1016/j.anifeedsci.2017.09.012 (2017).
Uyeno, Y., Shigemori, S. & Shimosato, T. Effect of Probiotics/Prebiotics on Cattle Health and Productivity. Microbes and Environments 30, 126–132, https://doi.org/10.1264/jsme2.ME14176 (2015).
Castro, J. J. et al. Changes in the intestinal bacterial community, short-chain fatty acid profile, and intestinal development of preweaned Holstein calves. 1. Effects of prebiotic supplementation depend on site and age. Journal of dairy science 99, 9682–9702, https://doi.org/10.3168/jds.2016-11006 (2016).
Malmuthuge, N., Griebel, P. J. & Guan, L. L. The Gut Microbiome and Its Potential Role in the Development and Function of Newborn Calf Gastrointestinal Tract. Frontiers in Veterinary Science 2, 36, https://doi.org/10.3389/fvets.2015.00036 (2015).
Conlon, M. A. & Bird, A. R. The Impact of Diet and Lifestyle on Gut Microbiota and Human Health. Nutrients 7, 17–44, https://doi.org/10.3390/nu7010017 (2015).
Lai, K. P., Chung, Y. T., Li, R., Wan, H. T. & Wong, C. K. Bisphenol A alters gut microbiome: Comparative metagenomics analysis. Environmental pollution (Barking, Essex: 1987) 218, 923–930, https://doi.org/10.1016/j.envpol.2016.08.039 (2016).
Callaway, T. R. et al. Evaluation of bacterial diversity in the rumen and feces of cattle fed different levels of dried distillers grains plus solubles using bacterial tag-encoded FLX amplicon pyrosequencing. Journal of animal science 88, 3977–3983, https://doi.org/10.2527/jas.2010-2900 (2010).
Meale, S. J. et al. Development of Ruminal and Fecal Microbiomes Are Affected by Weaning But Not Weaning Strategy in Dairy Calves. Frontiers in Microbiology 7, https://doi.org/10.3389/fmicb.2016.00582 (2016).
Meale, S. J., Chaucheyras-Durand, F., Berends, H., Guan, L. L. & Steele, M. A. From pre- to postweaning: Transformation of the young calf’s gastrointestinal tract1. Journal of Dairy Science 100, 5984–5995, https://doi.org/10.3168/jds.2016-12474 (2017).
Collado, M. C., Cernada, M., Bauerl, C., Vento, M. & Perez-Martinez, G. Microbial ecology and host-microbiota interactions during early life stages. Gut microbes 3, 352–365, https://doi.org/10.4161/gmic.21215 (2012).
Umetsu, D. T. Early exposure to germs and the Hygiene Hypothesis. Cell Research 22, 1210–1211, https://doi.org/10.1038/cr.2012.65 (2012).
Abecia, L., Martin-Garcia, A. I., Martinez, G., Newbold, C. J. & Yanez-Ruiz, D. R. Nutritional intervention in early life to manipulate rumen microbial colonization and methane output by kid goats postweaning. J Anim Sci 91, 4832–4840, https://doi.org/10.2527/jas.2012-6142 (2013).
Abecia, L. et al. Natural and artificial feeding management before weaning promote different rumen microbial colonization but not differences in gene expression levels at the rumen epithelium of newborn goats. PLOS ONE 12, e0182235, https://doi.org/10.1371/journal.pone.0182235 (2017).
Walsh, C. J., Guinane, C. M., O’Toole, P. W. & Cotter, P. D. Beneficial modulation of the gut microbiota. FEBS Letters 588, 4120–4130, https://doi.org/10.1016/j.febslet.2014.03.035 (2014).
Druart, C., Alligier, M., Salazar, N., Neyrinck, A. M. & Delzenne, N. M. Modulation of the Gut Microbiota by Nutrients with Prebiotic and Probiotic Properties. Advances in Nutrition: An International Review Journal 5, 624S–633S, https://doi.org/10.3945/an.114.005835 (2014).
Hemarajata, P. & Versalovic, J. Effects of probiotics on gut microbiota: mechanisms of intestinal immunomodulation and neuromodulation. Therapeutic Advances in Gastroenterology 6, 39–51, https://doi.org/10.1177/1756283X12459294 (2013).
Gomez, D. E., Arroyo, L. G., Costa, M. C., Viel, L. & Weese, J. S. Characterization of the Fecal Bacterial Microbiota of Healthy and Diarrheic Dairy Calves. J Vet Intern Med 31, 928–939, https://doi.org/10.1111/jvim.14695 (2017).
Weese, J. S. & Jelinski, M. Assessment of the Fecal Microbiota in Beef Calves. Journal of Veterinary Internal Medicine 31, 176–185, https://doi.org/10.1111/jvim.14611 (2017).
Carroll, I. M., Ringel-Kulka, T., Siddle, J. P., Klaenhammer, T. R. & Ringel, Y. Characterization of the Fecal Microbiota Using High-Throughput Sequencing Reveals a Stable Microbial Community during Storage. PLOS ONE 7, e46953, https://doi.org/10.1371/journal.pone.0046953 (2012).
Jami, E., Israel, A., Kotser, A. & Mizrahi, I. Exploring the bovine rumen bacterial community from birth to adulthood. The ISME Journal 7, 1069–1079, https://doi.org/10.1038/ismej.2013.2 (2013).
Li, R. W. et al. Characterization of the rumen microbiota of pre-ruminant calves using metagenomic tools. Environmental microbiology 14, 129–139, https://doi.org/10.1111/j.1462-2920.2011.02543.x (2012).
Mao, S., Zhang, M., Liu, J. & Zhu, W. Characterising the bacterial microbiota across the gastrointestinal tracts of dairy cattle: membership and potential function. Scientific reports 5, 16116, https://doi.org/10.1038/srep16116 (2015).
Wang, J., Fan, H., Han, Y., Zhao, J. & Zhou, Z. Characterization of the microbial communities along the gastrointestinal tract of sheep by 454 pyrosequencing analysis. Asian-Australasian Journal of Animal Sciences 30, 100–110, https://doi.org/10.5713/ajas.16.0166 (2017).
Gareau, M. G., Sherman, P. M. & Walker, W. A. Probiotics and the gut microbiota in intestinal health and disease. Nature reviews. Gastroenterology & hepatology 7, 503–514, https://doi.org/10.1038/nrgastro.2010.117 (2010).
Hemarajata, P. & Versalovic, J. Effects of probiotics on gut microbiota: mechanisms of intestinal immunomodulation and neuromodulation. Therapeutic Advances in Gastroenterology 6, 39–51, https://doi.org/10.1177/1756283X12459294 (2012).
Houghteling, P. D. & Walker, W. A. Why is initial bacterial colonization of the intestine important to the infant’s and child’s health? Journal of pediatric gastroenterology and nutrition 60, 294–307, https://doi.org/10.1097/MPG.0000000000000597 (2015).
McAllister, T. A. et al. Review: The use of direct fed microbials to mitigate pathogens and enhance production in cattle. Canadian Journal of Animal Science 91, 193–211, https://doi.org/10.4141/cjas10047 (2011).
Kiros, T. G. et al. Effect of live yeast Saccharomyces cerevisiae (Actisaf Sc 47) supplementation on the performance and hindgut microbiota composition of weanling pigs. Scientific reports 8, 5315, https://doi.org/10.1038/s41598-018-23373-8 (2018).
Sivieri, K. et al. Lactobacillus acidophilus CRL 1014 improved “gut health” in the SHIME(®) reactor. BMC Gastroenterology 13, 100–100, https://doi.org/10.1186/1471-230X-13-100 (2013).
Fomenky, B. E. et al. Impact of Saccharomyces cerevisiae boulardii CNCMI-1079 and Lactobacillus acidophilus BT1386 on total lactobacilli population in the gastrointestinal tract and colon histomorphology of Holstein dairy calves. Animal Feed Science and Technology 234, 151–161, https://doi.org/10.1016/j.anifeedsci.2017.08.019 (2017).
de Oliveira, M. N. et al. Characterizing the microbiota across the gastrointestinal tract of a Brazilian Nelore steer. Veterinary microbiology 164, 307–314, https://doi.org/10.1016/j.vetmic.2013.02.013 (2013).
Frey, J. C. et al. Comparative studies of microbial populations in the rumen, duodenum, ileum and faeces of lactating dairy cows. Journal of Applied Microbiology 108, 1982–1993, https://doi.org/10.1111/j.1365-2672.2009.04602.x (2010).
Malmuthuge, N., Griebel, P. J. & Guan le, L. Taxonomic identification of commensal bacteria associated with the mucosa and digesta throughout the gastrointestinal tracts of preweaned calves. Applied and environmental microbiology 80, 2021–2028, https://doi.org/10.1128/aem.03864-13 (2014).
Rosenberg, E. & Zilber-Rosenberg, I. Microbes Drive Evolution of Animals and Plants: the Hologenome Concept. mBio 7, e01395–01315, https://doi.org/10.1128/mBio.01395-15 (2016).
Booijink, C. C., Zoetendal, E. G., Kleerebezem, M. & de Vos, W. M. Microbial communities in the human small intestine: coupling diversity to metagenomics. Future microbiology 2, 285–295, https://doi.org/10.2217/17460913.2.3.285 (2007).
Meale, S. J. et al. Weaning age influences the severity of gastrointestinal microbiome shifts in dairy calves. Scientific reports 7, 198, https://doi.org/10.1038/s41598-017-00223-7 (2017).
Kim, M. et al. Investigation of bacterial diversity in the feces of cattle fed different diets. Journal of animal science 92, 683–694, https://doi.org/10.2527/jas.2013-6841 (2014).
Rey, M. et al. Establishment of ruminal bacterial community in dairy calves from birth to weaning is sequential. Journal of applied microbiology 116, 245–257, https://doi.org/10.1111/jam.12405 (2014).
Dias, J. et al. Effect of Pre-weaning Diet on the Ruminal Archaeal, Bacterial, and Fungal Communities of Dairy Calves. Frontiers in Microbiology 8, 1553, https://doi.org/10.3389/fmicb.2017.01553 (2017).
Fomenky, B. E. et al. Saccharomyces cerevisiae boulardii CNCM 1-1079 and Lactobacillus acidophilus BT1386 influences innate immune response and serum levels of acute-phase proteins during weaning in Holstein calves. Canadian Journal of Animal Science, https://doi.org/10.1139/CJAS-2017-0120 (2018).
Nigam, A., Gupta, D. & Sharma, A. Treatment of infectious disease: Beyond antibiotics. Microbiological Research 169, 643–651, https://doi.org/10.1016/j.micres.2014.02.009 (2014).
Willing, B. P., Russell, S. L. & Finlay, B. B. Shifting the balance: antibiotic effects on host-microbiota mutualism. Nature reviews. Microbiology 9, 233–243, https://doi.org/10.1038/nrmicro2536 (2011).
Ferrer, M., Martins dos Santos, V. A., Ott, S. J. & Moya, A. Gut microbiota disturbance during antibiotic therapy: a multi-omic approach. Gut microbes 5, 64–70, https://doi.org/10.4161/gmic.27128 (2014).
McFarland, L. V. Epidemiology, risk factors and treatments for antibiotic-associated diarrhea. Digestive diseases (Basel, Switzerland) 16, 292–307, 16879 (1998).
Backhed, F., Ley, R. E., Sonnenburg, J. L., Peterson, D. A. & Gordon, J. I. Host-bacterial mutualism in the human intestine. Science 307, 1915–1920, https://doi.org/10.1126/science.1104816 (2005).
Henderson, G. et al. Rumen microbial community composition varies with diet and host, but a core microbiome is found across a wide geographical range. Scientific reports 5, 14567, https://doi.org/10.1038/srep14567 (2015).
Kelly, T. N. et al. Gut Microbiome Associates With Lifetime Cardiovascular Disease Risk Profile Among Bogalusa Heart Study Participants. Circulation research 119, 956–964, https://doi.org/10.1161/circresaha.116.309219 (2016).
Hardie, J. M. & Whiley, R. A. Classification and overview of the genera Streptococcus and Enterococcus. Society for Applied Bacteriology symposium series 26, 1s–11s (1997).
Chung, H. et al. Gut immune maturation depends on colonization with a host-specific microbiota. Cell 149, 1578–1593, https://doi.org/10.1016/j.cell.2012.04.037 (2012).
Yasuda, M., Fujino, M., Nasu, T. & Murakami, T. Histological studies on the ontogeny of bovine gut-associated lymphoid tissue: appearance of T cells and development of IgG+ and IgA+ cells in lymphoid follicles. Developmental & Comparative Immunology 28, 357–369, https://doi.org/10.1016/j.dci.2003.09.013 (2004).
Luo, Y. et al. Genome Sequence and Analysis of Peptoclostridium difficile Strain ZJCDC-S82. Evolutionary Bioinformatics Online 12, 41–49, https://doi.org/10.4137/EBO.S32476 (2016).
Brousseau, J. P. et al. Effects of probiotics Pediococcus acidilactici strain MA18/5M and Saccharomyces cerevisiae subsp. boulardii strain SB-CNCM I-1079 on fecal and intestinal microbiota of nursing and weanling piglets. Journal of animal science 93, 5313–5326, https://doi.org/10.2527/jas.2015-9190 (2015).
Biddle, A., Stewart, L., Blanchard, J. & Leschine, S. Untangling the Genetic Basis of Fibrolytic Specialization by Lachnospiraceae and Ruminococcaceae in Diverse Gut Communities. Diversity 5, 627 (2013).
Crost, E. H. et al. Utilisation of mucin glycans by the human gut symbiont Ruminococcus gnavus is strain-dependent. PLoS One 8, e76341, https://doi.org/10.1371/journal.pone.0076341 (2013).
Kraatz, M., Wallace, R. J. & Svensson, L. Olsenella umbonata sp. nov., a microaerotolerant anaerobic lactic acid bacterium from the sheep rumen and pig jejunum, and emended descriptions of Olsenella, Olsenella uli and Olsenella profusa. International journal of systematic and evolutionary microbiology 61, 795–803, https://doi.org/10.1099/ijs.0.022954-0 (2011).
Dewhirst, F. E. et al. Characterization of novel human oral isolates and cloned 16S rDNA sequences that fall in the family Coriobacteriaceae: description of olsenella gen. nov., reclassification of Lactobacillus uli as Olsenella uli comb. nov. and description of Olsenella profusa sp. nov. International journal of systematic and evolutionary microbiology 51, 1797–1804, https://doi.org/10.1099/00207713-51-5-1797 (2001).
Gerritsen, J., Smidt, H., Rijkers, G. T. & de Vos, W. M. Intestinal microbiota in human health and disease: the impact of probiotics. Genes & Nutrition 6, 209–240, https://doi.org/10.1007/s12263-011-0229-7 (2011).
Duncan, S. H., Hold, G. L., Barcenilla, A., Stewart, C. S. & Flint, H. J. Roseburia intestinalis sp. nov., a novel saccharolytic, butyrate-producing bacterium from human faeces. International journal of systematic and evolutionary microbiology 52, 1615–1620, https://doi.org/10.1099/00207713-52-5-1615 (2002).
Stanton, T. B. & Savage, D. C. Roseburia cecicola gen. nov., sp. nov., a Motile, Obligately Anaerobic Bacterium from a Mouse Cecum. International journal of systematic and evolutionary microbiology 33, 618–627, https://doi.org/10.1099/00207713-33-3-618 (1983).
Tamanai-Shacoori, Z. et al. Roseburia spp.: a marker of health? Future Microbiol 12, 157–170, https://doi.org/10.2217/fmb-2016-0130 (2017).
Ryan, K. K. et al. FXR is a molecular target for the effects of vertical sleeve gastrectomy. Nature 509, 183–188, https://doi.org/10.1038/nature13135 (2014).
Martínez-Augustin, O. & de Medina, F. S. Intestinal bile acid physiology and pathophysiology. World Journal of Gastroenterology: WJG 14, 5630–5640, https://doi.org/10.3748/wjg.14.5630 (2008).
Ridlon, J. M., Kang, D. J., Hylemon, P. B. & Bajaj, J. S. Bile acids and the gut microbiome. Current opinion in gastroenterology 30, 332–338, https://doi.org/10.1097/mog.0000000000000057 (2014).
Bajaj, J. S. et al. The Cirrhosis Dysbiosis Ratio defines Changes in the Gut Microbiome Associated with Cirrhosis and its Complications. Journal of hepatology 60, 940–947, https://doi.org/10.1016/j.jhep.2013.12.019 (2014).
Kakiyama, G. et al. Modulation of the fecal bile acid profile by gut microbiota in cirrhosis. Journal of hepatology 58, 949–955, https://doi.org/10.1016/j.jhep.2013.01.003 (2013).
Begley, M., Gahan, C. G. M. & Hill, C. The interaction between bacteria and bile. FEMS Microbiology Reviews 29, 625–651, https://doi.org/10.1016/j.femsre.2004.09.003 (2005).
Rijkers, G. T. et al. Guidance for substantiating the evidence for beneficial effects of probiotics: current status and recommendations for future research. The Journal of nutrition 140, 671s–676s, https://doi.org/10.3945/jn.109.113779 (2010).
Tritsch, N. X. & Sabatini, B. L. Dopaminergic modulation of synaptic transmission in cortex and striatum. Neuron 76, 33–50, https://doi.org/10.1016/j.neuron.2012.09.023 (2012).
Mullur, R., Liu, Y.-Y. & Brent, G. A. Thyroid Hormone Regulation of Metabolism. Physiological Reviews 94, 355–382, https://doi.org/10.1152/physrev.00030.2013 (2014).
Alugongo, G. M. et al. Review: Utilization of yeast of Saccharomyces cerevisiae origin in artificially raised calves. Journal of Animal Science and Biotechnology 8, 34, https://doi.org/10.1186/s40104-017-0165-5 (2017).
Presley, L. L., Wei, B., Braun, J. & Borneman, J. Bacteria associated with immunoregulatory cells in mice. Applied and environmental microbiology 76, 936–941, https://doi.org/10.1128/aem.01561-09 (2010).
Rettedal, E. et al. Alteration of the Ileal Microbiota of Weanling Piglets by the Growth-Promoting Antibiotic Chlortetracycline. Applied and environmental microbiology 75, 5489–5495, https://doi.org/10.1128/aem.02220-08 (2009).
Wan, J. et al. The impact of dietary sn-2 palmitic triacylglycerols in combination with docosahexaenoic acid or arachidonic acid on lipid metabolism and host faecal microbiota composition in Sprague Dawley rats. Food & Function 8, 1793–1802, https://doi.org/10.1039/C7FO00094D (2017).
Heselmans, M. et al. Gut flora in health and disease: potential role of probiotics. Current issues in intestinal microbiology 6, 1–7 (2005).
Dumonceaux, T. J., Hill, J. E., Hemmingsen, S. M. & Van Kessel, A. G. Characterization of intestinal microbiota and response to dietary virginiamycin supplementation in the broiler chicken. Applied and environmental microbiology 72, 2815–2823, https://doi.org/10.1128/aem.72.4.2815-2823.2006 (2006).
Wise, M. G. & Siragusa, G. R. Quantitative analysis of the intestinal bacterial community in one- to three-week-old commercially reared broiler chickens fed conventional or antibiotic-free vegetable-based diets. J Appl Microbiol 102, 1138–1149, https://doi.org/10.1111/j.1365-2672.2006.03153.x (2007).
Knarreborg, A., Simon, M. A., Engberg, R. M., Jensen, B. B. & Tannock, G. W. Effects of dietary fat source and subtherapeutic levels of antibiotic on the bacterial community in the ileum of broiler chickens at various ages. Applied and environmental microbiology 68, 5918–5924 (2002).
Collier, C. T. et al. Molecular ecological analysis of porcine ileal microbiota responses to antimicrobial growth promoters. Journal of animal science 81, 3035–3045, https://doi.org/10.2527/2003.81123035x (2003).
Hiippala, K., Kainulainen, V., Kalliomäki, M., Arkkila, P. & Satokari, R. Mucosal Prevalence and Interactions with the Epithelium Indicate Commensalism of Sutterella spp. Frontiers in Microbiology 7, 1706, https://doi.org/10.3389/fmicb.2016.01706 (2016).
Waldor, M. K. et al. Where Next for Microbiome Research? PLOS Biology 13, e1002050, https://doi.org/10.1371/journal.pbio.1002050 (2015).
CCAC. Guidelines on the care and use of farm animals in research, teaching and testing. Canadian Council on Animal Care 2009. Documents/Standards/Guidelines/Farm_Animals.pdf) (2009).
Li, M. et al. Evaluation of QIAamp DNA Stool Mini Kit for ecological studies of gut microbiota. J Microbiol Methods 54, 13–20 (2003).
Klindworth, A. et al. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Research 41, e1–e1, https://doi.org/10.1093/nar/gks808 (2013).
Caporaso, J. G. et al. QIIME allows analysis of high-throughput community sequencing data. Nature methods 7, 335–336, https://doi.org/10.1038/nmeth.f.303 (2010).
Magoč, T. & Salzberg, S. L. FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics 27, 2957–2963, https://doi.org/10.1093/bioinformatics/btr507 (2011).
Edgar, R. C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461, https://doi.org/10.1093/bioinformatics/btq461 (2010).
Haas, B. J. et al. Chimeric 16S rRNA sequence formation and detection in Sanger and 454-pyrosequenced PCR amplicons. Genome research 21, 494–504, https://doi.org/10.1101/gr.112730.110 (2011).
Rognes, T., Flouri, T., Nichols, B., Quince, C. & Mahé, F. VSEARCH: a versatile open source tool for metagenomics. PeerJ 4, e2584, https://doi.org/10.7717/peerj.2584 (2016).
Caporaso, J. G. et al. PyNAST: a flexible tool for aligning sequences to a template alignment. Bioinformatics 26, 266–267, https://doi.org/10.1093/bioinformatics/btp636 (2010).
Price, M. N., Dehal, P. S. & Arkin, A. P. FastTree: computing large minimum evolution trees with profiles instead of a distance matrix. Molecular biology and evolution 26, 1641–1650, https://doi.org/10.1093/molbev/msp077 (2009).
Paulson, J. N., Stine, O. C., Bravo, H. C. & Pop, M. Differential abundance analysis for microbial marker-gene surveys. Nat Meth 10, 1200–1202, https://doi.org/10.1038/nmeth.2658. http://www.nature.com/nmeth/journal/v10/n12/abs/nmeth.2658.html#supplementary-information (2013).
Roesch, L. F. et al. Pyrosequencing enumerates and contrasts soil microbial diversity. Isme j 1, 283–290, https://doi.org/10.1038/ismej.2007.53 (2007).
McMurdie, P. J. & Holmes, S. phyloseq: An R Package for Reproducible Interactive Analysis and Graphics of Microbiome Census Data. PLOS ONE 8, e61217, https://doi.org/10.1371/journal.pone.0061217 (2013).
R Development Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2013; ISBN 3-900051-07-0 (2013).
Aguirre de Cárcer, D., Denman, S. E., McSweeney, C. & Morrison, M. Evaluation of Subsampling-Based Normalization Strategies for Tagged High-Throughput Sequencing Data Sets from Gut Microbiomes. Applied and environmental microbiology 77, 8795–8798, https://doi.org/10.1128/aem.05491-11 (2011).
Lozupone, C. & Knight, R. UniFrac: a New Phylogenetic Method for Comparing Microbial Communities. Applied and environmental microbiology 71, 8228–8235, https://doi.org/10.1128/aem.71.12.8228-8235.2005 (2005).
Anderson, M., Gorley, R. & Clarke, R. Permanova. Permutational multivariate analysis of variance, a computer program. Department of Statistics, University of Auckland 24 (2005).
Dhariwal, A. et al. MicrobiomeAnalyst: a web-based tool for comprehensive statistical, visual and meta-analysis of microbiome data. Nucleic acids research, https://doi.org/10.1093/nar/gkx295 (2017).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq. 2. Genome biology 15, 550 (2014).
Weiss, S. et al. Normalization and microbial differential abundance strategies depend upon data characteristics. Microbiome 5, 27, https://doi.org/10.1186/s40168-017-0237-y (2017).
Langille, M. G. I. et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat Biotech 31, 814–821, https://doi.org/10.1038/nbt.2676 http://www.nature.com/nbt/journal/v31/n9/abs/nbt.2676.html#supplementary-information (2013).
Kanehisa, M. & Goto, S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic acids research 28, 27–30 (2000).
Acknowledgements
Funding for this work was provided by Agriculture and Agri-Food Canada. We are grateful to the barn staff of Sherbrooke Research and Development Centre for their assistance during the animal phase of the project and to Lallemand Animal Nutrition for donating the DFMs used in this study.
Author information
Authors and Affiliations
Contributions
EMI-A conceived and designed the experiments with inputs from G.T., J.C., N.B., M.L. and Y.P.C. B.E.F. performed the experiments; B.E.F. and D.N.D. analyzed the data; B.E.F., D.N.D., G.T. and EMI-A interpreted the data, B.E.F. drafted the manuscript with input from D.N.D. all authors revised and approved the final draft.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Fomenky, B.E., Do, D.N., Talbot, G. et al. Direct-fed microbial supplementation influences the bacteria community composition of the gastrointestinal tract of pre- and post-weaned calves. Sci Rep 8, 14147 (2018). https://doi.org/10.1038/s41598-018-32375-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-32375-5
Keywords
This article is cited by
-
Bacteria colonization and gene expression related to immune function in colon mucosa is associated with growth in neonatal calves regardless of live yeast supplementation
Journal of Animal Science and Biotechnology (2024)
-
‘Multi-omics’ data integration: applications in probiotics studies
npj Science of Food (2023)
-
Host-specific probiotics feeding influence growth, gut microbiota, and fecal biomarkers in buffalo calves
AMB Express (2022)
-
Use of exogenous fibrolytic enzymes and probiotic in finely ground starters to improve calf performance
Scientific Reports (2022)
-
Probiotic Lactobacilli Administration Induces Changes in the Fecal Microbiota of Preweaned Dairy Calves
Probiotics and Antimicrobial Proteins (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
