
Abstract
Waves of highly infectious viruses sweeping through global honey bee populations have contributed to recent declines in honey bee health. Bees have been observed foraging on mushroom mycelium, suggesting that they may be deriving medicinal or nutritional value from fungi. Fungi are known to produce a wide array of chemicals with antimicrobial activity, including compounds active against bacteria, other fungi, or viruses. We tested extracts from the mycelium of multiple polypore fungal species known to have antiviral properties. Extracts from amadou (Fomes) and reishi (Ganoderma) fungi reduced the levels of honey bee deformed wing virus (DWV) and Lake Sinai virus (LSV) in a dose-dependent manner. In field trials, colonies fed Ganoderma resinaceum extract exhibited a 79-fold reduction in DWV and a 45,000-fold reduction in LSV compared to control colonies. These findings indicate honey bees may gain health benefits from fungi and their antimicrobial compounds.
Similar content being viewed by others
Introduction
The Western honey bee (Apis mellifera) is a critical component of crop production and food biosecurity worldwide. A. mellifera and other members of the genus Apis also play a key role in the ecological stability of wild plant communities within areas of endemism in Europe, Africa and Asia. Managed A. mellifera colonies are estimated to contribute over $15 billion annually to the US agricultural economy through the pollination of numerous fruits, nuts and vegetables1. The pollination of almonds in California alone requires relocating over 75% of the managed honey bee colonies (nearly 2 million) in the United States on this single crop during bloom. Over the past decade, beekeepers have experienced a dramatic increase in annual colony losses, typically averaging well over 30%2,3,4. This combination of high demand and reduced supply has led to expansive increases in pollination costs for growers, while beekeepers have been hard-pressed to maintain adequate numbers of healthy honey bee colonies to remain economically viable, even with the benefit of higher pollination service fees.
Two of the most important factors contributing to widespread colony losses are infestation of A. mellifera with the parasitic mite Varroa destructor and the suite of associated viruses5,6,7. The extent to which Varroa mites are involved in the amplification and dissemination of RNA viruses among honey bee populations has only recently become apparent, with Varroa infestation now known to be associated with at least 10 honey bee viruses8,9,10. Viruses are recognized to play a contributing role in widespread colony losses, especially deformed wing virus (DWV) and Varroa destructor virus-1 (VDV1)6,7,11,12,13. DWV is a devastating virus that causes shriveled wings, reduced worker life span, reduced foraging, and immunosuppression in honey bees14,15 (Fig. 1A). In addition to mite-mediated transmission, RNA viruses (including DWV) can also be transmitted among pollinators via pollen16,17. Another potentially problematic virus associated with honey bees and Varroa mites is the Lake Sinai virus group9,18. LSV was first identified in 201019 but is now widespread in US honey bee colonies.

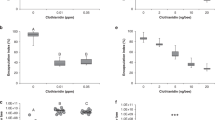
Extracts from fungi reduced deformed wing virus (DWV) in honey bees. (A) DWV shortens the lifespan of workers and causes developmental abnormalities in honey bees including deformed wings. (B) Honey bees forage on fungal mycelium where they ingest liquid exudates. (C) Bees fed extracts from polypore mycelium exhibited lower DWV virus titers. In three different trials, mixing fungal extracts into caged bees’ sucrose syrup food significantly reduced the levels of DWV as assayed by qPCR. Notably, the extracts of F. fomentarius mycelium reduced DWV more than 800-fold in the first trial (2-tailed t-test, n = 10, t = 1.12, p = 0.005). For each trial, qPCR ΔCt values are normalized to mean value of the respective control groups. Photos: Xolani90-https://commons.wikimedia.org/wiki/File:Deformed_Wing_Virus_in_worker_bee.JPG - [CC BY-SA 3.0 (https://creativecommons.org/licenses/by-sa/3.0)]-no changes (A), PES (B, Fomes, G. resinaceum), Titus Tscharntke – Wikimedia Commons (Betula), George Chernilevsky-Wikimedia Commons (G. applanatum), Norbert Nagel -https://commons.wikimedia.org/wiki/File:Trametes_versicolor_-_Coriolus_versicolor_-_Polyporus_versicolor_-_Schmetterlingstramete_-_Bunte_Tramete_-_Schmetterlingsporling_-_01.jpg [CC BY-SA 3.0 (https://creativecommons.org/licenses/by-sa/3.0)]-cropped (Trametes).
Currently, beekeepers are only able to indirectly control virus levels by using miticides to reduce mite infestation rates in managed honey bees. Overall, this effort has worked with only limited success, given the rapidity with which Varroa mites have developed resistance to synthetic miticides20,21. Another potential approach would be to reduce virus levels directly in honey bees by using a functional antiviral material, but no such products are currently available.
There is evidence that some fungi produce substances with demonstrable antiviral activity22,23,24,25. For example, alcohol or chloroform extracts from mycelial cultures and fruiting bodies of several polypore mushrooms (Order Polyporales) are known to have activity against pox virus, HIV-1 and H1N1 influenza24,25,26,27. Honey bees have been observed foraging directly on mycelium growing in outdoor beds28 (Fig. 1B), leading to speculation that they may be procuring a nutritional or medicinal gain. This behaviour may represent a novel facet of social immunity, given that a growing body of evidence indicates that honey bees self-medicate using plant-derived substances29,30,31. In this study, we evaluated extracts derived from the mycelia of several polypore mushroom species for activity against two major honey bee viruses in vivo in both laboratory and field studies. In both cases, reductions in DWV and LSV titers occurred in bees that were fed mycelial extracts in sucrose syrup.
Results and Discussion
Laboratory experiments with caged bees showed that oral treatment with mycelial extracts from wood conk species significantly reduced the level of DWV. The effect of treatment with fungal extracts, regardless of species, was highly significant compared to caged bees fed only sugar syrup (DWV sugar controls vs. all mycelium treatments: 2-tailed t-test, n = 58; t = 4.33, p = 0.0001, ΔΔCt fold change = 23.0). Fomes fomentarius (amadou conk) and Ganoderma applanatum (artist’s conk) extracts exhibited significant antiviral activity in a dose-dependent manner (Fig. 1C). The strongest performing extract, a 1% (v/v) F. fomentarius extract:sugar syrup mix, reduced DWV over 800-fold in caged honey bees compared to the sugar syrup control (2-tailed t-test, n = 10, t = 1.12, p = 0.005, ΔΔCt fold change = 879).
Like DWV, LSV is also highly prevalent in honey bees in the Americas and Europe9. We tested 36 individual bee abdomens sourced from the initial population and found 100% of individuals tested positive for both DWV and LSV (Fig. 2A). LSV showed higher levels of virus load than DWV, and LSV was frequently more highly expressed than the reference gene used for qPCR (Ribosomal protein 5). Treating bees in cages with fungal extracts reduced the levels of LSV (Fig. 2B), with F. fomentarius (2-tailed t-test, n = 8, t = 3.96, p = 0.009, ΔΔCt fold change = 5.40) and T. versicolor (2-tailed t-test, n = 8, t = 3.28, p = 0.0136, ΔΔCt fold change = 75) showing significant reductions. Ganoderma resinaceum demonstrated the greatest average reduction compared to control cages (2-tailed t-test, n = 8, t = 2.59, p = 0.084, ΔΔCt fold change = 499), but also the greatest variance among extracts tested. The overall effect of treatment with fungal extracts, regardless of species, was highly significant compared to caged bees fed only sugar syrup (LSV sugar controls vs. all mycelium treatments: 2-tailed t-test, n = 33, t = 3.98, p = 0.0004, ΔΔCt fold change = 21.1).

Lake Sinai virus (LSV) is reduced by fungal extract treatments. (A) All 36 individual bee abdomens from our source population tested positive for both DWV and LSV. LSV was more highly expressed than DWV, and more highly expressed than the honey bee RpS5 reference gene. (B) Fungal extracts reduced LSV titers in bees after 7 days of treatment. F. fomentarius significantly reduced LSV at a 1% dosage (2-tailed t-test, n = 8, t = 3.96, p = 0.009, ΔΔCt fold change = 5.40). The greatest fold change difference was seen with G. resinaceum extract where treatment reduced LSV levels in bees 499-fold compared to bees fed only sucrose syrup (2-tailed t-test, n = 8, t = 2.59, p = 0.084).
The two extracts exhibiting greatest fold reductions against DWV and LSV in the cage studies were selected for validation in a field trial. Small experimental colonies were treated once with 3 L of 1% fungal extract in 1:1 sucrose solution or sucrose solution only (Fig. 3A). Quantitative PCR analysis revealed that both DWV and LSV were reduced in treated colonies 12 days later (Fig. 3B). For DWV, hives treated with F. fomentarius extract showed a significant 79.7-fold reduction in viral levels (2-tailed t-test, n = 18, t = 6.58, p = 6.32 × 10−6, ΔΔCt fold change = 79.7), 44 times greater than in control colonies. G. resinaceum extracts also significantly decreased DWV levels (2-tailed t-test, n = 20, t = 9.75, p = 1.31 × 10−8, ΔΔCt fold change = 144) with treated colonies exhibiting 79.6-fold greater viral reductions than control colonies. The treatment effects from the fungal extracts were more pronounced with LSV. Although control colonies showed some reductions (2-tailed t-test, n = 18, t = 1.15, p = 0.267, ΔΔCt fold change = 82.3), F. fomentarius treatment lowered LSV levels 87.9 times greater than the control colonies (2-tailed t-test, n = 18, t = 2.50, p = 0.0238, ΔΔCt fold change = 7,380). The largest reduction in viral levels in these experiments were from G. resinaceum treatments where LSV levels were decreased 45,000 times greater than in control colonies (2-tailed t-test, n = 20, t = 6.37, p = 5.34 × 10−6, ΔΔCt fold change = 3.76 × 106).

Fungal extracts reduce DWV and LSV levels in field trials. (A) Fungal extracts were mixed into sucrose solution and fed to bees using in-hive feeders commonly used in beekeeping. (B) Both Fomes and Ganoderma extracts significantly reduced DWV and LSV levels in field trials using 5-frame colonies with bees divided from a common population. Bees were sampled for virus levels at the start of treatment and 12 days later. Colonies fed G. resinaceum extracts exhibited a 79-fold greater reduction in DWV and a 45,000-fold greater reduction in LSV compared to controls fed sugar syrup. Quantitative PCR ΔCt values are normalized to the mean starting virus levels across all groups. Photo: WSS.
In addition to the demonstrated antiviral activity of the polypore mushroom mycelial extracts, extracts from non-inoculated fungal growth substrate (birch wood) also showed some activity against DWV and LSV. Even though the birch sawdust did not show visible signs of fungal infection (See Supplemental Fig. S1), many or most free-living forest trees have cryptic endophytic and saprophytic fungal associates32,33 and many of these symbiotic endophytes provide fitness advantages to the host34,35. To evaluate this possibility, samples of the birch sawdust from this study were analyzed using next generation sequencing, and multiple species of fungi were found to be present. Three common birch associated fungi accounted for 99.5% of all mapped reads: Graphostroma platystoma, Chondrostereum purpureum, and Trametes versicolor. This raises the possibility that saprophytic and endophytic fungi, co-extracted with the birch wood, may have contributed to the activity found in the extracts, including in the uninoculated birch wood. Further studies are needed to evaluate the contributing role of these fungi relative to any activity toward viruses caused by intrinsic birch phytochemicals such as betulinic acid36,37.
This study demonstrates that extracts of several polypore mushrooms reduced RNA virus titers in honey bees in vivo. Viruses, including the DWV and LSV groups, have been reported to play a significant role in the global pattern of declining honey bee health7,9,10,11,12,13, but no approved antiviral materials are currently available for beekeepers. Viruses typically associated with honey bees are widespread among non-Apis wild pollinators38, highlighting the importance of developing a means to control virus infections in managed populations. The mycelial extracts tested here are orally active and readily consumed by bees, suggesting potential applications for beekeepers that provide critical pollination services. In addition to the potential direct impacts on honey bee health, the antiviral activity of fungal extracts can provide a research tool for the further exploration of the complex interactions between mites, viruses, and honey bee health.
Methods
Fungal mycelial extracts preparation
The following mycelial species/strains were collected and cultured by Paul Stamets:
Fomes fomentarius, Ithaca, New York
Ganoderma applanatum, Duckabush Valley, Washington
Trametes versicolor, Kamilche Point, Washington
The Ganoderma resinaceum culture originated from an anonymous source in Ontario, Canada.
Mycelial cultures were grown in sterile Petri dishes containing sterilized malt yeast agar (Fungi Perfecti, Shelton, WA). After three to four weeks of colonization in a clean room laboratory, the cultures were aseptically transferred into a 1000 mL Eberbach stirrer containing 800 mL of sterilized water. The mycelium was fragmented in the Eberbach container using a Waring blender base. The mycelial broth was then diluted 1:10 into sterilized water and transferred under sterile conditions into polypropylene incubation bags containing approximately 3 kg sterilized brown rice, which had been adjusted to approximately 45% moisture content prior to sterilization. An aliquot of 50–100 mL of diluted fluid was transferred into each of the 3 kg sterilized rice bags under sterile conditions. The fresh mycelial cultures were then incubated for 30–60 days, depending upon species, in a HEPA controlled clean room. Subsequently each mycofermented rice bag was distributed into 20 polypropylene bags of sterilized birch (Betula papyrifera) sawdust (2 kg each) and incubated for 30–60 days depending upon species.
Once colonization was determined to be sufficient (see Supplementary Fig. S1), the mycelium-colonized substrate was frozen to arrest growth, then transferred to HDPE containers for extraction. The myceliated substrate was mixed with a 50% ethanol/water solution (prepared by mixing equal weights of 95% organic ethyl alcohol and spring water) at twice the weight of the myceliated substrate, agitated, and then macerated at room temperature for 3–5 days. The mixture was pressure filtered and the supernatant decanted into containers for storage at 4 °C. The final product contains ethanol, fungal mycelial compounds and possibly unutilized growth substrate constituents. All extracts were prepared under a standardized manufacturing process and were used in subsequent tests as crude extracts of the solid substrate fermentation without further purification or characterization.
Cage and field treatments with fungal extract
For the cage studies, a large population cage was filled with approximately 10 kg of worker honey bees pooled from multiple colonies residing in a single apiary in Pullman, Washington, USA. Each experimental cage (n = 5 per treatment) was populated with approximately 300 worker bees each from the population cage and maintained in the laboratory with ad libitum sources of water and 1:1 sucrose/water syrup w/v or mycelial extracts (1%, 0.1% or 0.01% v/v in 1:1 sucrose/water syrup). Samples of 50 bees were collected from each replicated cage/treatment on day 7 and frozen until viral titer analysis. Cages where 50% or more of individuals died over the course of the experiment were not used for subsequent analyses. Three trials were run and individual bees were sampled from the population cage in the second cage trial to establish the individual virus levels in Fig. 2A.
For the field studies, 30 five-frame “nucleus” colonies, each with a laying queen and approximately 8,000 worker bees were established near Moscow, Idaho in September 2016. To create colonies with equalized starting viral levels, approximately 30 kg of worker honey bees were collected from multiple colonies residing in Washington State University experimental apiaries (Pullman, Washington) and placed in a large screened population cage. To stock the nucleus colonies, approximately 1 kg of adult worker bees was removed from the pooled population cage and added to each experimental colony. Nucleus colonies were maintained outside and allowed free flight and normal foraging throughout the experiment. All colonies were sampled before treatment to determine a viral baseline level and again 12 days following treatment. Ten replicate colonies were each fed mycelial extracts of F. fomentarius or G. resinaceum at a concentration of 0.01 (1% extract) in 3 liters of 1:1 sucrose/water syrup (extracts) or fed 1:1 sucrose syrup only (control).
Viruses - Sample processing, q-PCR, and statistical analyses
To analyze virus levels from a cage or nucleus colony, 50 mL of bees (~60–100 individuals) were collected onto dry ice and then stored at −80 °C. From each sample, 50 individual bees were then pooled for molecular analysis. Bees were homogenized in a disposable RNA extraction bag with 20 mL guanidine thiocyanate lysis (GITC) buffer, and nucleic acids were extracted using acid phenol protocol39. The nucleic acids were treated with DNase I at 37 °C for 1 h followed by 10 min at 75 °C. First-strand complementary DNA (cDNA) was generated from 2 μg total RNA using a master mix containing 50 U Superscript II (Invitrogen), random primer set (7-mer at 10- mM concentration), 2 nmol dNTP mix, 2 nmol polydT-18, and 0.1 nmol polydT (12–18). The cDNA synthesis was carried out at 42 °C for 50 min followed by 15 minutes at 70 °C37. Samples were screened for honey bee viruses via quantitative real-time PCR (1 ul cDNA template in a 20 ul reaction) using Bio-Rad SsoFast™ SYBR® Green Supermix, 96-well optical PCR plates, and a Bio-Rad CFX Connect™ thermal cycler. Positive controls (purified PCR product) and non-template controls (nuclease-free H2O) were included in each run. The thermocycler was programmed for enzyme activation at 95 °C for 30 seconds, followed by 50 cycles of denaturation at 95 °C for 5 seconds and annealing/extension 60 °C for 30 seconds.
Primer sequences used for qPCR are listed below. DWV and RpS5 primer sequences are from Englesdorp et al.38, and the sequence for LSV is from R. Cornman at the USDA-ARS (unpublished data). Both DWV and LSV primers are designed to target areas of genetic conservation in the viruses, allowing detection of the majority of genotypes.
A.mel-RpS5.F: AATTATTTGGTCGCTGGAATTG
A.mel-RpS5.R: TAACGTCCAGCAGAATGTGGTA
DWV.F: GAGATTGAAGCGCATGAACA
DWV.R: TGAATTCAGTGTCGCCCATA
LSVF: GTCATCCCAAGAGAACCACTYAC
LSVR: CRCACYGACATGAAGAAATGAGGTC
Viral levels were determined using the ∆Ct method40, normalizing the virus target expression to honey bee RpS5 reference gene expression. Statistics and standard errors were calculated using these ∆Ct values (which are log2 based). Fold change was then calculated using the ∆∆Ct method38. For cage trials, ∆∆Ct was calculated by directly comparing treatment cages to control cages. For field trials where a baseline viral level was established, ∆∆Ct was calculated within each treatment or control group and comparisons were made by dividing treatment ∆∆Ct by control ∆∆Ct. For graphical representation in Figs 1 and 2, ∆Ct values were normalized to the average viral values for the control colonies within that trial. Samples for which the RpS5 reference gene did not amplify by 40 Cts were removed. Because some hives cleared the virus to below qPCR detection levels, a value of Ct = 51 was assigned to any sample where the virus was not detected by the end of the qPCR run at Ct = 50. Student’s t-tests were performed using 2-tails and unequal variance in RStudio (RStudio Inc., Boston, MA, USA).
Sequencing
Because endophytes are common in mature trees, we tested uninoculated birch sawdust for the presence of endogenous fungi using next generation sequencing. This sawdust was used to make the birch wood extracts and served as the final growth substrate for the wood decay fungi used to make the fungal extracts. Authentechnologies (Richmond, CA) used proprietary universal primers for fungi to amplify 11,460 reads sequenced using an Ion Torrent Personal Genome Machine Next Generation DNA Sequencer.
Data Availability
The datasets generated during and/or analysed during the current study are available from the corresponding author on reasonable request.
References
Calderone, N. W. Insect pollinated crops, insect pollinators and US agriculture: Trend analysis of aggregate data for the period 1992–2009. PLOS ONE 7, e37235 (2012).
Seitz, N. et al. A national survey of managed honey bee 2014–2015 annual colony losses in the USA. J. Apic. Res. 54, 292–304 (2015).
Kulhanek et al. A national survey of managed honey bee 2015-2016 annual colony losses in the USA. J. Apic. Res. 56, 328–340 (2017).
vanEngelsdorp, D. et al. Colony Collapse Disorder: A Descriptive Study. PLOS ONE 4, e6481 (2009).
De la Rua, P. et al. Biodiversity, conservation and current threats to European honey bees. Apidologie 40, 263–284 (2009).
Martin, S. J. et al. Global honey bee viral landscape altered by a parasitic mite. Science 336, 1304–1306 (2012).
Wilfert, L. et al. Deformed wing virus is a recent global epidemic in honey bees driven by Varroa mites. Science 351, 594–597 (2016).
Chen, Y. P., Evans, J. & Feldlaufer, M. Horizontal and vertical transmission of viruses in the honey bee. Apis mellifera. Appl. Environ. Microbiol. 72, 606–611 (2006).
Daughenbaugh, K. F. et al. Honey bee infecting Lake Sinai viruses. Viruses 7, 3285–3309 (2015).
Ryabov, E. V. et al. Recent spread of Varroa destructor virus-1, a honey bee pathogen, in the United States. Scientific Reports 7, 17447 (2017).
McMahon, D. P. et al. Elevated virulence of an emerging viral genotype as a driver of honey bee loss. Proc. R. Soc. B 283, 20160811 (2016).
Natsopoulou, M. E. et al. The virulent, emerging genotype B of deformed wing virus is closely linked to overwinter honey bee worker loss. Scientific Reports 7, 5242 (2017).
Benaets, K. et al. Covert deformed wing virus infections have long-term deleterious effects on honey bee foraging and survival. Proc. R. Soc. B 284, 20162149 (2017).
Wells, T. et al. Flight performance of actively foraging honey bees is reduced by a common pathogen. Environ. Microbiol. Rep. 8, 728–737, https://doi.org/10.1111/1758-2229.12434. (2016).
Di Prisco, G. et al. A mutualistic symbiosis between a parasitic mite and a pathogenic virus undermines honey bee immunity and health. P. Natl. Acad. Sci. USA 113, 3203–3208 (2016).
Singh, R. et al. RNA viruses in hymenopteran pollinators: Evidence of inter-taxa virus transmission via pollen and potential impact on non-Apis hymenopteran species. PLOS ONE 5, e14357 (2010).
Mazzei, M. et al. Infectivity of DWV associated to flower pollen: Experimental evidence of a horizontal transmission route. PLOS ONE 9, e113448 (2014).
Cornman, R. S. et al. Pathogen webs in collapsing honey bee colonies. PLOS ONE 7, e43562 (2012).
Runkel, C. et al. Temporal analysis of the honey bee microbiome reveals four novel viruses and seasonal prevalence of known viruses, Nosema and Crithidia. PLOS ONE 6, e20656 (2011).
Sammataro, D., Untalan, P., Guerrero, F. & Finley, J. The resistance of Varroa mites (acari:varroidae) to acaracides and the presence of esterase. Internat. J. Acarol. 31, 67–74 (2005).
Kanga, L. H. B., Marshall, K. & Legaspi, J. Mechanisms of insecticide resistance in field populations of the Varroa mite (Acari: Mesostigmata: Varroidae) in Florida. Fla. Entomol. 99, 324–326 (2016).
Aoki, M. et al. Antiviral substances with systemic effects produced by Basidiomycetes such as Fomes fomentarius. Biosci. Biotech. Biochem. 57, 278–282 (1993).
Piraino, F. & Brandt, C. R. Isolation and partial characterization of an antiviral, RC-183, from the edible mushroom Rozites caperata. Antiviral Res. 43, 67–78 (1999).
Stamets, P. E. Antipox properties of Fomitopsis officinalis (ViLll.:Fr.) Bondartsev et Singer (Agarikon) from the pacific northwest of North America. Int. J. Med. Mushrooms 7, 495–506 (2005).
Sato, N., Zhang, G., Ma, C. M. & Hattori, M. Anti-human immunodeficiency virus-1 protease activity of new Lanostane-type triterpenoids from Ganoderma sinense. Chem. Pharm. Bull. 57, 1076–1080 (2009).
El-Mekkawy, S. et al. Anti-HIV-1 and anti-HIV-1-protease substances from Ganoderma lucidum. Phytochemistry 49, 1651–1657 (1998).
Krupodorova, T., Rybalko, S. & Barshteyn, V. Antiviral activity of Basidiomycete mycelia against influenza type A (serotype H1N1) and herpes simplex virus type 2 in cell culture. Virol. Sin. 29, 284–290 (2014).
Stamets, P. E. Growing Gourmet and Medicinal Mushrooms. (Ten Speed Press. New York 1993).
Simone-Finstrom, M. D. & Spivak, M. Increased resin collection after parasite challenge: a case of self-medication in honey bees? PLOS ONE 7, e34601 (2012).
Simone-Finstrom, M. D., Borba, R. S., Wilson, M. & Spivak, M. Propolis counteracts some threats to honey bee health. Insects 8, 46 (2012).
Tihelka, E. The immunological dependence of plant-feeding animals on their host’s medical properties may explain part of honey bee colony losses. Arthropod-Plant Inte. 12, 57–64, https://doi.org/10.1007/s11829-017-9553-1 (2018).
Aly, A. H., Debbab, A. & Proksch, P. Fungal endophytes: unique plant inhabitants with great promises. Appl. Microbiol. Biotechnol. 90, 1829–1845 (2011).
Atsatt, P. R. & Whiteside, M. D. Novel symbiotic protoplasts formed by endophytic fungi explain their hidden existence, lifestyle switching, and diversity within the plant kindgdom. PLOS ONE 9, e95266 (2014).
Rodriguez, R. & Redman, R. More than 400 million years of evolution and some plants still can’t make it on their own: plant stress tolerance via fungal symbiosis. J. Exp. Bot. 59, 1109–1114 (2008).
Doty, S. L. Growth-promoting endophytic fungi of forest trees in Endophytes of Forest Trees (eds Pirttila, A. M. & Frank, C.) 151–156 (Springer, Dordrecht, 2011).
Pavlova, N. I. et al. Antiviral activity of betulin, betulinic and betulonic acids against some enveloped and non-enveloped viruses. Fitoterapia 74, 489–492 (2003).
Navid, M. H., Laszczyk-Lauer, M. N., Reichling, J. & Schnitzler, P. Pentacyclic triterpenes in birch bark extract inhibit early step of herpes simplex virus type 1 replication. Phytomedicine 21, 1273–1280 (2014).
Galbraith, D. A. et al. Investigating the viral ecology of global bee communities with high-throughput metagenomics. Scientific Reports 8, 8879 (2018).
Evans, J. D. et al. Standard methods for molecular research in Apis mellifera. J. Apic. Res. 52, 1–54, https://doi.org/10.3896/ibra.1.52.4.11 (2013).
Livak, K. J. & Schmittgen, T. D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25, 402–408 (2001).
Acknowledgements
We thank the beekeepers and other colleagues who assisted in this undertaking through field and laboratory assistance or with valuable discussion: Philip Baker, Steve Cividanes, Renee Davis, James Gouin, William Hyde, Eric Olson, Louie Schwartzberg, Bulmaro Solano, Lee & June Stein, and Dusty Yao. We thank Bill Stamets for comments on the manuscript. NLN and JOH were supported by the WWW Foundation directed by Bryce, Emery and Adam Rhodes. WSS, LMC and BKH were supported in part by USDA-NIFA project WNP00604.
Author information
Authors and Affiliations
Contributions
P.E.S., N.L.N., J.D.E., J.O.H., B.K.H., A.W.T. and W.S.S. conceived and designed the experiments. P.E.S., H.M.M., R.N. and D.S. produced the mycelial extracts. N.L.N., J.D.E., J.O.H., B.K.H., D.L. and W.S.S. conducted the experiments. N.L.N., J.D.E. and J.O.H. analyzed the data. P.E.S., N.L.N., J.O.H., B.K.H. and W.S.S. wrote the manuscript. All co-authors contributed to data interpretation, and to writing of the manuscript.
Corresponding author
Ethics declarations
Competing Interests
P.E.S. holds patents pertaining to the use of fungal extracts for antiviral activity and honey bee health. W.S.S. received a research grant from Fungi Perfecti LLC to conduct cage trials.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Stamets, P.E., Naeger, N.L., Evans, J.D. et al. Extracts of Polypore Mushroom Mycelia Reduce Viruses in Honey Bees. Sci Rep 8, 13936 (2018). https://doi.org/10.1038/s41598-018-32194-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-32194-8
Keywords
This article is cited by
-
Bacterial and Fungal Symbionts in Pollen Provisions of a Native Solitary Bee in Urban and Rural Environments
Microbial Ecology (2023)
-
Antiviral Activities of a Medicinal Plant Extract Against Sacbrood Virus in Honeybees
Virology Journal (2021)
-
Enabling community-based metrology for wood-degrading fungi
Fungal Biology and Biotechnology (2020)
-
Beyond flowers: including non-floral resources in bee conservation schemes
Journal of Insect Conservation (2020)
-
Research News
IMA Fungus (2018)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.

{kind=link}
{kind=link}