
Abstract
The plant hormone auxin regulates numerous aspects of the plant life cycle. Auxin signalling is mediated by auxin response factors (ARFs) that dimerise with modulating Aux/IAA repressors. ARF3 (ETTIN or ETT) is atypical as it does not interact with Aux/IAA repressors. It is proposed to be a non-canonical auxin sensor, regulating diverse functions essential for development. This sensing ability relies on a unique C-terminal ETT specific domain (ES domain). Alignments of ETT orthologues across the angiosperm phylum revealed that the length and sequence identities of ES domains are poorly conserved. Computational predictors suggested the ES domains to be intrinsically disordered, explaining their tolerance of insertions, deletions and mutations during evolution. Nevertheless, five highly conserved short linear motifs were identified suggesting functional significance. High-throughput library screening identified an almost full-length soluble ES domain that did not bind auxin directly, but exhibited a dose-dependent response in a yeast two-hybrid system against the Arabidopsis INDEHISCENT (IND) transcription factor. Circular dichroism confirmed the domain was disordered. The identification and purification of this domain opens the way to the future characterisation of the ETT auxin-sensing mechanism in planta and an improved understanding of auxin-mediated regulation.
Similar content being viewed by others

Introduction
The plant hormone auxin (indole-3-acetic acid; IAA) is involved in almost every aspect of the plant life cycle and guides crucial developmental decisions during organ growth and differentiation1,2. Canonical nuclear auxin signalling, which provides interpretation of auxin cellular concentration and translation of the latter into a precise transcriptional outcome, relies on the activity of the auxin response factors (ARFs), which in turn are regulated by the Aux/IAA repressors3. At low cellular concentrations of auxin, the ARFs are kept in an inactive state through heterodimerisation with the Aux/IAA repressors. When auxin cellular concentrations increase, the Aux/IAA repressors are ubiquitinated and degraded, thus releasing the ARFs to regulate transcription of their target genes.
The Arabidopsis genome encodes 23 ARFs with a conserved domain organisation: a DNA binding domain (DBD), located at the N terminus4, and the PB1 (Phox and Bem1) domain at the C terminus that mediates binding to the Aux/IAA repressor and other ARF members4,5. Crystal structures of the highly conserved ARF1 and ARF5 DBDs of A. thaliana have been determined, the former in complex with its auxin response DNA element. The DBDs were observed to homodimerise with specificity of different ARFs conferred by half-site spacing4.
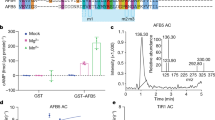
Three atypical ARFs: ARF3, ARF13 and ARF17, exhibit a different domain arrangement since they lack the PB1 domain5,6. This suggests they do not interact with Aux/IAA repressors7 and may thus function differently from canonical auxin signalling. However, in contrast to ARF13 and ARF17 that are simply truncated ARFs6, ARF3, also known as ETTIN (ETT), possesses a C-terminal domain that is not observed in other eukaryotic or prokaryotic proteins8. An alignment of ETT and its closest homologue ARF4 reveals the canonical ARF family DBD (residues 1–391) and this ETT-specific domain (ES domain; residues 392–608) (Fig. 1A).

Analyses of ETT domains, order-disorder prediction and secondary structure prediction. (A) Protein sequence alignment by SIM between ETT/ARF3 and its closest homologue ARF4. Different colours indicate percentage similarity. Below, schematic representation of the ETT showing limit of DNA binding domain (cyan) as determined by alignment with ARF4 and the following ETT-Specific (ES) domain (dark green). (B) Y2H assay between the ETT and IND, an A. thaliana transcription factor previously identified as ETT interactor8, and ETT-ES domain and IND. Interaction is observed on the selective plate -W-L-H-A. Yeast growth is inhibited by IAA in the media (lower panel). (C) A disorder prediction and putative protein binding regions for full-length ETT by PrDos and DISOPRED3 servers. (D) Secondary structure prediction by SOPMA.
The ES domain has been proposed recently to facilitate the ETT-mediated response to auxin cellular concentration8. The molecular mechanism remains to be unravelled; however, several independent methods have revealed that interaction between ETT and a range of transcription factors is affected by the presence of auxin. The interaction between ETT and INDEHISCENT (IND) is inhibited by the addition of IAA in a yeast-two-hybrid (Y2H) assay8. Interestingly, this effect was specific to the naturally occurring IAA molecule, since synthetic auxin analogues NAA (1-naphthaleneacetic acid) and 2,4D (2,4-dichlorophenoxyacetic acid) did not affect ETT heterodimerisation by Y2H8. In A. thaliana, mutational disruption of the ES domain induces developmental aberrations in different plant organs (lateral roots, branches, ovules and carpel) suggesting that ETT-mediated auxin-sensing is essential for correct organ morphogenesis8.
The direct effect of auxin on ETT heterodimerisation observed in Y2H assays suggested that ETT may interact with the IAA molecule, perhaps in a similar manner to ligand-binding by nuclear steroid hormone receptors in animals9. In order to investigate this hypothesis, we sought to carry out a detailed bioinformatics and biophysical characterisation of purified recombinant ETT protein combined with Y2H assay. The results revealed that the ES domain is predominantly intrinsically unstructured, but bears striking motifs that are almost universally conserved, both in sequence and in linear organisation, across evolutionarily diverse ETT homologues. The identification of these motifs allowed us to interpret A. thaliana phenotypic studies on ETT deletion and point mutants. A 27 kDa engineered form of the ES domain was identified by the high throughput ESPRIT method10,11 that could be produced and purified at multi-milligram quantities, and yet exhibited identical auxin responses to the wild-type sequence in Y2H assay. This opens the way for structural and biophysical studies into mechanisms of non-canonical auxin-mediated plant responses following identification of the molecular partners that interact with the conserved linear motifs in the ES domain.
Results
Bioinformatic analysis of domain composition in ETT
Analysis of ETT (UniProt O23661) using the Basic Local Alignment Search Tool (BLAST) revealed a clear sequence conservation in the N-terminal DBD region across ARF family members, but not in the C-terminal ES domain (Fig. 1A). The ES domain is both necessary and sufficient to mediate the IAA-sensitive interaction with the protein partner IND (Fig. 1B). Predictors of disorder were used to assess the foldedness of the ES domain. PrDOS12, DISOPRED313, IUPred14 all predicted high levels of disorder, in contrast to the upstream DBD (Fig. 1C and Supp. Fig. 1A). DISOPRED3 and IUPred further predicted short regions within the ES domain with characteristics found in protein-interacting motifs (Fig. 1C and Suppl. Fig. 1A). A secondary structure analysis using PSIPRED15 predicted only two short helices in the whole ES domain (residues 392–608) (Supp. Fig. 1B) with 196/217 (90%) of residues annotated as random coil. Similarly, another secondary structure predictor, SOPMA16 predicted low secondary structure content (Fig. 1D). The Phyre217 server attempts to predict structural similarities in distantly related proteins; here it identified no such similarities with the ES domain, predicting 76% of this region to be disordered (Supp. Fig. 1C). Phyre2 generates an alignment of distantly related sequences identified via PSI-BLAST18 that revealed five short highly conserved regions within the ETT C-terminal domain that otherwise varied highly in length and sequence identity (Fig. 2A–C). We used this PSI-BLAST output to guide the manual alignment of ETT members from a diverse evolutionary collection of angiosperm species, both mono- and dicotyledon (Fig. 2C,D). A broader comparison of annotated ETT protein sequences reveals these regions to be almost completely conserved in otherwise highly variant sequences including Amborella tricopoda (Fig. 2C,D), the only living species of a sister clade to extant flowering plants19.

Four motifs are conserved in the ES domain across angiosperms. (A,B) Schematic representation of the conserved motifs identified in the ES domain (A) and their sequence and possible function (B). (C,D) Alignment of ES domain from different angiosperm species (C) and their phylogenetic tree (D). Motif colour code: red, NLS; yellow, Motif 1A and 1B; blue, Ser patch; purple, Motif 2. (E) Alignment between ETT/ARF3 and ARF4 highlighting conservation of NLS, Motif 1A and 1B and Motif 2 between the two proteins.
We attempted to predict the functions of these regions using the Eukaryotic Linear Motif (ELM) database20 which identified the KRx(K/R) motif as a classic mono-partite nuclear localisation signal (NLS) and the SSPSSVL motif, that we called the Ser patch, as the site of phosphorylation by multiple kinases (Fig. 2B). The tandem repeated VLQGQE motif, called Motif 1A and 1B, and the downstream CRLFGFSLTE motif (Motif 2) were not identified by ELM and we could not find functional descriptions of these motifs elsewhere. Across the twenty-three Arabidopsis ARF family members, the predicted NLS is found in a similar position in 14 ARFs (Supp. Fig. 2). The closest ARF family member to ETT is ARF4: this contains a shorter version of the Motif 2 and the tandem repeated Motif 1A and Motif 1B (Fig. 2E) but lacks the full NLS motif and Ser patch. The identification of highly conserved motifs across ETT homologues in evolutionary distant plant species suggests that the auxin-sensing ability of ETT is of ancient origin.
Strategy for random truncation libraries of ETT
Although the approximate region of the ETT domains seems clear, precise definition of well-behaving soluble ETT constructs, especially of the ES domain, is complicated by the lack of similar proteins for sequence alignments. Our initial attempts to sub-clone constructs yielded only insoluble material, while the full-length protein was poorly expressed, insoluble and toxic. We therefore applied the ESPRIT (expression of soluble proteins by random incremental truncation) method10,11 to identify a soluble and well-expressing fragment of ETT containing most or all of the ES domain. Three different fixed C termini were defined as possible end points of ETT against which the N terminus would be randomly truncated. These comprised the native C terminus (residue 608) and two short deletions terminating at residues 602 and 594 that truncated a predicted 11 residue α-helix (Fig. 3A). DNA fragments encoding ETT 1–608, 1–602 and 1–594 were subcloned into pESPRIT00211, a reduced-size pET-derivative, downstream of an N-terminal TEV protease-cleavable hexahistidine tag and upstream and in-frame with a C-terminal biotin acceptor peptide (BAP) used as a marker of protein solubility and stability10,21. The three sequence-verified plasmid constructs were pooled at equimolar concentrations, then plasmids linearised and truncated with exonuclease III and mung bean nuclease as described11. Two sub-libraries were generated based on their size range: a medium sub-library (250–1200 bp) that spanned the suspected ES domain, and a large sub-library (800–1850 bp) that additionally included the region encoding the DBD (Fig. 3B). Following transformation of a highly competent cloning strain (MACH1), colony PCR of 34 randomly selected clones from each sub-library revealed an even distribution of construct sizes (Supp. Fig. 3). Sub-library plasmids were then used to transform E. coli BL21 AI RIL for protein expression analysis, and a total of 15975 colonies (8550 for the medium sub-library and 7425 for the large sub-library) were picked into 384-well plates, comprising an approximate three-fold oversampling of constructs.

Screening for expression and solubility from a ETT random truncation library. (A) Schematic representation of the three ETT C termini used in N-terminal truncation experiments: aa 1–608, 1–602 and 1–594. (B) Schematic representation of the strategy for identifying the soluble ES domain of ETT. From a high throughput screen of 15,975 clones, 12 clones were selected for 1 litre scale-up. (C) Summary of sequenced ETT constructs yielding purifiable fragments after 4 mL scale-up. Construct boundaries are shown with the predicted molecular weights including 5 kDa from the hexahistidine and biotin tags. (D) Fluorescent streptavidin blot against the C-terminal BAP for protein fragments summarised in (C). Uncropped gel shown in Supp. Fig. 4A,B. (E) CD analysis of the ETT_388-594 construct showing characteristic spectrum of an unstructured protein.
High throughput expression and purification screening for soluble constructs
Clones from each sub-library were robotically arrayed as inocula from 384-well plates onto nitrocellulose membranes laid over LB agar trays. Protein expression in growing colonies was induced by placing the membranes on agar trays containing L-arabinose and biotin only or L-arabinose, biotin and IAA, the latter to see whether IAA enhanced protein expression through ligand-induced stabilisation. Clones expressing putatively soluble constructs were identified by hybridising the membranes simultaneously with Alexa488-streptavidin against the biotinylated C-terminal tag, and with a monoclonal anti-hexahistidine tag antibody with associated Alexa532 mouse secondary antibody, against the N-terminal hexahistidine tag. No obvious difference was observed when comparing signals in the presence and absence of IAA. Pixel intensity values from C-terminal biotin and N-terminal hexahistidine tags were extracted for each arrayed colony and imported into a spreadsheet for analysis. Clones with no detectable hexahistidine signal were discarded and remaining clones were ranked according to their biotin intensity. Data from colony filters screened both with and without IAA was treated separately and the highest ranking 95 clones, almost all from the medium sub-library, were isolated from the library (Fig. 3B). The 95 selected ETT clones from IAA-minus and IAA-plus screens were expressed in 4 mL liquid cultures in 24-well plates along with a positive control expressing maltose-binding protein. Cells were lysed and the hexahistidine-tagged proteins purified with Ni2+-NTA agarose beads in a 96-well filter plate. Imidazole elution fractions were analysed by fluorescent streptavidin blot after SDS-PAGE and transfer to nitrocellulose membranes (Supp. Fig. 4A,B). Based on the quantity and level of proteolysis of purified proteins, 48 clones (27 from IAA-minus and 21 from IAA_plus screens) exhibiting sizes of 15–35 kDa were isolated, plasmids extracted and sequenced. Interestingly, all 48 selected clones harboured the truncated aa 594 C terminus, indicating that the deletion of the small C-terminal helix was necessary to confer soluble expression of the ETT ES-domain in E. coli. Colony PCR of randomly selected clones prior to screening steps confirmed the presence of all three fixed C-termini in the library (Supp. Fig. 4C).
Characterisation of constructs
Twelve constructs spanning 19–28 kDa (Fig. 3C) were selected for larger scale expression trials. Streptavidin blot of Ni2+-NTA small-scale purified constructs showed a continuum of sizes, consistent with a lack of compact globular structure (Fig. 3D). Based upon these preliminary results, 3 L cultures were performed for selected clones: ETT_439-594 of 21.6 kDa, ETT_388-594 of 27.6 kDa and ETT_373-594 of 28.8 kDa. All three constructs yielded approximately 3 mg of protein after Ni2+-NTA affinity purification. The longest protein, ETT_388-594, contained the entire predicted ES domain except for the C-terminal α-helix, and was selected for subsequent studies following further optimisation of the purification protocol (see methods and Supp. Fig. 5A).
Analyses by Y2H revealed the ETT_388-594 construct retained full responsiveness to increases in auxin concentration (Fig. 4A; ETT-BD and ES-BD and ETT-388-594-BD rows) confirming it to be a stable and purifiable form of the ETT auxin-responsive domain capable of binding the Arabidopsis INDEHISCENT (IND) transcription factor used as the bait in the assay. To probe the auxin sensing ability of the ES domain, random mutations were introduced into the ES domain sequence by error prone mutagenesis. A pool of ES mutants was cloned and tested in Y2H assay in the presence of IAA to isolate viable clones exhibiting IAA insensitivity. Three ES domain variants were isolated (Fig. 4A, clones ES_EP1-3 BD) harbouring amino acid substitutions at the beginning (Fig. 4A, I393T, I424T, F445I) and the end (Fig. 4A, T562A, Y597H, D601G) of the ES domain. Mutations were also identified in proximity to the Motif 1B (Fig. 4A, Q436L) and in the Ser patch (Fig. 4A, S520T) which impair the auxin sensing property of the ETT-IND dimer. In parallel to the error-prone approach, we also introduced manually point mutations into the Ser patch by site-specific mutagenesis (S523T, S S524T, S526T and S527T; Fig. 4a). Interestingly, these specific mutations within the Ser patch strongly inhibited auxin perception.

ETT 388–594 is the auxin-responsive domain. (A) Schematic representation of ETT variants and their behaviour in Y2H assay in combination with IND and in the absence and presence of auxin at different concentrations. Clones EP1, 2 and 3 were isolated from a random mutagenesis analysis of the ES domain; clones called ES_SP, referring to the Ser patch, are serine to threonine mutations introduced by site-specific mutagenesis. (B–E) Mapping and Scanning electron microscopy images of gynoecium of annotated A. thaliana wild-type (B) and ett mutant: arf3-1 (D), arf3-2 (E) and ett-3 (F). (G–J) Mapping and optical microscopy images of gynoecium of Brassica rapa wild type (G) and ett mutant: BraA.ett.a Stop220 (I) and BraA.ett.a Gln480Stop (J). Abbreviations: rp, replum; sg, stigma; st, style, va, valves. Scale bars: 100 μm (B,D,E), 1 mm (G,I,J).
In order to characterise the ES domain in purified form, an expression and purification protocol was developed yielding soluble protein at the multi-milligram levels required for structural and biophysical techniques. Six milligrams of purified protein was obtained from 2 L of bacterial culture with limiting amounts of Ni2+-NTA agarose. Elution fraction 10 comprised a strong major species and a minor component of lower molecular weight (Supp. Fig. 5A). Analysis by LC ESI-TOF mass spectrometry revealed the major species (27,192 Da) had the expected weight of ETT_388-594 (including hexahistidine and biotin peptide tags), minus the N-terminal methionine. The minor lower molecular weight species shifted in size upon TEV digestion (Supp. Fig. 5B), identifying it as a truncated form of the ES domain; the mass (18,014 Da) corresponded closely to hexahistidine-tagged ES-388-530 minus the N-terminal methionine. Analysis of protein in fraction 10 by circular dichroism (CD) revealed only unstructured polypeptide (Fig. 3E) in agreement with the bioinformatic predictions. Furthermore, the purified ES domain was unaffected by incubation at 95 °C for 1 h (Supp. Fig. 5B), a well-known characteristic of some intrinsically disordered proteins (IDPs)22. The clear absence of structure, and therefore potential for an auxin binding pocket, explained the failure to detect auxin binding by ITC in preliminary experiments (not shown).
Biological function of the ES domain
The female reproductive organ, the gynoecium, develops at the centre of the flower and it is composed of several different tissues with specific function (Fig. 4B,G): the style and stigma at the top, fundamental for pollen recognition and germination; two elongated valves that protect and enclose the developing ovules; and the replum, which separates the two valves and offers mechanical support. This precise tissue and organ sub-division is completely lost in A. thaliana ett mutants that harbour premature translational interruption in the DNA binding domain and thus delete the region identified in this study (Fig. 4C,D). Indeed, gynoecia from the arf3-1 mutant allele (A. thaliana)23 (Fig. 4D) develop with reduced valve length and width, an absence of replum and conspicuous growth of stigmatic tissue. Consistent with a role for ETT in the specification of tissue identity and patterning in female reproductive structures, ectopic growth of stigmatic tissue and irregularities at the gynoecium top can be observed along the style of the Bra.A.ett.a Stop22023 mutant (Brassica rapa, accession R-o-18) (Fig. 4H,I). Similar phenotypes are also observed in ett mutants that harbour a premature stop codon or T-DNA insertion in the ES domain. For example, gynoecia from arf3-223, ett-38,24 (Fig. 4E,F) and Bra.A.ett.a-Gln480Stop mutants (Fig. 4J) typically show severe and extreme phenotypes such as poor tissue identity and definition and ectopic growth of stigmatic tissue. This genetic evidence supports a fundamental role in the biological and functional contribution of the ES domain and motifs therein to the complete spectrum of ETT activities.
Discussion
The Auxin Response Factor ETT (ARF3) of A. thaliana is essential for correct female reproductive structure development8,25. For decades ETT has been proposed to act as an interpreter of auxin concentration in the female reproductive organ, but only recently has a molecular mechanism been proposed whereby auxin affects the interaction between ETT and protein partners, including transcription factors, leading to changes in the expression of downstream targets8,25. Such a mechanism may involve ETT binding directly or indirectly to auxin in a concentration-dependent manner. To study this interaction in vitro using purified components, we previously attempted to produce recombinant full-length ETT but encountered problems of low yield, insolubility and toxicity to host organisms. Similar problems occurred when attempting to express the ES domain alone using E. coli, yeast, insect cells and cell-free expression systems. One complicating factor in expressing the ES domain is that design of expression constructs proved difficult since BLAST yielded no similar sequences, thus sequence alignments could not be assembled for domain identification. To overcome this problem, we used ESPRIT, a random-based library construct screening process, to identify regions of ETT that could be over-expressed in E. coli. The usefulness of this method for empirical dissection of intractable proteins into manageable folded domains has been demonstrated for a diverse collection of proteins11,26 including the A. thaliana SEP3 transcription factor27. During the course of the ESPRIT screen, we performed a deeper bioinformatic appraisal of the ES domain of ETT. Our analysis using a range of structural prediction servers indicated that ETT belongs to the class of intrinsically disordered proteins (IDPs), with approximately 90% of its residues present as random coil i.e. lacking significant secondary and tertiary structure. This was subsequently confirmed by CD spectroscopic and thermal stability analyses on purified ES domain. Despite their lack of structure, IDPs are often functional and have diverse roles in forming protein-protein interactions in processes such as signalling and gene regulation28,29,30. A common characteristic of IDPs is the presence of short linear motifs (SLiMs) of typically fewer than 10 amino acids that form stable or transient complexes with other biomolecules31. Analysis of functionally equivalent ETT proteins from many plant species revealed that the length and sequence identities of their ES domains were poorly conserved, consistent with the prediction that these regions possess no folded domains and therefore tolerate sequence insertions, deletions and mutations during evolution. However, four SLiMs were identified as being almost invariably present and always in the same linear order (Fig. 2).
ESPRIT has been used previously on IDPs to express long unstructured polypeptides (100–200 residues) for biophysical and NMR studies32,33. Thus, although the experiment here was originally conceived to identify folded ES domain constructs, the results obtained are consistent with the predictions of intrinsic disorder, notably that a continuum of construct lengths were obtained, all being soluble and purifiable (Fig. 3C,D and Suppl. Fig. 4A,B). The design of the experiment, in which all 5′ unidirectional truncations of the ETT coding region were generated against three fixed 3′ ends, revealed that only those lacking a predicted 11 residue C-terminal α-helix produced protein, suggesting that deletion of this helix is necessary to obtain the ETT ES domain in E. coli at levels above the threshold of the high-throughput screen. Combining the results of the screen with the informatic predictions, a 27 kDa ETT variant ETT_388-594, encompassing the entire predicted ES domain (except the short C-terminal truncation) and including the four identified SLiMs, was selected for further functional studies.
Circular dichroism and thermal stability analyses confirmed the bioinformatic predictions of near-complete disorder of the ES domain, explaining earlier failed efforts to detect direct binding of auxin and analogues to purified ETT_388-594 in vitro using ITC (data not shown). This may also suggest why auxin presence during the ESPRIT screen or scale-up expression elicited no difference in construct behaviour (Supp. Fig. 4A,B). However, once cloned into the Y2H system, the ETT_388-594 construct exhibited full responsiveness to exogenous auxin (Fig. 4A). Thus, auxin sensing is an inherent property of the ETT_388-594 fragment in this system, and presumably in planta. The inability to detect direct auxin binding to purified ETT in vitro may be expected since an IDP will not contain a classical ligand binding pocket in isolation. By contrast, the auxin-sensing protein complex constituted in the Y2H assay requires heterodimerisation with transcription factors (e.g. IND, Fig. 1B) to elicit a response. Based on our results, we suggest a model of auxin-sensing in which ETT heterodimerises with partners, including Arabidopsis transcription factors belonging to bHLH, HOMEOBOX, AP2-Domain and YABBY families, to generate an auxin-sensing conformation. The molecular-level details remain unclear, but we speculate that it might involve the formation of an auxin-binding pocket, either in the heterodimer interfacial region, or via an allosteric mechanism whereby an auxin-binding conformation in one partner is induced upon binding the other.
Regarding the functions of the SLiMs, the prediction of a NLS by ELM is unsurprising due to the nuclear localisation of the ETT transcription factor8. The Ser patch is predicted to be a phosphorylation site of multiple kinases. A functional significance of this motif is suggested by specific and random mutagenesis results where substitution of serine to threonine induced auxin insensitivity8. The functions of the highly conserved Motif 1A, 1B and Motif 2 remain unclear, but clues about their function may be provided from Arabidopsis and Brassica rapa ett mutants with partial truncations of the ES domain that exclude the Ser patch and Motif 2; these result in phenotypic changes during gynoecium development (Fig. 4B–J). Moreover, auxin-insensitive ETT variants can be obtained by introducing specific point mutations in the ES domain8 (Fig. 4A). Therefore, the newly identified ETT 388_594 domain appears to be the same entity that mediates the effect of IAA on ETT-protein interactions. It seems possible that some of the SLiMs are direct docking sites for plant transcription factors, but further investigation will be required involving interaction screens to identify partners, mutational analyses in vitro and phenotypic studies in planta.
In this study, we have advanced the knowledge on the auxin sensitivity mechanism by delineating a functional region of ETTIN (ETT_388-594) that exhibits full sensitivity in Y2H. It can be over-expressed and purified, providing a useful tool for future structural and biophysical studies. This region in isolation is predicted to be intrinsically unstructured and is poorly conserved across species, but contains four highly conserved linear motifs arranged in a constant order. We propose that these are involved in interactions with importins (NLS), kinases (Ser patch) and maybe transcription factors in promoter complexes of auxin-sensitive genes implicated in floral organ morphogenesis.
Methods
Amplification and sub-cloning of the ETT gene
The ETT coding sequence was amplified by PCR from Arabidopsis thaliana Col-0 inflorescence cDNA with primers ETTfor (5′-ATGCA TGCGG CGCGC CTAGA TGGGT GGTTT AATCG ATCTG AACGT GATG-3′) and ETT-FLrev (5′-ATGCA TGCAT CCATT GAGAG CAATG TCTAG CAACA TGTCT CTCAT TGC-3′), ETT-MRMD-rev (5′-ATGCA TGCAT GCATT CATGT CTCTC ATTGC ATAGA TGTCC CTTCC TTG-3′) and ETT-SQGRrev (5′-ATGCA TGCAT GCATT CCTTC CTTGC GAATG ATGAT GATTG CTTTG-3′) to obtain full-length ETT (ending 608) and the two ETT truncated variants ETT-MRMD (ending 602) and ETT-SQGR (ending 594) respectively. Internal NsiI sites were silenced by site-directed mutagenesis. The fragments were cloned in the pGEM-T-easy vector (Promega) and sequenced. Subsequently, fragments were excited from the pGEM-T vectors by digestion with AscI and NsiI and inserted in the pESPRIT002 vector11, designed for the 5′ truncation of the gene, and resulting in inserts fused to an N-terminal TEV-cleavable hexahistidine tag (MGHHHHHHDYDIPTTENLYFQG) and a C-terminal biotin acceptor peptide (SNNGSGGGLNDIFEAQKIEWHE).
ESPRIT library screening for soluble domains of ETT
Full methods and materials for ESPRIT have been detailed recently11. Briefly, 5′ deletion libraries of the ETT gene were constructed on an equimolar pool of the three plasmids above using exonuclease III and mung bean nuclease and recovered into E. coli MACH1 (Thermo Fisher Scientific). The linear distribution of insert sizes was confirmed by colony PCR with insert-flanking primers. Plasmid DNA was purified from medium and large insert sub-libraries and used to transform E. coli BL21 AI RIL for expression screening. Sub-libraries were titrated on agar trays and 15,975 colonies (8550 for the medium sub-library and 7425 for the large sub-library) were picked robotically into 384-well plates, corresponding to a three-fold oversample of each clone, then arrayed onto nitrocellulose membranes over LB agar with antibiotics. Once visible colonies were apparent, membranes were transferred to LB agar containing antibiotics, arabinose and biotin for protein expression for 4 h at 30 °C. Membrane-supported colonies were lysed using NaOH and membranes neutralised and blocked with Superblock (Thermo Fisher Scientific). Membranes were hybridised with fluorescent probes: streptavidin Alexa 488 to detect in vivo biotinylation of the C-terminal biotin acceptor peptide on the target proteins, and antihexahistidine mouse monoclonal followed by Alexa532 labelled rabbit anti-mouse secondary antibody to detect the N-terminal hexahistidine tags. Quantitative analysis of fluorescence intensities of both signals per colony resulted in 95 putatively soluble clones re-arrayed into a destination plate. These were grown at 4 mL scale and hexahistidine-tagged proteins purified in a 96-well plate format with His6-MBP-BAP as a positive control. Eluted proteins were analysed by fluorescent streptavidin blot with fluorimagery, and InstantBlue (Merck) stained SDS-PAGE. The plasmid inserts from clones yielding visible purified proteins were DNA sequenced.
Expression and purification of selected ETT clones
Clones were grown in 1 L LB medium (kanamycin 50 μg/mL and chloramphenicol 30 μg/mL) at 37 °C until OD600 0.8. The temperature was reduced to 18 °C, and cultures were induced with 0.2% w/v L-arabinose overnight. Cells were harvested by centrifugation (11,000 g for 20 min at 4 °C) and the pellet frozen at −70 °C. Five grams of wet cell pellet were lysed by sonication in lysis buffer (8 M aqueous urea, 1 mM tris (2-carboxyethyl)phosphine (TCEP)), then centrifuged at 20,000 g, 40 min, 4 °C. Ni2+-NTA agarose resin (1.5 mL of 50% slurry) was added to the supernatant and incubated for 30 min at room temperature, then loaded on a gravity column. Five 50 mL wash steps were performed with buffers containing decreasing urea concentrations (8, 4, 2 and 1 M), 20 mM imidazole and 1 mM TCEP. A final wash was performed in equilibration buffer (50 mM Na phosphate pH 7, 50 mM NaCl, 20 mM imidazole, 1 mM TCEP), then purified proteins eluted in 1 mL fractions with elution buffer (50 mM Na phosphate pH 7, 50 mM NaCl, 300 mM imidazole, 1 mM TCEP). Total, soluble and unbound lysate samples, plus 10 elution fractions were analysed by 15% SDS-PAGE with staining by InstantBlue. For CD and thermal stability analyses, 1 mL of fraction 10 was dialysed twice against 1 L of Tris 5 mM pH 8.5 1 mM TCEP.
Characterisation of purified ETT C-terminal domain
LC ESI-TOF analysis was performed by the IBS-ISBG mass spectrometry platform on an Agilent Technologies 6210 spectrometer coupled to a capillary HPLC system (1100 series). The far-UV CD spectrum (from 190 nm to 260 nm) was collected on a JASCO J-810 CD spectrophotometer. The ETT 388_594 sample at 6.5 µM in Tris 5 mM pH 8.5 1 mM TCEP was scanned ten times at 20 °C in a 1 mm path-length cuvette. After subtraction of baseline buffer values, the CD signal, expressed in mdeg, was converted to mean molar residue ellipticity (expressed in deg.cm2.dmol-1). Data were plotted using Prism (GraphPad Software, San Diego). The thermal stability analysis involved heating ETT 388_594 in the same buffer used for CD for 20, 40 and 60 min, followed by centrifugation at 13,000 g, 10 min. Supernatants were loaded on SDS-PAGE with an unheated centrifuged reference sample, electrophoresed and analysed after staining with InstantBlue.
Yeast two-hybrid (Y2H) assay
The Y2H assays were performed at 28 °C in the yeast strain AH109 (Clontech) using the co-transformation technique34. Coding sequences were cloned into the Gateway vector GAL4 system (pGADT7 and pGBKT7; Clontech) via pDONR207 (Life Technologies). Strength of interaction was tested on selective yeast synthetic dropout medium (YSD) lacking Leu (L), Trp (W), Adenine (A), and His (H). IAA was dissolved in ethanol and added at the desired concentration directly to the cooling media.
Plant Materials and Growth Conditions
Arabidopsis plants were grown on soil in long-day conditions (16 h light/8 h dark). T-DNA insertional mutants23 were obtained from the Nottingham Arabidopsis Stock Centre. Brassica rapa plants were grown in soil in 1 litre pots under long-day conditions (16 h light/8 h dark). Mutant lines ett-3, arf3-1, arf3-2, Bra.A.ett.a.Stop220 have been described previously24,35,36. Bra.A.ett.a.Stop220 and Bra.A.ett.a.Gln480Stop mutants can be found at http://revgenuk.jic.ac.uk/, codes JI32298-B and JI33329-B respectively.
Scanning electron microscopy
Whole gynoecia and flowers were fixed 16 h at 25 °C in 3.7% formaldehyde, 5% glacial acetic acid, and 50% ethanol. After a complete dehydration through an ethanol series until 100%, gynoecia were critical point dried. Samples were dissected, mounted on metal stubs and coated with gold and examined under Zeiss Supra 55VP field emission scanning electron microscope using an acceleration voltage of 3 kV.
Error-prone screening of ETT ES domain
Error-prone PCR mix was composed of G2-polymerase buffer (Promega) and G2-polymerase, 5 mM MgCl2, 0.15 mM MnCl2, unbalanced dNTP mix (2.5 mM dATP, 2.5 mM dGTP, 10 mM dCTP, 10 mM dTTP). The pool of PCR fragments was incubated with the pGADT7-rec plasmid (Clontech) following the co-transformation protocol described in the “Matchmaker Library Construction & Screening User Manual (Clontech)”. Cells were plated on YSD selective media lacking Trp (W), Leu (L), Ade (A) and His (H) and supplemented with 100 μM IAA. Plasmids were recovered from those colonies able to grow in presence of IAA and sequenced.
References
Benjamins, R. & Scheres, B. Auxin: The Looping Star in Plant Development. Annual Review of Plant Biology 59, 443–465 (2008).
Vanneste, S. & Friml, J. Auxin: A Trigger for Change in Plant Development. Cell 136, 1005–1016 (2009).
Calderón Villalobos, L. I. A. et al. A combinatorial TIR1/AFB–Aux/IAA co-receptor system for differential sensing of auxin. Nature Chemical Biology 8, 477–485 (2012).
Boer, D. R. et al. Structural basis for DNA binding specificity by the auxin-dependent ARF transcription factors. Cell 156, 577–589 (2014).
Guilfoyle, T. J. The PB1 Domain in Auxin Response Factor and Aux/IAA Proteins: A Versatile Protein Interaction Module in the Auxin Response. The Plant Cell Online 27, 33–43 (2015).
Guilfoyle, T. J. & Hagen, G. Auxin response factors. Current Opinion in Plant Biology 10, 453–460 (2007).
Vernoux, T. et al. The auxin signalling network translates dynamic input into robust patterning at the shoot apex. Molecular Systems Biology 7, 508–508 (2014).
Simonini, S. et al. A noncanonical auxin-sensing mechanism is required for organ morphogenesis in. Arabidopsis. Genes & Development 30, 2286–2296 (2016).
Molecular Biology of Steroid and Nuclear Hormone Receptors, https://doi.org/10.1007/978-1-4612-1764-0 (Birkhäuser Boston, 1998).
Yumerefendi, H., Tarendeau, F., Mas, P. J. & Hart, D. J. ESPRIT: an automated, library-based method for mapping and soluble expression of protein domains from challenging targets. J Struct Biol 172, 66–74 (2010).
Mas, P. J. & Hart, D. J. ESPRIT: A Method for Defining Soluble Expression Constructs in Poorly Understood Gene Sequences. Methods Mol. Biol. 1586, 45–63 (2017).
Ishida, T. & Kinoshita, K. PrDOS: prediction of disordered protein regions from amino acid sequence. Nucleic Acids Research 35, W460–W464 (2007).
Jones, D. T. & Cozzetto, D. DISOPRED3: precise disordered region predictions with annotated protein-binding activity. Bioinformatics 31, 857–863 (2015).
Dosztanyi, Z., Csizmok, V., Tompa, P. & Simon, I. IUPred: web server for the prediction of intrinsically unstructured regions of proteins based on estimated energy content. Bioinformatics 21, 3433–3434 (2005).
Buchan, D. W. A., Minneci, F., Nugent, T. C. O., Bryson, K. & Jones, D. T. Scalable web services for the PSIPRED Protein Analysis Workbench. Nucleic Acids Res 41, W349–W357 (2013).
Geourjon, C. & Deléage, G. SOPMA: significant improvements in protein secondary structure prediction by consensus prediction from multiple alignments. Comput. Appl. Biosci. 11, 681–684 (1995).
Kelley, L. A., Mezulis, S., Yates, C. M., Wass, M. N. & Sternberg, M. J. The Phyre2 web portal for protein modelling, prediction and analysis. Nat Protoc 10, 845–858 (2015).
Altschul, S. F. et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25, 3389–3402 (1997).
Amborella Genome Project. The Amborella genome and the evolution of flowering plants. Science 342, 1241089 (2013).
Dinkel, H. et al. ELM 2016—data update and new functionality of the eukaryotic linear motif resource. Nucleic Acids Res 44, D294–D300 (2016).
Tarendeau, F. et al. Structure and nuclear import function of the C-terminal domain of influenza virus polymerase PB2 subunit. Nat. Struct. Mol. Biol 14, 229–233 (2007).
Kalthoff, C. A novel strategy for the purification of recombinantly expressed unstructured protein domains. Journal of Chromatography B 786, 247–254 (2003).
Alonso, J. M. et al. Genome-wide insertional mutagenesis of Arabidopsis thaliana. Science 301, 653–657 (2003).
Sessions, A. et al. ETTIN patterns the Arabidopsis floral meristem and reproductive organs. Development 124, 4481–4491 (1997).
Simonini, S., Bencivenga, S., Trick, M. & Østergaard, L. Auxin-Induced Modulation of ETTIN Activity Orchestrates Gene Expression in Arabidopsis. Plant Cell 29, 1864–1882 (2017).
Hart, D. J. & Waldo, G. S. Library methods for structural biology of challenging proteins and their complexes. Curr. Opin. Struct. Biol. 23, 403–408 (2013).
Puranik, S. et al. Structural Basis for the Oligomerization of the MADS Domain Transcription Factor SEPALLATA3 in Arabidopsis. Plant Cell 26, 3603–3615 (2014).
Dunker, A. K., Silman, I., Uversky, V. N. & Sussman, J. L. Function and structure of inherently disordered proteins. Current Opinion in Structural Biology 18, 756–764 (2008).
Tompa, P. Unstructural biology coming of age. Current Opinion in Structural Biology 21, 419–425 (2011).
Wright, P. E. & Dyson, H. J. Intrinsically Disordered Proteins in Cellular Signaling and Regulation. Nat Rev Mol Cell Biol 16, 18–29 (2015).
Davey, N. E. et al. Attributes of short linear motifs. Mol. BioSyst. 8, 268–281 (2011).
Desravines, D. C. et al. Structural Characterization of the SMRT Corepressor Interacting with Histone Deacetylase 7. Sci Rep 7, 3678 (2017).
Sukackaite, R. et al. Structural and Biophysical Characterization of Murine Rif1 C Terminus Reveals High Specificity for DNA Cruciform Structures. J. Biol. Chem. 289, 13903–13911 (2014).
Egea-Cortines, M. Ternary complex formation between the MADS-box proteins SQUAMOSA, DEFICIENS and GLOBOSA is involved in the control of floral architecture in Antirrhinum majus. The EMBO Journal 18, 5370–5379 (1999).
Okushima, Y. Functional Genomic Analysis of the AUXIN RESPONSE FACTOR Gene Family Members in Arabidopsis thaliana: Unique and Overlapping Functions of ARF7 and ARF19. The Plant Cell 17, 444–463 (2005).
Simonini, S., Stephenson, P. & Østergaard, L. A molecular framework controlling style morphology in Brassicaceae. Development 145 (2018).
Acknowledgements
SS is supported by grants BBSRC BB/M004112/1 (to LØ) and BB/P013511/1 (to the John Innes Centre), a JIC Institute Development Grant (16.03.3), an EMBO Short Term fellowship (ASTF 585-2015) and a grant from Instruct-ERIC (PID1526). It used the platforms of the Grenoble Instruct-ERIC centre (ISBG; UMS 3518 CNRS-CEA-UGA-EMBL) with support from FRISBI (ANR-10-INBS-05-02) and GRAL (ANR-10-LABX-49-01) within the Grenoble Partnership for Structural Biology (PSB). We thank F. Gerard for assistance with biophysical analyses, and T. Gibson (EMBL) for discussion of sequence alignments.
Author information
Authors and Affiliations
Contributions
S.S., D.H. and L.O. designed experiments. S.S., P.M. and C.M. performed experimental steps and prepared the figures. All authors participating in analysis of data. S.S. and D.H. wrote the manuscript with input from P.M. and L.O.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Simonini, S., Mas, P.J., Mas, C.M.V.S. et al. Auxin sensing is a property of an unstructured domain in the Auxin Response Factor ETTIN of Arabidopsis thaliana. Sci Rep 8, 13563 (2018). https://doi.org/10.1038/s41598-018-31634-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-31634-9
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
