
Abstract
Galectin-9 (Gal9) has been postulated to have anti-inflammatory properties based on the ability of exogenous Gal9 to induce apoptosis in synovial fibroblasts in animal models of rheumatoid arthritis (RA). Here we aimed to assess the potential role of endogenous Galectins, including Gal9, in the inflammatory pathology of the RA synovium in humans. Firstly expression of Galectins 1–9 was determined in synovial fibroblasts (RASF) and dermal fibroblasts (DF) isolated from RA patients, the latter representing a non-inflamed site. We then further challenged the cells with pro-inflammatory TLR agonists and cytokines and assessed Galectin expression. Gal9 was found to be differentially and abundantly expressed in RASF compared to DF. Agonists of TLR3 and TLR4, along with IFNgamma were also found to induce Gal9 expression in RASF. siRNA was then used to knock-down Gal9 expression in RASF and the effects of this on apoptosis and cell viability were assessed. Increased apoptosis was observed in RASF following Gal9 knock-down. We conclude that, unlike exogenous Gal9, endogenous Gal9 is protective against apoptosis and enhances synovial fibroblast viability suggesting that its role in RA is both pathogenic and pro-inflammatory.
Similar content being viewed by others


Introduction
Rheumatoid arthritis (RA) is a chronic inflammatory disease which leads to destruction of the joint and expansion of the synovial membrane leading to an “activated” synovial membrane. Multiple cell types are recruited to the inflammatory milieu which develops within the normally acellular synovium. Although the exact mechanisms driving the onset and maintenance of inflammation in RA are still being determined, it is known that fibroblasts, macrophages and pro-inflammatory B and T cell populations significantly contribute to RA disease pathology1,2,3,4,5,6 including IL-6, IL-23p19, CCL-20, and GM-CSF7,8,9. Their presence within the RA synovium is thus thought to be an important driver of the chronicity of inflammation1,10,11,12.
Galectins are an evolutionarily conserved family of immunomodulatory animal lectins which are expressed in a number of immune cell populations including macrophages, T cells and fibroblasts13. Eleven galectins have been described in humans and they have a broad range of actions14. In addition, several galectins have been implicated in the regulation of cell death, notably galectins -1 (T and B lymphocytes), -7 (keratinocytes and carcinomas), -8 (carcinomas), -9 (thymocytes) and -12 (adipocytes) (reviewed in15). Galectin-9 (Gal9) was first described as a selective chemoattractant for eosinophils16, but is now known to have a wider function including as a urate transporter in the kidney17. Gal9 functions as a voltage-sensitive channel that mediates transport of urate, a product of purine metabolism that is elevated in the serum of individuals with renal dysfunction and associated with development of gout.
In the immune system Gal9 has been shown to regulate interactions between thymic epithelial cells and thymocytes and promotes apoptosis of immature thymocytes when applied exogenously18. Similar to galectin-1, Gal9 induces cell death of mature activated T cells through caspase and calpain-dependent pathways19. The surface receptor that is crucial for exogenous Gal9 function is TIM3 (T cell immunoglobulin- and mucin domain containing molecule) expressed on differentiated T helper cells20,21. Dysregulation of this signalling pathway has been documented in autoimmune diseases such as multiple sclerosis22. Activation of the Gal9/TIM3 pathway has also been shown to be important in graft rejection23. In addition to pro-apoptotic activity, Gal9 induces differentiation of naïve T cells into a regulatory phenotype while inhibiting their development into Th17 cells2 and stimulates maturation of dendritic cells with production of Th1-type cytokines24. Gal9 has been shown to be expressed in RA synovium and it has been reported that a mutant form of Gal9 was able to induce apoptosis in synovial fibroblasts when added exogenously to cell cultures25. Furthermore, Gal9 was shown to have a beneficial effect on mouse collagen-induced arthritis when given exogenously25. However, it is clear that galectins often have different functions in the extracellular and intracellular context14,26 and the role of endogenous Gal9 in synovial fibroblasts and RA pathogenesis has not been established. With an emerging role for galectins in immune modulation and inflammatory diseases the present study examined galectin expression in the specific context of RA.
Results
Galectin 9 is differentially expressed in RA synovial tissue
Analysis of galectin expression by real time PCR in synovial fibroblasts and matched skin fibroblasts showed expression of Gal1, -3, -4, -8 and -9 in both synovial and dermal fibroblasts (Fig. 1). However, Gal9 was the only galectin that showed increased expression in the synovial compared to the dermal fibroblasts (p < 0.01). Immunofluorescence staining of synovial tissue sections from RA patients confirmed extensive Gal9 expression. Co-staining with the synovial fibroblast markers, podoplanin and fibroblast activation protein (FAP) demonstrated that Gal9 was expressed largely by fibroblasts (Fig. 2). Given these data, we focussed on the regulation and role of Gal9 in synovial fibroblasts.

Galectin expression was assessed in synovial and dermal fibroblasts from RA patients by real-time PCR. Data are mean ± SD and * indicates p < 0.05, n = 6.

Synovial membrane sections were stained for Podoplanin (green), FAP (red), galectin-9 (blue) and nuclei using DAPI (white). Co-localisation of podoplanin and galectin-9 can be seen as cyan, co-localisation of FAP and galectin-9 appears magenta. Scale bar 50 μm.
Regulation of Gal9 expression
Toll-Like Receptor effects on Gal9 Expression
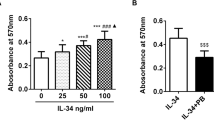
In order to assess which factors might modulate Gal9 expression we first considered whether toll-like receptor (TLR) activation could affect expression of Gal9. RA synovial fibroblasts were stimulated with the TLR ligands bLP, poly(I:C) and LPS. No significant effects on Gal9 mRNA expression were observed following stimulation with bLP, but stimulation with poly(I:C) (a TLR3 agonist) gave a significant (p < 0.01) 25-fold increase in Gal9 mRNA. LPS (a TLR4 agonist) stimulation led to a smaller, but significant (p < 0.05) 10-fold increase in Gal9 mRNA (Fig. 3A). Thus, Gal9 expression can be induced by activation of the TLR3 and TLR4 pathways. By contrast Gal3 expression could not be induced by any of the TLR ligands used here, suggesting some selectivity for TLR3 and TLR4 signalling towards induction of Gal9 (data not shown).

RA synovial fibroblasts were stimulated with (A) TLR agonists, TNFα, IL-1β, bLP, poly(I:C), LPS and (B) pro-inflammatory cytokines –IL-1β, IL-4, IL-6, IL-17, IFNγ, TNFα and TGFβ. mRNA was extracted and fold-changes were plotted. (C) The increase in Gal9 expression following IFNγ stimulation was confirmed at the protein level by Western blotting. Long (39 KDa) and short (35 KDa) isoforms of Gal9 are shown. The short isoform was induced by IFNγ stimulation whereas the long isoform is constitutively expressed. *p < 0.05, **p < 0.01. Full uncropped versions of the blots are shown in the supplementary data.
Cytokine effects on Gal9 expression
The RA synovial environment contains many pro-inflammatory cytokines, including IL-1β, IL-4, IL-6, IL-17, IFNγ, TNFα and TGFβ which mediate different aspects of disease pathology. In order to determine whether they play a role in the upregulation of Gal9 expression, RA synovial fibroblasts and matched skin fibroblasts were stimulated with each of the aforementioned cytokines. Only IL-1β (4-fold) and IFNγ (35-fold) were found to induce significant (p < 0.05) increases in Gal9 mRNA expression (Fig. 3B). The effects of IFNγ were studied further with increases in Gal9 protein expression in synovial fibroblasts in response to IFNγ stimulation confirmed by Western blot (Fig. 3C, upper panel). Two isoforms of Gal9 exist -a long (39 KDa) and short (35 KDa) form. The long isoform was constitutively expressed in RA synovial fibroblasts and expression was increased in a time-dependent manor following IFNγ stimulation for 24–48 hours. However IFNγ stimulation induced a more profound effect on the short isoform, which was not readily detectable in untreated cells but was evident after 24 hours. Increased expression of the short isoform could be inhibited by an anti-IFNγ antibody (Fig. 3C, lower panel).
Effect of Galectin 9 Silencing on synovial fibroblasts
To determine whether the increased expression of Gal9 might affect synovial fibroblast proliferation and apoptosis RA synovial fibroblasts were transfected with Gal9 siRNA, or a scrambled siRNA (Scr) sequence and a control group of cells were left un-transfected. Effective silencing of Gal9 expression using RNA interference was confirmed by Western blotting (Fig. 4A). In order to assess whether knockdown of Gal9 affected fibroblast proliferation, XTT reduction to formazan was measured at 48, 72 and 96 h post Gal9 siRNA transfection. Gal9 knockdown was shown to have no effect on fibroblast proliferation stimulated with TNFα and IL-1β (10 ng/ml each) (Fig. 4B). AnnexinV binds to phosphatidylserine exposed on the cell membranes of apoptotic cells. Staining with annexin V-FITC showed greater binding of annexin V to the Gal9 siRNA transfected population compared to the scr siRNA population (Fig. 3C), suggesting that loss of Gal9 leads to increased apoptosis in these cells. This finding was confirmed using JC-1 staining to measure mitochondrial membrane depolarisation as a second measure of apoptosis, (Fig. 4D), consistent with increased apoptosis after Gal9 knock down. The Gal9 siRNA transfected fibroblasts also displayed visual signs of cell death 24 hours post-transfection with marked cell cytoplasm shrinkage, rounding up of cells and loss of classic fibroblast morphology (Fig. 5A).

(A) Gal9 knock-down following siRNA transfection was confirmed by Western blotting. (B) Cell proliferation in the presence of TNFα and IL-1β (10 ng/ml of each) was shown to be unaffected by siRNA transfection. Data are mean ± SD. Apoptosis in each fibroblast population was measured by (C) annexin V staining and (D) JC-1 mitochondrial membrane depolarisation staining. *p < 0.05, ***p < 0.001. Full uncropped versions of the blots are shown in the supplementary data.

(A) Fibroblasts were visualised by light microscopy following transfection with scr siRNA, Gal9 siRNA or treatment with Lipofectamine only (no RNAi control). Original magnification x40. Images are representative of 2 populations from a total of 5 primary RA synovial fibroblast populations observed. (B) Cell counts of n = 5 primary human synovial fibroblast populations in a 12-well plate following Gal9 siRNA transfection. Data are pooled from three separate experiments. Statistical significance was determined using a one-way ANOVA with Tukey post-hoc test.***p < 0.001.
Discussion
Galectins 1 and 3 have previously been described as mediators of inflammatory processes27,28,29 where they act as chemoattractants for neutrophils, and facilitate recognition and killing of bacteria in pathogen-mediated inflammation10. In the present study we assessed galectin expression in fibroblasts from synovial tissue and matched non-inflamed skin of patients with RA and showed that expression of Gal9 was unique in being differentially increased in the synovial fibroblasts. Gal9 has been reported to be expressed at high levels within the RA synovium and previous reports have shown exogenous Gal9 to have a pro-apoptotic role within this tissue25. The very low level of Gal9 in skin fibroblasts in the present study suggests that the raised Gal9 could be a consequence of the local inflammatory environment. Fibroblasts play a central role in the “activated” synovium, recruitment of immune cells to the inflammatory milieu, and thus maintenance of persistent inflammation1,30. Their survival and proliferation within the inflamed joint may, therefore, represent a key early step in the transition to chronic inflammation. Knockdown of Gal9 by siRNA induced apoptosis in RA synovial fibroblasts, suggesting that endogenous Gal9, in contrast to exogenous Gal925, protects against rather than induces apoptosis.
The induction of apoptosis following siRNA knockdown of Gal9 compared with the pro-survival effects of exogenous Gal9 shown previously by Seki and colleagues25 indicates that endogenous and exogenous forms of Gal9 play opposing roles in regulating cell death. In the RA synovium, fibroblast populations are maintained, in part due to the cytokine and chemokine repertoire produced by the inflammatory milieu. We have demonstrated here that activation of the TLR3 and TLR4 pathways in synovial fibroblasts lead to increased protection from apoptosis of synovial fibroblasts through induction of Gal9 expression. TLR expression in the RA synovium has been well characterised31 and it is known that TLRs in concert with pattern recognition receptors (PPRs), in particular nucleotide-binding oligomerisation domain 2 (NOD-2) activate synovial fibroblasts and promote and maintain inflammatory mediator expression32.
In the current study, IFNγ was the most potent inducer of Gal9 expression (Fig. 3B). Additionally, our data suggest that an inflammatory environment containing IFNγ would profoundly upregulate the short isoform of Gal9. Therefore, it could be that this short isoform is of functional significance in protection of synovial fibroblasts against apoptosis during inflammation and may represent an important therapeutic target.
We have shown that endogenous Gal9 expression also maintains the fibroblast population through protection against apoptosis, and thus contributes directly to the maintenance of persistent inflammation within the RA joint. Understanding the endogenous and exogenous roles of Gal9 is integral to understanding how to manipulate it for use as a therapeutic target. Endogenous Gal9 has been implicated in prevention of malignant tumour progression through disruption of CD44-hyaluronan interactions33, acting as a potent IgE antagonist preventing degranulation of mast cells34 and as an inducer of immunosuppressive macrophages which ameliorate T cell-driven inflammation35. Exogenous Gal9, however, has been shown to induce apoptosis in synovial fibroblasts, T cells25,36 and other immune cell lineages18 to prevent Th17 differentiation2 and to reduce inflammation in psoriasis patients37. Unlike Gal3, Gal9 is largely intracellular in vivo due to its lack of a signal peptide for secretion38. There is currently no evidence that fibroblasts are able to secrete the Gal9 which they produce, unlike T cells which readily secrete it upon T cell receptor (TCR) stimulation39. Therefore, it is possible that intracellular Gal9 effects may dominate extracellular Gal9 effects that are generated through cell surface receptors such as TIM3. The nature of Gal9 intracellular function could be similar to that observed for intracellular Gal3, which is known to interact with members of the anti-apoptotic Bcl-2 family40. There are, therefore, two possible means of exploiting Gal9 as a therapeutic: firstly removing blockade of apoptosis through blockade of endogenous, intracellular Gal9. Secondly, through administration of exogenous Gal9 which can signal through membrane bound receptors to induce apoptosis. We conclude that Gal9 represents an important and novel target for treatment of RA and potentially other auto-inflammatory diseases.
Methods
Galectin expression in synovial tissue and fibroblasts
RA synovial tissue was obtained from biopsies from RA patients recruited from Sandwell and West Birmingham Hospitals NHS Trust and University Hospitals Birmingham NHS Foundation Trust UK. All patients fulfilled the 1987 ACR classification criteria for RA with symptom duration of greater than 3 months, were CCP+, RHF+ and underwent synovial biopsy of the knee. All patients provided written, informed consent (West Midlands Black Country Research Ethics Committee Approval 07/H1203/57). Frozen RA tissues were cut in 6 μm thick sections, fixed with acetone and frozen prior to use. Slides were washed in Phosphate Buffered Saline (PBS), incubated for 30 min in 10% Horse serum/PBS and incubated overnight at 4circC with primary antibodies to Galectin-9 (Abcam, Cambridge, UK), Fibroblast Activation Protein, FAP (Invitrogen, Waltham, MA, USA, clone F11-24) and Podoplanin (Invitrogen, clone NZ-1.3). After washing with PBS, slides were incubated with secondary antibodies anti-mouse IgG1 Alexa 546 (Life Technologies, Paisley, Scotland), anti-rabbit IgG Alexa 488 (Life Technologies) and anti-rat IgG Alexa 647 (Jackson ImmunoResearch, Cambridge, UK) for 30 min, counterstained with DAPI (Life Technologies) and mounted in aqueous mounting media. Tissues were imaged using a Zeiss LSM 780 confocal microscope and processed using ZEN black (ZEISS, Cambridge, UK). Fibroblasts were isolated by explant culture from synovial tissue and skin from the same patients as previously described41 and grown to confluence to determine expression of galectins 1, 3, 4, 8 and 9 by real-time PCR. All patients provided written, informed consent (West Midlands Black Country Research Ethics Committee Approval 07/H1204/191).
Real-Time PCR
RNA was prepared using an RNeasy Mini Kit (Qiagen, Manchester, UK) and reverse transcription was carried out using Superscript Vilo (Life Technologies, Paisley, UK) according to the manufacturer’s instructions. Real-time PCR was carried out using FAM-labelled TaqMan® Human Gene Expression assays for the galectins of interest as well as IL-1β, IL-4, IL-6, IL-17, IFNγ, TNFα, TGFβ and β-actin control (Life Technologies, Paisley, UK). All samples were analysed using a 7900HT real-time PCR machine (Life Technologies, Paisley, UK). Data were expressed as ΔΔCt values.
Cell stimulation
RA synovial fibroblasts were seeded into 24 well plates and incubated at 37 °C, 5% CO2 until confluent. The culture medium was replaced with 500 μl fresh complete RPMI 1640 medium (10% FCS, 400 μM L-glutamine, 2,000 U/ml penicillin, 2 mg/ml streptomycin, 1% v/v NEAA, 1% v/v sodium pyruvate, all from Sigma Aldrich, Gillingham, UK) prior to treatment with 10 ng/ml of each of the cytokines IL-1β, IL-17, IFNγ, TNFα, TGFβ, IL-4 (all R&D Systems, Abingdon, UK), or IL-6 (Peprotech, New Jersey, USA) (20 ng/ml), or TLR agonists 300 ng/ml bacterial lipoprotein (bLP; InvivoGen, San Diego, US), 10 μg/ml poly(I:C) (InvivoGen, San Diego, US), or 100 ng/ml LPS (List Biological Laboratories, Campbell, USA). The cells were incubated for 24 hours at 37 °C, 5% CO2 after which RNA was extracted for Gal9 expression as determined by Real time PCR.
Western blotting
Synovial fibroblasts were washed with PBS and lysed with SDS loading buffer (0.125 M Tris pH 6.8, 20% glycerol, 2% SDS, 5% 2-mercaptoethanol and 25 mg/ml bromophenol blue). Lysates were then heated for 10 minutes at 100 °C. Samples were run on a 12% SDS-PAGE gel followed by transfer to PVDF membrane. Gal9 was detected using an anti-Gal9 primary antibody (GalPharma) and anti-mouse secondary antibody (ThermoFisher, Uk). Blots were developed with ECL before transfer to x-ray film (Kodak). Densitometry was determined using ImageJ (NIH).
Galectin 9 silencing
Synovial fibroblasts at passage 3–6 were seeded into 6-well culture plates and incubated in complete RPMI 1640 at 37 °C to 50% confluence. Fibroblasts were transfected using Lipofectamine 2000 (Life Technologies, Paisley, UK) following the manufacturers protocol. siRNA was used at a final concentration of 90 nM for both Scr and Gal9.
Fibroblast Proliferation Assay
Fibroblast proliferation was assessed using XTT (Cell Proliferation Kit; Biological Industries, Kibbutz Beit-Haemek, Israel) following Gal9 knockdown. XTT is reduced to formazan by live cells and the intensity of absorption at 450 nm is directly proportional to the number of live metabolically active cells present42. Fibroblasts were cultured in 96-well plates at a density of 8 × 104 cells/100 μl culture medium. As a positive control, fibroblasts were stimulated with 10 ng/ml TNFα and 10 ng/ml IL-1β. Cell proliferation was assessed at 48, 72 and 96 h post-stimulation. XTT was prepared according to the manufacturer’s instructions and was added to the fibroblasts. Fibroblasts were incubated for 3 h in the presence of XTT at 37 °C and the plate was read at 450 nm with a reference wavelength of 620 nm.
Fibroblast Apoptosis Assays
Synovial fibroblast apoptosis was assessed using two methods, annexin V binding43 and JC-1 staining44. For Annexin V staining fibroblasts were trypsinised and resuspended in RPMI 1640 and combined with the cell culture supernatants containing non-adherent dead cells and washed in PBS (300 xg, 5 minutes). The resulting pellet was resuspended in annexin V buffer (10 mM HEPES, pH7.4; 140 mM NaCl; 2.5 mM CaCl2) and stained with anti-annexin V-FITC and propidium iodide (PI) (eBioscience). Annexin V positive cells were detected using a CyAn ADP flow cytometer (Beckman Coulter, High Wycombe, UK).
For JC-1 staining fibroblasts were stained with 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethyl-imidacarbocyanine iodide (JC-1) (Sigma Aldrich, Dorset, UK). In live cells, JC-1 exists in both its monomeric and aggregated forms which emit fluorescence at 530 nm (green) and 590 nm (red). In apoptosing or dead cells, only the monomeric form exists. RA synovial fibroblasts transfected with Gal9 siRNA, Scr siRNA and the control group were stained with JC-1 following the manufacturer’s instructions, and the shift from red to green fluorescence was measured using an Accuri C6 flow cytometer (Becton Dickinson, Oxford, UK).
Statistical Analysis
Non-parametric distribution was assumed for all assays. For comparison of two groups the Mann-Whitney U test was used. Analysis of Gal9 expression in multiple groups of fibroblasts was performed with Kruskal-Wallis one-way analysis of variance and Dunn’s post-test. All analyses were carried out using the Graphpad Prism 5 statistical software package.
References
Bradfield, P. F. et al. Rheumatoid fibroblast-like synoviocytes overexpress the chemokine stromal cell-derived factor 1 (CXCL12), which supports distinct patterns and rates of CD4+ and CD8+ T cell migration within synovial tissue. Arthritis rheumatism 48, 2472–2482 http://www.ncbi.nlm.nih.gov/pubmed/13130466., https://doi.org/10.1002/art.11219 (2003).
Seki, M. et al. Galectin-9 suppresses the generation of Th17, promotes the induction of regulatory T cells, and regulates experimental autoimmune arthritis. Clin. immunology 127, 78–88, https://doi.org/10.1016/j.clim.2008.01.006 (2008).
McGettrick, H. M., Buckley, C. D., Filer, A., Rainger, G. E. & Nash, G. B. Stromal cells differentially regulate neutrophil and lymphocyte recruitment through the endothelium. Immunol. 131, 357–370 http://www.ncbi.nlm.nih.gov/pubmed/20518822., https://doi.org/10.1111/j.1365-2567.2010.03307.x (2010).
Yeo, L. et al. Cytokine mRNA profiling identifies B cells as a major source of RANKL in rheumatoid arthritis. Annals rheumatic diseases 70, 2022–2028 http://www.ncbi.nlm.nih.gov/pubmed/21742639., https://doi.org/10.1136/ard.2011.153312 (2011).
Corvaisier, M. et al. IL-26 is overexpressed in rheumatoid arthritis and induces proinflammatory cytokine production and Th17 cell generation. PLoS Biol 10, e1001395, https://doi.org/10.1371/journal.pbio.1001395 (2012).
Yarilina, A., Xu, K., Chan, C. & Ivashkiv, L. B. Regulation of inflammatory responses in tumor necrosis factor-activated and rheumatoid arthritis synovial macrophages by JAK inhibitors. Arthritis rheumatism 64, 3856–3866 http://www.ncbi.nlm.nih.gov/pubmed/22941906., https://doi.org/10.1002/art.37691 (2012).
Kim, H. R. et al. Up-regulation of IL-23p19 expression in rheumatoid arthritis synovial fibroblasts by IL-17 through PI3-kinase-, NF-kappaB- and p38 MAPK-dependent signalling pathways. Rheumatol. 46, 57–64 http://www.ncbi.nlm.nih.gov/pubmed/16772307., https://doi.org/10.1093/rheumatology/kel159 (2007).
Kawashiri, S. Y. et al. Proinflammatory cytokines synergistically enhance the production of chemokine ligand 20 (CCL20) from rheumatoid fibroblast-like synovial cells in vitro and serum CCL20 is reduced in vivo by biologic disease-modifying antirheumatic drugs. The J. rheumatology 36, 2397–2402 http://www.ncbi.nlm.nih.gov/pubmed/19797510., https://doi.org/10.3899/jrheum.090132 (2009).
Hidalgo, E. et al. The response of T cells to interleukin-6 is differentially regulated by the microenvironment of the rheumatoid synovial fluid and tissue. Arthritis rheumatism 63, 3284–3293 http://www.ncbi.nlm.nih.gov/pubmed/22038403., https://doi.org/10.1002/art.30570 (2011).
Filer, A. et al. Galectin 3 induces a distinctive pattern of cytokine and chemokine production in rheumatoid synovial fibroblasts via selective signaling pathways. Arthritis rheumatism 60, 1604–1614 http://www.ncbi.nlm.nih.gov/pubmed/19479862., https://doi.org/10.1002/art.24574 (2009).
Buckley, C. D. Why does chronic inflammation persist: An unexpected role for fibroblasts. Immunol. letters 138, 12–14 http://www.ncbi.nlm.nih.gov/pubmed/21333681., https://doi.org/10.1016/j.imlet.2011.02.010 (2011).
Sanchez-Pernaute, O. et al. Citrullination enhances the pro-inflammatory response to fibrin in rheumatoid arthritis synovial fibroblasts. Annals rheumatic diseases 72, 1400–1406, https://doi.org/10.1136/annrheumdis-2012-201906 (2013).
Asakura, H. et al. Selective Eosinophil Adhesion to Fibroblast Via IFN–Induced Galectin-9. The J. Immunol., https://doi.org/10.4049/jimmunol.169.10.5912 (2002).
Liu, F. T. & Rabinovich, G. A. Galectins as modulators of tumour progression. Nat Rev Cancer 5, 29–41, https://doi.org/10.1038/nrc1527 (2005).
Hernandez, J. D. & Baum, L. G. Ah, sweet mystery of death! Galectins and control of cell fate. Glycobiol. 12, 127R–36R (2002).
Matsumoto, R. et al. Human ecalectin, a variant of human galectin-9, is a novel eosinophil chemoattractant produced by T lymphocytes. The J. biological chemistry 273, 16976–16984 (1998).
Lipkowitz, M. S., Leal-Pinto, E., Cohen, B. E. & Abramson, R. G. Galectin 9 is the sugar-regulated urate transporter/channel UAT. Glycoconj J 19, 491–498, https://doi.org/10.1023/B:GLYC.0000014078.65610.2f (2004).
Wada, J., Ota, K., Kumar, A., Wallner, E. I. & Kanwar, Y. S. Developmental regulation, expression, and apoptotic potential of galectin-9, a beta-galactoside binding lectin. J Clin Invest 99, 2452–2461, https://doi.org/10.1172/jci119429 (1997).
Kashio, Y. et al. Galectin-9 induces apoptosis through the calcium-calpain-caspase-1 pathway. J. Immunology 170, 3631–3636 (2003).
van de Weyer, P. S. et al. A highly conserved tyrosine of Tim-3 is phosphorylated upon stimulation by its ligand galectin-9. Biochem. Biophys Res Commun 351, 571–576, https://doi.org/10.1016/j.bbrc.2006.10.079 (2006).
Zhu, C. et al. The Tim-3 ligand galectin-9 negatively regulates T helper type 1 immunity. Nat. Immunology 6, 1245–1252, https://doi.org/10.1038/ni1271 (2005).
Koguchi, K. et al. Dysregulated T cell expression of TIM3 in multiple sclerosis. The J. experimental medicine 203, 1413–1418, https://doi.org/10.1084/jem.20060210 (2006).
Naka, E. L., Ponciano, V. C., Cenedeze, M. A., Pacheco-Silva, A. & Camara, N. O. Detection of the Tim-3 ligand, galectin-9, inside the allograft during a rejection episode. Int Immunopharmacol 9, 658–662, https://doi.org/10.1016/j.intimp.2008.11.013 (2009).
Dai, S. Y. et al. Galectin-9 induces maturation of human monocyte-derived dendritic cells. J. Immunol. 175, 2974–2981 (2005).
Seki, M. et al. Beneficial effect of galectin 9 on rheumatoid arthritis by induction of apoptosis of synovial fibroblasts. Arthritis rheumatism 56, 3968–3976 http://www.ncbi.nlm.nih.gov/pubmed/18050192., https://doi.org/10.1002/art.23076 (2007).
Nangia-Makker, P., Nakahara, S., Hogan, V. & Raz, A. Galectin-3 in apoptosis, a novel therapeutic target. J. Bioenerg Biomembr 39, 79–84, https://doi.org/10.1007/s10863-006-9063-9 (2007).
Almkvist, J. & Karlsson, A. Galectins as inflammatory mediators. Glycoconj J 19, 575–581, https://doi.org/10.1023/B:GLYC.0000014088.21242.e0 (2004).
Ohshima, S. et al. Galectin 3 and its binding protein in rheumatoid arthritis. Arthritis rheumatism 48, 2788–2795, https://doi.org/10.1002/art.11287 (2003).
Neidhart, M. et al. Galectin-3 is induced in rheumatoid arthritis synovial fibroblasts after adhesion to cartilage oligomeric matrix protein. Annals rheumatic diseases 64, 419–424, https://doi.org/10.1136/ard.2004.023135 (2005).
Lee, D. M. et al. Cadherin-11 in synovial lining formation and pathology in arthritis. Science 315, 1006–1010, https://doi.org/10.1126/science.1137306 (2007).
Huang, Q. Q. & Pope, R. M. The role of toll-like receptors in rheumatoid arthritis. Curr Rheumatol Rep 11, 357–364 (2009).
Ospelt, C. et al. Expression, regulation, and signaling of the pattern-recognition receptor nucleotide-binding oligomerization domain 2 in rheumatoid arthritis synovial fibroblasts. Arthritis rheumatism 60, 355–363, https://doi.org/10.1002/art.24226 (2009).
Nobumoto, A. et al. Galectin-9 suppresses tumor metastasis by blocking adhesion to endothelium and extracellular matrices. Glycobiol. 18, 735–744 http://www.ncbi.nlm.nih.gov/pubmed/18579572., https://doi.org/10.1093/glycob/cwn062 (2008).
Niki, T. et al. Galectin-9 is a high affinity IgE-binding lectin with anti-allergic effect by blocking IgE-antigen complex formation. The J. biological chemistry 284, 32344–32352, https://doi.org/10.1074/jbc.M109.035196 (2009).
Arikawa, T. et al. Galectin-9 expands immunosuppressive macrophages to ameliorate T-cell-mediated lung inflammation. Eur. journal Immunol. 40, 548–558 http://www.ncbi.nlm.nih.gov/pubmed/19902429., https://doi.org/10.1002/eji.200939886 (2010).
Makishi, S. et al. A modified version of galectin-9 induces cell cycle arrest and apoptosis of Burkitt and Hodgkin lymphoma cells. Br J Haematol 142, 583–594, https://doi.org/10.1111/j.1365-2141.2008.07229.x (2008).
Niwa, H. et al. Stable form of galectin-9, a Tim-3 ligand, inhibits contact hypersensitivity and psoriatic reactions: a potent therapeutic tool for Th1- and/or Th17-mediated skin inflammation. Clin. immunology 132, 184–194 http://www.ncbi.nlm.nih.gov/pubmed/19464955., https://doi.org/10.1016/j.clim.2009.04.012 (2009).
Hughes, R. C. Secretion of the galectin family of mammalian carbohydrate-binding proteins. Biochimica et biophysica acta 1473, 172–185 (1999).
Oomizu, S. et al. Cell surface galectin-9 expressing Th cells regulate Th17 and Foxp3+ Treg development by galectin-9 secretion. PloS one 7, e48574, https://doi.org/10.1371/journal.pone.0048574 (2012).
Matarrese, P. et al. Galectin-3 overexpression protects from apoptosis by improving cell adhesion properties. Int J Cancer 85, 545–554 (2000).
Filer, A. et al. Differential survival of leukocyte subsets mediated by synovial, bone marrow, and skin fibroblasts: site-specific versus activation-dependent survival of T cells and neutrophils. Arthritis rheumatism 54, 2096–2108, https://doi.org/10.1002/art.21930 (2006).
Roehm, N. W., Rodgers, G. H., Hatfield, S. M. & Glasebrook, A. L. An improved colorimetric assay for cell proliferation and viability utilizing the tetrazolium salt XTT. J. immunological methods 142, 257–265 (1991).
Vermes, I., Haanen, C., Steffens-Nakken, H. & Reutelingsperger, C. A novel assay for apoptosis. Flow cytometric detection of phosphatidylserine expression on early apoptotic cells using fluorescein labelled Annexin V. J. immunological methods 184, 39–51 (1995).
Salvioli, S., Ardizzoni, A., Franceschi, C. & Cossarizza, A. JC-1, but not DiOC6(3) or rhodamine 123, is a reliable fluorescent probe to assess ΔΨ changes in intact cells: implications for studies on mitochondrial functionality during apoptosis. FEBS Letters 411, 77–82, https://doi.org/10.1016/s0014-5793(97)00669-8 (1997).
Acknowledgements
MAB was supported by a Marie Curie EST fellowship (TRIFID; Ref 20996-EST), MJP, AF and AJN were supported by Arthritis Research UK (Ref 18547). CW was supported by a Deutsche Forschungsgemeinschaft (DFG) Fellowship (Ref 319464273) The authors thank Hema Chahal and Hannah Smith for technical assistance and confirm that there is no conflict of interest associated with this work. The authors acknowledge the support of Euro-TEAM.
Author information
Authors and Affiliations
Contributions
M.J.P., M.A.B., C.O., A.J.N. and C.W., designed the study, performed experiments and analysed the data, S.W.J., C.D.B., S.G., A.F. and J.M.L. designed the study. M.J.P., M.A.B., S.W.J. and J.M.L. wrote the manuscript and all authors read and approved the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Pearson, M.J., Bik, M.A., Ospelt, C. et al. Endogenous Galectin-9 Suppresses Apoptosis in Human Rheumatoid Arthritis Synovial Fibroblasts. Sci Rep 8, 12887 (2018). https://doi.org/10.1038/s41598-018-31173-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-31173-3
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
