
Abstract
Work-exacerbated asthma (WEA) is defined as preexisting asthma that worsens with exposure to irritants [e.g., chlorine (Cl2) derivatives] in the workplace. The maximum allowable concentration in the workplace of Cl2 exposure is 3 mg/ m3 (described in OSHA). We investigated in an experimental asthma model in mice the effects of a single exposure to a sodium hypochlorite dose with this allowed chlorine concentration and a tenfold higher dose. Acute chlorine exposure at 3.3 mg/m3 in the OVA-sensitized group increased eosinophils in the peribronquial infiltrate, cytokine production, nasal mucus production and the number of iNOS positive cells in the distal lung compared to only sensitized mice. The exposure to a higher dose of 33.3 mg/m3 in the OVA-sensitized group resulted in an increase in respiratory system elastance, in the total and differential numbers of inflammatory cells in bronchoalveolar lavage fluid, IL-4, IL-5, and IL-17 in the lungs, eosinophils in peribronquial infiltrate and mucus content in nasal compared to non-exposed and sensitized animals. In this asthma model, chorine exposures at an allowable dose, contributed to the potentiation of Th2 responses. The functional alterations were associated with increased iNOS and ROCK-2 activation in the distal lung.
Similar content being viewed by others

Introduction
Allergic respiratory conditions are among the most prevalent disorders in Western populations1,2. The exact cause of the increase in the prevalence of these conditions remains unclear1,2.
Because of its wide inner surface, the respiratory system is usually exposed to particles and gases, and it is also easily exposed to allergens and irritant compounds3,4. The airway epithelium works as a barrier that regulates water and ion transport and, through mucociliary clearance, transports inhaled particles. Furthermore, the epithelium participates in inflammation, immune defense against pathogens, tissue remodeling, and cytokine production4,5. This complex role allows epithelial cells to communicate with mesenchymal cells in the airway and transfer signals to other cells, such as immune cells, which are capable of producing inflammatory mediators and amplifying the injury process4.
Once exposed to inhaled substances, such as air pollutants, allergens or pathogens, epithelial damage and cell disruption occur4, and the airway afferent nerve promotes the release of mediators and stimulates changes in the respiratory response6. Some of these mediators are the inducible nitric oxide (iNOS) enzyme7 and Rho-kinase8. Changes in ROCK-2, which modulates contraction, affect smooth muscle and alter the actin cytoskeleton, cell adhesion, motility, migration and contraction8,9,10.
The response to an allergen is primarily mediated by the interaction between dendritic cells (DCs) and lymphocytes, whereas irritant agents might cross the cell membrane and react with local proteins, creating or revealing new epitopes11 and generating oxygen free radicals12, ultimately resulting in respiratory injury13,14. This injury can increase epithelial permeability, allowing access by allergens to the subepithelial DCs15, thus becoming an efficient adjuvant to present these proteins to macrophages and DCs, releasing a wide range of inflammatory cytokines16.
Animal studies have also shown that exposure to respiratory irritants can increase pulmonary responsiveness17 and activate immune cells, thus contributing to the release of inflammatory cytokines (e.g., IL-4 and IL-17) in the lungs and the production of reactive oxygen (ROS) and reactive nitrogen species (RNS)18,19,20. This status might also contribute to DNA damage18,21,22,23.
Among environmental factors, exposure to disinfectants based on chlorine products has been identified as a source of irritation and airway inflammation24. Products that contain Cl2 derivatives, such as sodium hypochlorite, are widely used by cleaning workers and are directly linked to respiratory illness25,26,27,28.
These situations are defined as work-related asthma (WRA), which represents a health problem with significant potential for acute morbidity, long-term disability, and severe social and economic impacts29,30.
Occupational asthma (OA) is the most common form of WRA and accounts for approximately 15% of all adult-onset asthma31,32. OA has emerged as a relevant focus of investigations related to asthma physiopathology31. The Cl2 time-weighted average exposure to be considered occupational exposure is 0.5 ppm over 8 h or 1 ppm over 10 minutes33. The Occupational Safety and Health Administration (OSHA) recommends that the allowable permissible exposure level (PEL) for chlorine is 1 ppm or 3 mg/m3 34.
However, the mechanisms that mediate the induction or facilitation of allergic conditions in WRA remain unclear. A growing number of animal studies have been performed, but the results remain controversial35,36,37. Hox et al. demonstrated that chronic nasal instillation of NaClO with 3 ppm of active Cl2 before ovalbumin (OVA) sensitization resulted in airway hyperresponsiveness (AHR) without affecting sensitization to the antigen36. In contrast, Kim et al., who also used an experimental OVA model, found that exposure to chronic low doses of chlorine through a vaporized 5% NaClO solution aggravated allergen-induced airway inflammation37.
To our knowledge, no previous study has addressed whether acute exposure to chlorine in a background of pre-existing allergic lung inflammation could affect the distinctive features of asthma. Thus, we first assessed, in naïve animals and afterwards in an asthma model, the effects of a single exposure to a sodium hypochlorite dose with the maximum allowable concentration and a tenfold higher dose described in the OSHA to assess lung function, inflammation, remodeling, the oxidative stress pathway and ROCK-2 expression.
Results
Phase I: Chlorine gas exposure in naïve animals and its effects on pulmonary responsiveness and lung inflammation
Acute chlorine exposure increases airway bronchoconstriction and inflammatory cells in naive mice
In the first phase of this experiment, we evaluated chlorine gas exposure by inhalation of increasing concentrations of sodium hypochlorite. We analyzed the enhanced pause (Penh) to the dose-response curve (Fig. 1a) of each group and observed an increase in lung responsiveness when mice were exposed to 33.3 Cl2 compared to the SAL dose (p < 0.001) and compared to the 3.3 Cl2 dose (p < 0.001).

Enhanced pause (Penh) and inflammatory cells in health mice. These values are expressed as the mean ± SE (n = 6). (a) Penh: *significantly different compared to the others groups (p < 0.001). (b) Inflammatory cells in the BALF: *significantly different when total cells (p = 0.024), neutrophils (p = 0.026) and lymphocytes (p = 0.031) in the Cl2 exposed animals were compared to the SAL group.
When inflammatory cells (Fig. 1b) were analyzed, we observed an increase in total cells (p = 0.024), neutrophils (p = 0.026) and lymphocytes (p = 0.031) in Cl2-exposed animals compared to the SAL group.
Phase II: Chlorine gas exposure in a model of allergic pulmonary inflammation
Acute chlorine exposure exacerbates respiratory system responsiveness
Compared to the SAL group (Fig. 2), respiratory system resistance (Rrs) (Fig. 3a) and elastance (Ers) (Fig. 3b) were exacerbated in OVA-sensitized animals (p = 0.009 and p = 0.013, respectively).

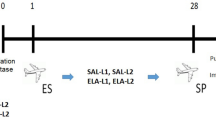
Timeline of the experimental protocol. On days 0 and 14 mice of OVA, OVA +3.3 Cl2, OVA + 33.3 Cl2 received intraperitoneal (I.P.) injections of OVA solution with vehicle, while the SAL group received only vehicle (open triangles). Aerosol challenges with a 1% OVA solution were performed four times on days 22, 24, 26, and 28 (closed triangles) and animals in the SAL group received only aerosolized saline. A single Cl2 exposure was performed on day 29 (closed circle). After Cl2 exposure, lung mechanics were measured, and the animals were euthanized.

Resistance (Rrs) and elastance (Ers) of the respiratory system. These values are expressed as the mean ± SE (n = 6). (a) Rrs: *significantly different compared to the Saline group (p < 0.009 for OVA group and p < 0.001 for OVA + 33.3Cl2). (b) Ers *significantly different compared to the Saline group (p = 0.013 for OVA group and p < 0.001 for both Cl2 exposed groups) and #significantly different compared to the OVA group (p < 0.001).
Furthermore, compared to OVA-sensitized animals, only the OVA + 33.3 Cl2-exposed animals exhibited an increase in Ers (p < 0.001).
Acute exposure to the higher dose of chlorine increases pulmonary inflammation
Figure 4 shows the effects of acute sodium hypochlorite exposure on the number of leukocytes in the BALF.

Counting of inflammatory cells in the BALF. The values are expressed as the mean ± SE (n = 6). (a) Total cells: *significantly different compared to the Saline group (p = 0.037 for OVA group, p = 0.011 for OVA + 3.3 Cl2 group and p < 0.001 for OVA + 33.3 Cl2 group), #significantly different compared to the OVA group (p < 0.001) and ##significantly different compared to the OVA + 3.3 Cl2 group (p < 0.001). (b) Lymphocytes: *significantly different compared to the Saline group (p < 0.001), #significantly different compared to the OVA group (p < 0.001) and ##significantly different compared to the OVA + 3.3 Cl2 exposed group (p < 0.001). (c) Eosinophils: *significantly different compared to the Saline group (p < 0.001), #significantly different compared to the OVA group (p < 0.001) and ##significantly different compared to the OVA + 3.3 Cl2 exposed group (p < 0.006). (d) Neutrophils: *significantly different compared to the Saline group (p < 0.001), #significantly different compared to the OVA group (p = 0.005) and ##significantly different compared to the OVA + 33.3 Cl2 exposed group (p = 0.009).
Compared to the SAL group, OVA-sensitized animals exhibited an increase in the number of total cells (Fig. 4a) when not exposed to Cl2 (p = 0.037). Furthermore, animals in the OVA + 33.3 Cl2 group showed a higher number of inflammatory cells compared to the OVA and OVA + 3.3 Cl2 groups (p < 0.001).
Figure 4b shows that exposure to a higher dose of chlorine in OVA-sensitized animals resulted in an increased number of lymphocytes compared to the OVA and OVA + 3.3 Cl2 groups (p < 0.001).
Exposure to a higher dose of chlorine resulted in an increased number of eosinophils (Fig. 4c) compared to the OVA (p < 0.001) and OVA + 3.3 Cl2 (p < 0.006) groups.
Furthermore, Fig. 4d shows that exposure to the maximal allowable dose of chlorine in OVA-sensitized animals resulted in an increased number of neutrophils compared to the OVA and OVA + 33.3 Cl2 groups (p = 0.005 and p = 0.009, respectively).
When macrophages were analyzed, no differences between groups were observed. The number of macrophages in each experimental group was as follows (mean ± SE): SAL group (0.16 ± 0.11 × 104/mL), OVA group (1.32 ± 0.27 × 104/mL), OVA + 3.3 Cl2 (1.50 ± 0.66 × 104/mL) and OVA + 33.3 Cl2 group (1.65 ± 0.37 × 104/mL).
iNOS expression is only increased by chlorine gas in the distal lung but not in the peribronchial infiltrate
As shown in Fig. 5a, quantification of the number of iNOS-positive cells in the peribronchial infiltrate was increased only in OVA-sensitized animals not exposed to Cl2 (p < 0.005) compared to the SAL group.

iNOS positive cells. The values are expressed as the mean ± SE (n = 6). (a) Positive cells in the airways: *significantly different compared to Saline group (p < 0.005 for OVA group and p < 0.014 for OVA + 33.3Cl2). (b) Positive cells in the lung parenchyma: *significantly different compared to Saline group (p < 0.001 for all groups), #significantly different compared to the OVA group (p = 0.008 and p < 0.001, respectively) and ##significantly different compared to the OVA + 3.3 Cl2 group (p = 0.001).
On the other hand, analyses in the distal lung (Fig. 5b) revealed an increase in the number of iNOS-positive cells in the OVA-sensitized group compared to the SAL group (p < 0.001) and an increase in both groups exposed to Cl2 (p = 0.008 and p < 0.001, respectively). Furthermore, animals exposed to the high dose of chlorine differed compared to the group that received the maximum allowable concentration of chlorine (p = 0.001).
Acute chlorine exposure to a high dose of Cl2 increased ROCK-2-positive cells in the distal lung but not in the peribronchial infiltrate
The cellular expression of ROCK-2 in the peribronchial infiltrate is shown in Fig. 6a. We observed increased ROCK-2 expression in OVA-sensitized animals not exposed to Cl2 (p < 0.007) compared to the SAL group.

ROCK-2 positive cells. The values are expressed as the mean ± SE (n = 6). (a) Positive cells in the airways: *significantly different compared to Saline group (p = 0.007). (b) Positive cells in the distal lung: *significantly different compared to Saline group (p < 0.001 for all groups), #significantly different compared to the OVA group (p < 0.001) and ##significantly different compared to the OVA + 3.3 Cl2 group (p < 0.001).
In the lung parenchyma (Fig. 6b), this increase was observed in the OVA-sensitized group compared to the SAL group (p < 0.001) and in OVA + 33.3 Cl2 animals compared to the OVA group (p < 0.001) and the group that received the maximum allowable concentration of chlorine (p < 0.001).
Acute chlorine exposure increases the number of eosinophils in the peribronchial infiltrate
Figure 7 shows the effects of Cl2 exposure on the number of eosinophils in the peribronchial infiltrate. Acute chlorine exposure at both concentrations tested significantly increased the number of eosinophils in OVA-sensitized animals compared to the OVA group (p = 0.002 and p = 0.008, respectively).

Eosinophils cells in the peribronchial space. The values are expressed as the mean ± SE (n = 6). *significantly different compared to Saline group (p < 0.001 for both Cl2 groups). #Significantly different compared to the OVA group (p = 0.002 for OVA + 3.3 Cl2 and p = 0.008 for OVA + 33.3 Cl2). Photomicrography panel of peribronchial infiltrate. All figures are presented at a magnification of 400×, scale bars = 50 µm with an insert at a magnification of 1000×, scale bar = 5 µm. (a) SAL group, (b) OVA group, (c) OVA + 3.3 Cl2 and (d) OVA + 33.3 Cl2.
Acute chlorine exposure did not increase the number of neutrophils in the peribronchial infiltrate
To exclude the possibility that cells observed in the peribronchial infiltrate were neutrophils, we performed an immunohistochemical analysis. The cell count of the experimental groups was as follows (mean ± SE): SAL group (0.75 ± 0.31 cells/104 µm2), OVA group (0.34 ± 0.14 cells/104 µm2), OVA + 3.3 Cl2 (1.14 ± 0.33 cells/104 µm2) and OVA + 33.3 Cl2 (0.78 ± 0.30 cells/104 µm2). No significant differences between experimental groups were observed.
Acute chlorine exposure did not enlarge the mean linear intercept (Lm)
The Lm was analyzed to evaluate lung tissue destruction; no differences between groups were observed. The Lm of each experimental group was as follows (mean ± SE): SAL group (27.35 ± 2.16 µm), OVA group (30.52 ± 0.99 µm), OVA + 3.3 Cl2 (30.66 ± 1.27 µm) and OVA + 33.3 Cl2 (32.92 ± 1.76 µm).
Interstitial edema is not increased by chlorine exposure
Interstitial edema, which was evaluated by score, was low in all experimental groups and did not present differences between groups. The results are as follows (mean ± SE): SAL group (4.11 ± 0.81), OVA group (5.27 ± 0.89), OVA + 3.3 Cl2 (6.38 ± 0.45) and OVA + 33.3 Cl2 (3.77 ± 0.65).
Acute chlorine exposure increases lung cytokine levels
By analyzing lung tissue homogenates, we found that chlorine exposure at both concentrations tested significantly increased the lung levels of IL-4, IL-5, and IL-17 in all groups (Fig. 8a–c, respectively).

Cytokine levels in lung homogenates. The values are expressed as the mean ± SE (n = 6). (a) IL-4 levels: *significantly different compared to Saline group (p = 0.007 for OVA group and p < 0.001 for both Cl2 groups). #Significantly different compared to the OVA group (p < 0.001). (b) IL-5 levels: *significantly different compared to the Saline group (p < 0.001 for both Cl2 groups). #Significantly different compared to OVA group (p = 0.001). (c) IL-17 levels: *significantly different compared to the Saline group (p = 0.035 for OVA group and p < 0.001 for both Cl2 groups). #Significantly different compared to the OVA group (p < 0.001).
The OVA group not exposed to Cl2 exhibited increased IL-4 levels compared to the SAL group (p = 0.007), and the OVA groups exposed to both concentrations showed an increase compared to OVA non-hypochlorite-exposed mice (p < 0.001).
Similarly, IL-5 lung levels showed a significant increase in OVA mice exposed to 3.3 or 33.3 Cl2 compared to the OVA group not exposed (p < 0.018 and p < 0.001, respectivily). Additionally, both concentrations tested increased the IL-17 levels relative to the OVA group not exposed (p < 0.001), and the OVA-sensitized group had a higher level of pulmonary IL-17 compared to the SAL group (p = 0.035).
Acute chlorine exposure increases the acid mucus content in the nasal epithelium
In this experimental model, we observed that the OVA group not exposed to Cl2 was significantly different compared to the SAL group (p < 0.001). We measured both neutral (data not shown) and acid mucus substances, but the analysis of neutral mucus per se did not show a significant difference between groups (p > 0.05). However, when neutral and acid mucus were analyzed together, we observed an increase in total mucus content (Fig. 9).

Volume proportion of airway mucus. The values are expressed as the mean ± SE (n = 6). *Significantly different compared to the Saline group (p < 0.001 for all groups). Photomicrography panel of airway epithelium. All figures are presented at a magnification of 1000×, scale bars = 10 µm. (a) SAL group, (b) OVA group, (c) OVA + 3.3 Cl2 and (d) OVA + 33.3 Cl2.
We also observed an increase in acid mucus in the nasal epithelium (Fig. 10) in the OVA-sensitized group compared to the SAL group (p < 0.001). Furthermore, both groups exposed to Cl2 exhibited an increase in acid mucus compared to the OVA group (p = 0.015 and p < 0.001, respectively), and animals exposed to the higher dose differed compared to animals that received the maximum allowable concentration of chlorine (p < 0.001).

Volume proportion of nasal acid mucus. The values are expressed as the mean ± SE (n = 6). *Significantly different compared to the Saline group (p < 0.001 for all groups). #Significantly different compared to the OVA group (p = 0.015 for OVA + 3.3 Cl2 and p < 0.001 for OVA + 33.3 Cl2) ##significantly different compared to the OVA + 3.3 Cl2 group (p < 0.001). Photomicrography panel of nasal epithelium. All figures are presented at a magnification of 1000×, scale bars = 10 µm. (a) SAL group, (b) OVA group, (c) OVA + 3.3 Cl2 and (d) OVA + 33.3 Cl2.
Discussion
This study was performed to provide experimental evidence related to a common work situation referred to as WEA, which is defined as current asthma that worsens in response to various work-related factors, such as aeroallergens, changes in temperature, exercise, and exposure to irritants29,30,38,39,40,41,42,43,44,45. Among the environmental irritant factors, exposure to chlorine derivatives, especially disinfection products, has been highlighted29,30.
Our results demonstrated, for the first time, that acute chlorine exposure at 3.3 mg/m3 in the OVA-sensitized group increased eosinophils in the peribronchial space, cytokine production, nasal mucus production and iNOS-positive cells in the distal lung compared to only sensitized mice. Exposure to a higher dose (33.3 mg/m3 Cl2) in the OVA-sensitized group resulted in an increase in respiratory system elastance, total and differential numbers of inflammatory cells in the BALF, IL-4, IL-5, and IL-17 in the lungs, eosinophils in the peribronchial space, mucus content in the nasal epithelium and the number of iNOS- and ROCK-2-positive cells in the distal lung compared to non-exposed and sensitized animals. Furthermore, naïve mice exposed to increased doses of chlorine showed alterations in respiratory mechanics and a higher number of neutrophils and lymphocytes in the BALF.
AHR is usually employed to investigate the functional status in many pulmonary conditions, including those involving toxic irritants and allergens17,36,46. In this study, we showed that OVA-sensitized and OVA-challenged mice acutely exposed to chlorine at concentrations of 33.3 mg/m3 (tenfold higher than the maximum allowable concentration of chlorine in the workplace) exhibited exacerbated elastance of the respiratory system.
Our results agree, in part, with those reported by Hox et al., who observed a slight increase in the resistance of the respiratory system in OVA-sensitized mice and those chronically exposed to nasal instillations of NaClO at 3 ppm36 and by Kim et al.37, who observed airway hyperresponsiveness in sensitized animals and challenged mice exposed to naturally vaporized 5% sodium hypochlorite during four weeks. In our experimental model, we found increased elastance of the respiratory system in OVA-sensitized mice acutely exposed to the higher dose of Cl2 and in the Penh of naïve mice exposed to increasing doses of chlorine.
In addition to hyperresponsiveness and transient bronchospasm, chlorine gas inhalation can lead to temporary mucous membrane irritation and pulmonary edema due to an increase in vascular permeability, which allows an influx of eosinophils, neutrophils and lymphocytes into the lungs, causing enhanced pulmonary inflammation1,15,47. We observed a higher influx of eosinophils in the peribronchovascular space in animals acutely exposed to chlorine at both concentrations tested. This inflammation may be related to the enhancement of the lung mechanical response in our experiment.
Additionally, the influx of inflammatory cells into the injury site promotes the release of inflammatory cytokines11,48,49. In experimental asthma models and clinical studies of asthmatic populations, increases in pro-inflammatory cytokines, such as IL-4 and IL-5, are directly related to the differentiation, proliferation, recruitment, and survival of inflammatory cells in allergic inflammation50,51,52,53.
In our experimental model, 24 h after the last challenge, the OVA groups exposed to both chlorine concentrations showed bronchial eosinophilia and bronchial Th2 cytokine production, as previously published17,46,49,50. This eosinophilia was confirmed by immunohistochemical analysis using an antibody specific for neutrophils, and no differences between experimental groups were observed, suggesting that the majority of these cells were eosinophils.
Interestingly, acute Cl2 application had an additional major impact on the allergic response, as evaluated by measuring inflammatory cells in the BALF, with an increase in the number of total cells, neutrophils, eosinophils and lymphocytes, and by measuring IL-4, IL-5, and IL-17 levels in homogenized lung tissue.
Curiously, some studies have also shown that other inflammatory factors may be activated more directly by irritating substances through the activation of pro-inflammatory signaling pathways within cells, as occurs in the bronchial epithelium54,55,56. The iNOS enzyme synthesizes L-arginine to produce nitric oxide (NO) in response to pro-inflammatory mediators, including cytokines, in chronic diseases such as asthma57,58. High levels of chlorine (100 ppm) have already been described as a mechanism to induce iNOS production by recruiting neutrophils14,59.
Our results are consistent with this idea, where OVA-sensitized animals exhibited an increase in oxidative stress markers following iNOS production in the lungs60. However, OVA-sensitized and chlorine-exposed animals did not show an increase in iNOS-positive cells in the airways, which can be explained by the chlorine dose used in this study. On the other hand, the number of positive cells in the lung parenchyma were increased in the OVA group and in both OVA groups exposed to chlorine.
Another signaling pathway that may increase inflammatory cytokines, the infiltration of inflammatory cells into airways and changes in the respiratory response is the Ro-kinase pathway. Righetti et al.8 demonstrated that the inhibition of Ro-kinase reduced distal lung tissue responsiveness and the release of inflammatory mediators. Although the inflammatory response in the BALF showed an increase in the number of inflammatory cells compared to the low dose of chlorine, cytokine analyses and the number of eosinophils in the peribronchial infiltrate did not differ between chlorine groups. Because the Ers value is dependent on the distal airway and distal lung hyperresponsiveness, we believed it was important to evaluate oxidative stress, which is associated with the Rho-kinase system8.
In our experimental study, we observed an increase in the number of ROCK-2-positive cells in the airways and in the lung parenchyma of OVA-sensitized animals. Furthermore, animals exposed to an acute high dose of Cl2 exhibited an increase in ROCK-2 expression in the lung parenchyma. Changes in respiratory elastance were accompanied with an increase in ROCK-2 expression, suggesting the influence of this mediator in the pulmonary response.
The airways are normally protected with a thin layer of mucus, a hydrogel that protects the epithelium from harmful particles, pathogens and chemicals61. They are transported out of the lungs by this gel formed with water and mucins (glycoproteins)62.
In some diseases, acidification of the mucus alters its properties, increasing its viscosity and impairing mucociliary clearance63,64. Because of this increased viscosity, the acid mucus is normally present in the nasal epithelium, acting as a barrier against pathogens and allergens. Daily, the nasal epithelium produces 1.5 to 2 L of mucus that is renewed approximately every 20 minutes. By contrast, the particles reach the airway lining in 15 minutes to 2 h after inhalation, and the amount of mucus secreted each day is approximately 10 mL61.
The Th2 cytokines, such as IL-4, that were increased in the present study may stimulate mucin production61. We believe that the systemic effects of Th2 cytokines that were potentialized by chlorine exposure contributed to the results observed in the nasal mucus. Therefore, this evidence contributes to a better understanding of the differences between the nasal and airway mucus results.
Here, we show that single chlorine exposure induced an increase in mucus content and the nasal epithelium in the OVA groups. These results may be partially explained by the oxidative modification of free functional groups of proteins and the induction of lipid peroxidation, which are caused by chlorine and result in increased vascular permeability, the release of inflammatory cytokines, the influx of inflammatory cells, and increased mucus production11,48,65.
A limited number of studies have reported the quantitative exposure assessment to chlorine in the workplace66,67. Since most respiratory toxicity appears to be related to chlorine released from a sodium hypochlorite solution, we determined the chlorine levels once this could be related to the PEL of 1 ppm or 3 mg/m3 provided by OSHA34 and a tenfold higher dose. Even at allowable chlorine concentrations, we observed an increase in inflammatory cells in the airway walls, increased cytokine production, and increased nasal mucus production when allergic inflammation was combined with acute exposure to irritant products.
We are also aware of the limitations of the animal models of asthma, including some differences in the immune responses53. However, the OVA model used in our study is a well-established model of allergic asthma that we have used to study another acute condition46,50,53,56,68,69,70. Furthermore, more experimental and clinical research addressing acute or chronic chlorine provided from sodium hypochlorite exposure should be performed to understand the signaling pathways, remodeling and oxidative stress in an allergic or health condition.
On the other hand, no previous work has been performed using acute exposure to the accepted levels of chlorine in a model of asthma that has reported increased inflammation even without changes in pulmonary function, suggesting the importance of the evaluation of inflammation in asthmatic patients without changes in pulmonary function, as well as a review of the permitted level of chlorine exposure in this group of people.
In conclusion, this experimental asthma model revealed that chorine exposure, at an allowable dose, contributed to the potentiation of Th2 responses. Furthermore, the functional alterations were associated with increased iNOS and ROCK-2 activation in the distal lung.
Materials and Methods
The present study was submitted and approved by the Review Board for Human and Animal Studies of the School of Medicine of the University of Sao Paulo (n° 029-10). Male BALB/c mice (25–30 g, 6 weeks old, specific pathogen-free [SPF]) were obtained from the Animal Facility of the School of Medicine of the University of Sao Paulo. Animals were maintained in controlled conditions of temperature (22 ± 2 °C), humidity (70–75%), and dark/light cycle (12 h; lights on at 06:00 am) and allowed food and water ad libitum. All animal care and experimental procedures followed the Guide for the Care and Use of Laboratory Animals71.
Phase I: Chlorine gas exposure in naïve animals and its effects on pulmonary responsiveness and lung inflammation
Animals and experimental groups
Twelve male BALB/c mice were divided into two groups as follows (n = 6 mice/group): (1) naïve mice exposed to saline (SAL); (2) naïve mice exposed to increasing concentrations of chlorine, 3.3 and 33.3 mg/m3 (Cl2).
Whole-body plethysmography
In the first phase of experiments, we determined the minimal dose of chlorine to be used in the second phase protocol. Animals were sensitized to ovalbumin by placing naive mice in a chamber and evaluating the pulmonary mechanics to increasing doses of sodium hypochlorite (0.00%, 0.03% [3.3 mg/m3] and 0.3% [33.3 mg/m3]) using a whole-body plethysmography system (BUXCO, Winchester, UK). Briefly, data acquisition was performed using a transducer connected to a computer system that continuously measured the box pressure–time wave, which calculated the Penh, which is related to air bronchoconstriction3,72.
After measuring the baseline Penh, animals received aerosolized saline followed by two sodium hypochlorite concentrations (0.03% and 0.3%) through an inlet in the chamber for 3 minutes. The Penh values were collected for 5 minutes and averaged. Next, animals were anesthetized (thiopental sodium, 33 mg/kg i.p.) and tracheotomized for bronchoalveolar lavage fluid collection, and the numbers of total and differential cells were counted.
Total and differential cell counts in the BALF
Three instillations of 0.5 mL of 0.9% NaCl were performed via a tracheal cannula, and the lungs were gently washed. All washes were combined, processed, and analyzed. The total cell number was counted in a Neubauer hemocytometer chamber, and 300 cells (macrophages, lymphocytes, eosinophils and neutrophils) were counted in each Diff Quick-stained cytospin slide in a blinded fashion.
Chlorine level determination
To determine the chlorine levels in the exposure atmosphere, samples of filter paper (55-mm diameter, Whatman 1, Sigma-Aldrich Brazil Ltda., Sao Paulo, Brazil) were nebulized with NaClO. The chlorine level was determined by neutron activation analysis. Filter paper samples were folded and placed in polyethylene envelopes for irradiation in the IEA-R1 Nuclear Research Reactor of Nuclear and Energy Research Institute along with the synthetic standard of chlorine.
The chlorine mass in the synthetic standard was 500 µg, and the irradiation time was 25 s under a thermal neutron flux of 1.9 × 1012 n cm−2 s−1. After a decay time of 5 minutes, gamma ray activities were measured using a hyperpure Ge detector coupled to a Digital Spectrum Analyzer DSA 1000 (Canberra, Meriden, CT, USA). The gamma spectrum was processed using Genie 2000 software version 3.1 (Canberra). Chlorine was identified by the 38Cl peak with a half-life of 37.24 minutes and a gamma ray energy of 1642.69 keV. The chlorine mass was calculated by the comparative method73 after discounting chlorine present in the filter paper (blank value).
After the procedures, the chlorine masses per filter paper area were 3.3 (an accepted level of chlorine) and 33.3 mg/m3 (10-times higher), respectively.
Phase II: Chlorine exposure in a model of allergic pulmonary inflammation
Animals and experimental groups
Twenty-four male BALB/c mice were divided into four groups as follows (n = 6 mice/group): (1) mice that were non-sensitized and not exposed to chlorine (SAL); (2) mice that were OVA-sensitized and OVA-challenged but not exposed to chlorine (OVA); (3) mice that were OVA-sensitized and OVA-challenged and exposed to 3.3 mg/m3 of chlorine (OVA + 3.3 Cl2); and (4) mice that were OVA-sensitized and OVA-challenged and exposed to 33.3 mg/m3 of chlorine (OVA + 33.3 Cl2).
OVA model of allergic pulmonary inflammation
BALB/c mice were sensitized with an intraperitoneal (i.p.) injection of 50 µg of OVA with aluminum hydroxide (vehicle) on days 0 and 14, while the SAL group received vehicle only. Mice were exposed to aerosolized OVA (1%) or saline (0.9% NaCl) for 30 minutes on days 22, 24, 26 and 28 (Fig. 2)17,56.
On day 29, animals were anesthetized (thiopental sodium, 33 mg/kg i.p.), tracheotomized, and placed in a plethysmograph chamber connected to a small animal ventilator (Harvard Apparatus, South Natick, MA, USA).
Respiratory system mechanical assessment
Differential pressure transducers (Honeywell, Freeport, IL, USA) were applied to measure the whole-body plethysmography pressure (from which changes in lung volume were derived) and tracheal pressure. Volume and tracheal pressure signals were acquired at 200 Hz over 10 s with a 12-bit analog-to-digital converter (DT 01-EZ, Data Translation, Marlboro, MA, USA) and stored (LABDAT, RHT-InfoData, Inc., Montreal, QC, Canada) in a microcomputer. Airflow changes were obtained via electronic derivation of the volume signal. Rrs and Ers were obtained by applying the single compartment model of the respiratory system to the experimental data (pressure, volume, and flow signals). All data were analyzed using computer software for the assessment of respiratory mechanics (ANADAT 4.0, RHT-InfoData, Inc., Montreal, QC, Canada).
Animals were anesthetized and tracheotomized and connected to the ventilator. Next, baseline Ers and Rrs values were collected, and chlorine gas exposure was performed by nebulization of 0.03% (3.3 mg/m3) and 0.3% (33.3 mg/m3) NaClO solutions (Sigma Chemical Co., St. Louis, MO, USA). An ultrasonic device (Respira Max, NS, LTDA., Sao Paulo, Brazil) nebulized either aerosolized saline or NaClO solutions through the air inlet of the ventilator for 2 minutes. The respiratory mechanics data were assessed after 30 s and after 1, 2, and 3 minutes. The total duration of lung mechanics evaluation was approximately 12 minutes for each animal, and the results are expressed as the maximum responses of Rrs and Ers. Animals in the SAL and OVA groups received aerosolized saline only.
After these procedures, the animals were euthanized by rapid exsanguination of the abdominal aorta. The bronchoalveolar fluid (BALF) was then collected, and lungs were excised.
Total and differential cell counts in the bronchoalveolar lavage fluid (BALF)
Same as in phase I.
Evaluation of iNOS, ROCK-2 and neutrophils positive cells in the peribronchial infiltrate
Immunohistochemistry for iNOS, ROCK-2 and neutrophil detection was performed using a protocol modified from Martins-Oliveira et al.74. Lung sections (5-µm-thick) were deparaffinized and hydrated. Antigen retrieval was performed, and sections were washed in phosphate-buffered saline (PBS) and blocked with 3% hydrogen peroxide at room temperature. Next, sections were incubated with rabbit antibody anti-iNOS (cod. RB-9242-P; LabVision, Neo-Markers, Fremont, CA, USA) at a dilution of 1:300, goat anti-ROCK-2 (cod. sc-1851; Santa Cruz Biotechnology, Santa Cruz, CA, USA) at a dilution of 1:50 and rabbit anti-neutrophil elastase (cod. ab21595, Abcam, Cambridge, MA, USA) at a dilution of 1:1500. All primary antibodies were diluted in bovine serum albumin (BSA) overnight (16–18 h) in a humid chamber at 4–8 °C. Subsequently, sections were washed in PBS and incubated with a secondary antibody (Vector ABCElite, horseradish peroxidase [HRP]; Vector Laboratories, Burlingame, CA, USA, cods. PK-6101 anti-rabbit and PK-6105 anti-goat) at 37 °C in a humidified chamber. Three additional 5-minutes washes in PBS were performed and samples were revealed with 3,3-diaminobenzidine (DAB) (cod. K3468, Dako Citomation, Fort Collins, CO, USA) for 5 minutes. Subsequently, tissues were washed with tap water and counterstained with Harris hematoxylin74. Cell density was assessed as the number of cells divided by the respective edema area (104 cells/µm2) in five peribronchovascular structures. The analysis was performed by an optical microscope provided with an integrating eyepiece containing a known area (104 μm2 at a magnification of 1000×) of 50 lines and 100 points75.
Evaluation of eosinophils in the peribronchial infiltrate
Sections (5-µm-thick) were stained with hematoxylin and eosin (H&E) and used to measure the edema area (data not shown) and eosinophil density; counts were performed at a magnification of 1000× in the peribronchovascular space using the point-counting technique. Cell density was assessed as described above75.
Evaluation of iNOS- and ROCK-2-positive cells in the distal lung
Using the point-counting technique, we quantified the number of iNOS- and ROCK-2-positive cells divided by the number of points hitting the alveolar septa wall area in each field; 10–15 random non-overlapping fields were analyzed, and the results are expressed as 104 cells/area8.
Mean linear intercept (Lm)
The Lm was measured using a microscope with an integrating eyepiece containing a known area (50 lines and 100 points) at 200× magnification and was calculated as the number of times the reticular lines intercepted the alveolar walls in the distal lung parenchyma. For each animal, 20 non-overlapping fields were randomly analyzed. Values are expressed in micrometers. Sections (5-µm-thick) were stained with H&E56,76.
Morphometric analysis for interstitial edema (IE) evaluation
We used a weighted scoring system (scale 0 to 4) to quantify interstitial edema, where 0 represents no visible evidence and 4 represents complete involvement of each field. Briefly, by using a light microscope, the extent of each scored characteristic per field of view (score 0–4) was determined by four non-overlapping fields of view at 100× (0–16), 200× (0–16) and 400× (0–16) magnification77. Final score for each animal was determined as an average of the score at the 3 magnifications (IE = score at 100×+ score at 200×+ score at 400×/3) ranging from 0 to 16.
Measurement of cytokines in the lung homogenate
After removal, the right lung was homogenized and centrifuged at 900 × g for 7 minutes at 4 °C (Powerlyzer, Mo Bio Laboratories, Carlsbad, CA, USA), and the supernatant was stored at −70 °C until subsequent analysis. The levels of IL-4, IL-5 and IL-17 in the lung homogenate were measured using ELISA Duo Set Kits (R&D Systems, Minneapolis, MN, USA) according to the manufacturer’s instructions.
Measurement of lung mucus
After formalin fixation, the left lung was embedded in paraffin, and 5-µm-thick sections were stained with Schiff Periodic Acid and Alcian Blue (PAS-AB) at a pH of 2.578. An optical microscope with an integrating eyepiece (Weibel reticle)75 containing a known area (104 μm2 at a magnification of 1000×) of 50 lines and 100 points was used to measure the total (neutral and acidic) mucus content and the total airway epithelial area of non-cartilaginous airways (data not shown). The volume proportions of total mucus were obtained by dividing the total number of points hitting the positive mucus area by the number of total points hitting the airway epithelial area17. Morphometric measures were performed in five airways per animal according to previous studies17. The airways were chosen randomly according to optical scanning of each slide. All morphometric measurements were performed in a blinded fashion.
Measurement of nasal epithelium acid mucus
The nasal cavity was flushed through the nasopharyngeal orifice with 5 mL of 10% neutral-buffered formalin in a retrograde manner, and the head was fixed in formalin for 24 h and then decalcified in 5% ethylenediamine tetraacetic acid (EDTA) for two weeks. Next, 5-μm-thick sections were taken immediately from the nasal cavity from the posterior region to the upper incisor teeth79 and stained with PAS-AB78. The region of the nasal respiratory epithelium that was analyzed included the epithelium lining the nasal septum. Ten randomly selected fields from each slide were studied, and the number of points corresponding to the total area of the epithelium in each field was calculated. The acidic mucous substance was quantified as described above75,80.
Statistical analysis
Comparisons of the BALF from the first phase were performed using a t-test. One-way analysis of variance followed by the Holm-Sidak test was performed for other analyses for multiple comparisons17.
The significance level was adjusted to 5% (p < 0.05). Sigma Stat 3.5 software (San Jose, CA, USA) was used for all statistical analyses. All values are expressed as the mean ± standard error (SE).
Availability of materials and data
All data generated or analyzed during this study are included in this published article.
Change history
12 December 2018
A correction to this article has been published and is linked from the HTML and PDF versions of this paper. The error has not been fixed in the paper.
References
Woolcock, A. J. & Peat, J. K. Evidence for the increase in asthma worldwide. Ciba Foundation symposium 206, 122–134; discussion 134-129, 157-129 (1997).
Upton, M. N. et al. Intergenerational 20 year trends in the prevalence of asthma and hay fever in adults: the Midspan family study surveys of parents and offspring. Bmj 321, 88–92 (2000).
Avila, L. C. et al. Effects of High-Intensity Swimming on Lung Inflammation and Oxidative Stress in a Murine Model of DEP-Induced Injury. PloS one 10, e0137273, https://doi.org/10.1371/journal.pone.0137273 (2015).
Hiemstra, P. S., McCray, P. B. Jr. & Bals, R. The innate immune function of airway epithelial cells in inflammatory lung disease. The European respiratory journal 45, 1150–1162, https://doi.org/10.1183/09031936.00141514 (2015).
Parker, D. & Prince, A. Innate immunity in the respiratory epithelium. American journal of respiratory cell and molecular biology 45, 189–201, https://doi.org/10.1165/rcmb.2011-0011RT (2011).
Erle, D. J. & Sheppard, D. The cell biology of asthma. The Journal of cell biology 205, 621–631, https://doi.org/10.1083/jcb.201401050 (2014).
Prado, C. M., Martins, M. A. & Tiberio, I. F. Nitric oxide in asthma physiopathology. ISRN allergy 2011, 832560, https://doi.org/10.5402/2011/832560 (2011).
Righetti, R. F. et al. Effects of Rho-kinase inhibition in lung tissue with chronic inflammation. Respiratory physiology & neurobiology 192, 134–146, https://doi.org/10.1016/j.resp.2013.12.012 (2014).
Pigati, P. A. et al. Y-27632 is associated with corticosteroid-potentiated control of pulmonary remodeling and inflammation in guinea pigs with chronic allergic inflammation. BMC pulmonary medicine 15, 85, https://doi.org/10.1186/s12890-015-0073-4 (2015).
Taki, F. et al. Effects of Rho-kinase inactivation on eosinophilia and hyper-reactivity in murine airways by allergen challenges. Clinical and experimental allergy: journal of the British Society for Allergy and Clinical Immunology 37, 599–607, https://doi.org/10.1111/j.1365-2222.2007.02693.x (2007).
Hammerschmidt, S. & Wahn, H. The oxidants hypochlorite and hydrogen peroxide induce distinct patterns of acute lung injury. Biochimica et biophysica acta 1690, 258–264, https://doi.org/10.1016/j.bbadis.2004.07.003 (2004).
Winder, C. The toxicology of chlorine. Environmental research 85, 105–114, https://doi.org/10.1006/enrs.2000.4110 (2001).
Woods, C. G. et al. Dose-dependent transitions in Nrf2-mediated adaptive response and related stress responses to hypochlorous acid in mouse macrophages. Toxicology and applied pharmacology 238, 27–36, https://doi.org/10.1016/j.taap.2009.04.007 (2009).
Martin, J. G. et al. Chlorine-induced injury to the airways in mice. American journal of respiratory and critical care medicine 168, 568–574, https://doi.org/10.1164/rccm.200201-021OC (2003).
Bernard, A. Chlorination products: emerging links with allergic diseases. Current medicinal chemistry 14, 1771–1782 (2007).
Prokopowicz, Z. M. et al. Hypochlorous acid: a natural adjuvant that facilitates antigen processing, cross-priming, and the induction of adaptive immunity. Journal of immunology 184, 824–835, https://doi.org/10.4049/jimmunol.0902606 (2010).
Arantes-Costa, F. M. et al. Effects of residual oil fly ash (ROFA) in mice with chronic allergic pulmonary inflammation. Toxicologic pathology 36, 680–686, https://doi.org/10.1177/0192623308317427 (2008).
Cao, D. et al. Diesel exhaust particulate-induced activation of Stat3 requires activities of EGFR and Src in airway epithelial cells. American journal of physiology. Lung cellular and molecular physiology 292, L422–429, https://doi.org/10.1152/ajplung.00204.2006 (2007).
Takano, H. et al. Diesel exhaust particles enhance lung injury related to bacterial endotoxin through expression of proinflammatory cytokines, chemokines, and intercellular adhesion molecule-1. American journal of respiratory and critical care medicine 165, 1329–1335, https://doi.org/10.1164/rccm.2108122 (2002).
Halonen, J. I. et al. Urban air pollution, and asthma and COPD hospital emergency room visits. Thorax 63, 635–641, https://doi.org/10.1136/thx.2007.091371 (2008).
Park, E. J. et al. Biological responses to diesel exhaust particles (DEPs) depend on the physicochemical properties of the DEPs. PloS one 6, e26749, https://doi.org/10.1371/journal.pone.0026749 (2011).
Patel, H., Eo, S. & Kwon, S. Effects of diesel particulate matters on inflammatory responses in static and dynamic culture of human alveolar epithelial cells. Toxicology letters 200, 124–131, https://doi.org/10.1016/j.toxlet.2010.11.007 (2011).
Zhao, H., Barger, M. W., Ma, J. K., Castranova, V. & Ma, J. Y. Cooperation of the inducible nitric oxide synthase and cytochrome P450 1A1 in mediating lung inflammation and mutagenicity induced by diesel exhaust particles. Environmental health perspectives 114, 1253–1258 (2006).
Weisel, C. P. et al. Childhood asthma and environmental exposures at swimming pools: state of the science and research recommendations. Environmental health perspectives 117, 500–507, https://doi.org/10.1289/ehp.11513 (2009).
Helenius, I. J. et al. Respiratory symptoms, bronchial responsiveness, and cellular characteristics of induced sputum in elite swimmers. Allergy 53, 346–352 (1998).
Langdeau, J. B. et al. Airway hyperresponsiveness in elite athletes. American journal of respiratory and critical care medicine 161, 1479–1484, https://doi.org/10.1164/ajrccm.161.5.9909008 (2000).
Racioppi, F. et al. Household bleaches based on sodium hypochlorite: review of acute toxicology and poison control center experience. Food and chemical toxicology: an international journal published for the British Industrial Biological Research Association 32, 845–861 (1994).
Gorguner, M., Aslan, S., Inandi, T. & Cakir, Z. Reactive airways dysfunction syndrome in housewives due to a bleach-hydrochloric acid mixture. Inhalation toxicology 16, 87–91, https://doi.org/10.1080/08958370490265004 (2004).
Tarlo, S. M. et al. Diagnosis and management of work-related asthma: American College Of Chest Physicians Consensus Statement. Chest 134, 1S–41S, https://doi.org/10.1378/chest.08-0201 (2008).
Henneberger, P. K. et al. An official american thoracic society statement: work-exacerbated asthma. American journal of respiratory and critical care medicine 184, 368–378, https://doi.org/10.1164/rccm.812011ST (2011).
Lemière, C., Boulet, L. P. & Cartier, A. Reactive airways dysfunction syndrome and irritant induced asthma, http://www.uptodate.com/contents/reactive-airways-dysfunction-syndrome-and-irritant-induced-asthma (2016).
Pawankar, R., Canonica, G. W., Holgate, S. T., Lockey, R. F. & Blaiss, M. World Allergy Organization (WAO) White Book on Allergy. (2013).
Quirce, S. & Barranco, P. Cleaning agents and asthma. Journal of investigational allergology & clinical immunology 20, 542-550; quiz 542p following 550 (2010).
OSHA. Occupational Safety and Health Administration, https://www.osha.gov/dts/chemicalsampling/data/CH_226500.html.
Medina-Ramon, M. et al. Asthma, chronic bronchitis, and exposure to irritant agents in occupational domestic cleaning: a nested case-control study. Occupational and environmental medicine 62, 598–606, https://doi.org/10.1136/oem.2004.017640 (2005).
Hox, V. et al. Airway exposure to hypochlorite prior to ovalbumin induces airway hyperreactivity without evidence for allergic sensitization. Toxicology letters 204, 101–107, https://doi.org/10.1016/j.toxlet.2011.04.017 (2011).
Kim, S. H., Park, D. E., Lee, H. S., Kang, H. R. & Cho, S. H. Chronic low dose chlorine exposure aggravates allergic inflammation and airway hyperresponsiveness and activates inflammasome pathway. PloS one 9, e106861, https://doi.org/10.1371/journal.pone.0106861 (2014).
Cartier, A. & Sastre, J. Clinical assessment of occupational asthma and its differential diagnosis. Immunology and allergy clinics of North America 31, 717–728, vi, https://doi.org/10.1016/j.iac.2011.07.005 (2011).
Kenyon, N. J., Morrissey, B. M., Schivo, M. & Albertson, T. E. Occupational asthma. Clinical reviews in allergy & immunology 43, 3–13, https://doi.org/10.1007/s12016-011-8272-0 (2012).
Cowl, C. T. Occupational asthma: review of assessment, treatment, and compensation. Chest 139, 674–681, https://doi.org/10.1378/chest.10-0079 (2011).
Goe, S. K. et al. A descriptive study of work aggravated asthma. Occupational and environmental medicine 61, 512–517 (2004).
Occupational asthma-identification, management and prevention: evidence based review and guidelines. British Occupational Health Research Foundation, http://www.bohrf.org.uk/downloads/OccupationalAsthma (2010).
Berger, Z. et al. Prevalence of workplace exacerbation of asthma symptoms in an urban working population of asthmatics. Journal of occupational and environmental medicine/American College of Occupational and Environmental Medicine 48, 833–839, https://doi.org/10.1097/01.jom.0000225169.45337.97 (2006).
Fishwick, D. et al. Standards of care for occupational asthma: an update. Thorax 67, 278–280, https://doi.org/10.1136/thoraxjnl-2011-200755 (2012).
Jares, E. J., Baena-Cagnani, C. E. & Gomez, R. M. Diagnosis of occupational asthma: an update. Current allergy and asthma reports 12, 221–231, https://doi.org/10.1007/s11882-012-0259-2 (2012).
Hizume, D. C. et al. Cigarette smoke dissociates inflammation and lung remodeling in OVA-sensitized and challenged mice. Respiratory physiology & neurobiology 181, 167–176, https://doi.org/10.1016/j.resp.2012.03.005 (2012).
Deschamps, D., Soler, P., Rosenberg, N., Baud, F. & Gervais, P. Persistent asthma after inhalation of a mixture of sodium hypochlorite and hydrochloric acid. Chest 105, 1895–1896 (1994).
Schraufstatter, I. U. et al. Mechanisms of hypochlorite injury of target cells. The Journal of clinical investigation 85, 554–562, https://doi.org/10.1172/JCI114472 (1990).
Vieira, R. P. et al. Aerobic conditioning and allergic pulmonary inflammation in mice. II. Effects on lung vascular and parenchymal inflammation and remodeling. American journal of physiology. Lung cellular and molecular physiology 295, L670–679, https://doi.org/10.1152/ajplung.00465.2007 (2008).
Vieira, R. P. et al. Aerobic exercise decreases chronic allergic lung inflammation and airway remodeling in mice. American journal of respiratory and critical care medicine 176, 871–877, https://doi.org/10.1164/rccm.200610-1567OC (2007).
Hewitt, M., Estell, K., Davis, I. C. & Schwiebert, L. M. Repeated bouts of moderate-intensity aerobic exercise reduce airway reactivity in a murine asthma model. American journal of respiratory cell and molecular biology 42, 243–249, https://doi.org/10.1165/rcmb.2009-0038OC (2010).
Robinson, D. S. et al. Predominant TH2-like bronchoalveolar T-lymphocyte population in atopic asthma. The New England journal of medicine 326, 298–304, https://doi.org/10.1056/NEJM199201303260504 (1992).
Bruggemann, T. R. et al. Effects of Swimming on the Inflammatory and Redox Response in a Model of Allergic Asthma. International journal of sports medicine. https://doi.org/10.1055/s-0035-1549904 (2015).
Schwarze, P. E. et al. Inflammation-related effects of diesel engine exhaust particles: studies on lung cells in vitro. BioMed research international 2013, 685142, https://doi.org/10.1155/2013/685142 (2013).
Chaudhuri, N. et al. Diesel exhaust particles override natural injury-limiting pathways in the lung. American journal of physiology. Lung cellular and molecular physiology 299, L263–271, https://doi.org/10.1152/ajplung.00297.2009 (2010).
Camargo, L. D. N. et al. Frontiers in immunology 8, 1835, https://doi.org/10.3389/fimmu.2017.01835 (2017). Effects of Anti-IL-17 onInflammation, Remodeling, and Oxidative Stress in an Experimental Model of Asthma Exacerbated by LPS.
Leppanen, T., Tuominen, R. K. & Moilanen, E. Protein kinase C and its inhibitors in the regulation of inflammation: inducible nitric oxide synthase as an example. Basic & clinical pharmacology & toxicology 114, 37–43, https://doi.org/10.1111/bcpt.12139 (2014).
Pautz, A. et al. Regulation of the expression of inducible nitric oxide synthase. Nitric oxide: biology and chemistry 23, 75–93, https://doi.org/10.1016/j.niox.2010.04.007 (2010).
White, C. W. & Martin, J. G. Chlorine gas inhalation: human clinical evidence of toxicity and experience in animal models. Proceedings of the American Thoracic Society 7, 257–263, https://doi.org/10.1513/pats.201001-008SM (2010).
Possa, S. S. et al. Rho-kinase inhibition attenuates airway responsiveness, inflammation, matrix remodeling, and oxidative stress activation induced by chronic inflammation. American journal of physiology. Lung cellular and molecular physiology 303, L939–952, https://doi.org/10.1152/ajplung.00034.2012 (2012).
Taherali, F., Varum, F. & Basit, A. W. A slippery slope: On the origin, role and physiology of mucus. Advanced drug delivery reviews 124, 16–33, https://doi.org/10.1016/j.addr.2017.10.014 (2018).
Fahy, J. V. & Dickey, B. F. Airway mucus function and dysfunction. The New England journal of medicine 363, 2233–2247, https://doi.org/10.1056/NEJMra0910061 (2010).
Seriani, R. et al. Diesel exhaust particulates affect cell signaling, mucin profiles, and apoptosis in trachea explants of Balb/C mice. Environmental toxicology 30, 1297–1308, https://doi.org/10.1002/tox.22000 (2015).
Lemos, M. et al. Quantitative pathology of nasal passages in rats exposed to urban levels of air pollution. Environmental research 66, 87–95 (1994).
Fliss, H. Oxidation of proteins in rat heart and lungs by polymorphonuclear leukocyte oxidants. Molecular and cellular biochemistry 84, 177–188 (1988).
Siracusa, A. et al. Asthma and exposure to cleaning products - a European Academy of Allergy and Clinical Immunology task force consensus statement. Allergy 68, 1532–1545, https://doi.org/10.1111/all.12279 (2013).
Bello, A., Quinn, M. M., Perry, M. J. & Milton, D. K. Quantitative assessment of airborne exposures generated during common cleaning tasks: a pilot study. Environmental health: a global access science source 9, 76, https://doi.org/10.1186/1476-069X-9-76 (2010).
Dahlen, B. & Dahlen, S. E. Leukotrienes as mediators of airway obstruction and inflammation in asthma. Clinical and experimental allergy: journal of the British Society for Allergy and Clinical Immunology 25(Suppl 2), 50–54 (1995).
James, A. L., Pare, P. D. & Hogg, J. C. The mechanics of airway narrowing in asthma. The American review of respiratory disease 139, 242–246, https://doi.org/10.1164/ajrccm/139.1.242 (1989).
O’Byrne, P. M. Leukotrienes in the pathogenesis of asthma. Chest 111, 27S–34S (1997).
National Research Council of The National Academies: Guide for the care and use of laboratory animals. 8th edn, (Natl Acad Press, 2011).
Adler, A., Cieslewicz, G. & Irvin, C. G. Unrestrained plethysmography is an unreliable measure of airway responsiveness in BALB/c and C57BL/6 mice. Journal of applied physiology 97, 286–292, https://doi.org/10.1152/japplphysiol.00821.2003 (2004).
De Soete, D., Gijbels, R. & Hoste, J. Neutron activation analysis. (Wiley-Interscience, 1972).
Martins-Olivera, B. T. et al. The Plant-Derived Bauhinia bauhinioides Kallikrein Proteinase Inhibitor (rBbKI) Attenuates Elastase-Induced Emphysema in Mice. Mediators of inflammation 2016, 5346574, https://doi.org/10.1155/2016/5346574 (2016).
Weibel, E. R., Kistler, G. S. & Scherle, W. F. Practical stereological methods for morphometric cytology. The Journal of cell biology 30, 23–38 (1966).
Lourenco, J. D. et al. The tick-derived rBmTI-A protease inhibitor attenuates the histological and functional changes induced by cigarette smoke exposure. Histology and histopathology 33, 289–298, https://doi.org/10.14670/HH-11-927 (2018).
Kiss, T. et al. Comparison of different degrees of variability in tidal volume to prevent deterioration of respiratory system elastance in experimental acute lung inflammation. British journal of anaesthesia 116, 708–715, https://doi.org/10.1093/bja/aew093 (2016).
Jones, R. & Reid, L. Secretory cell hyperplasia and modification of intracellular glycoprotein in rat airways induced by short periods of exposure to tobacco smoke, and the effect of the antiinflammatory agent phenylmethyloxadiazole. Laboratory investigation; a journal of technical methods and pathology 39, 41–49 (1978).
Herbert, R. A. & Leininger, J. R. In Pathology of the Mouse: Reference and Atlas (ed R. R. Maronpot) 259–292 (1999).
Cruz-Orive, L. M. & Weibel, E. R. Recent stereological methods for cell biology: a brief survey. The American journal of physiology 258, L148–156 (1990).
Acknowledgements
This work was supported by Sao Paulo Research Foundation, (FAPESP) grant 2009/53904-9 and grant 2012/15165-2, Experimental Therapeutic Laboratory I (LIM 20), School of Medicine, University of Sao Paulo, School of Medicine Foundation (FFM/USP) and Financial Assistance to Educational Project or Research (AUXPE/PROAP) grant 2696/2013.
Author information
Authors and Affiliations
Contributions
I.G., F.A., D.K., H.M., R.S., R.V., M.M., I.T., F.C., B.R., contributed to design, data analysis, wrote and prepared the manuscript. I.G., F.A., J.C., R.L., R.R., R.V., M.S. contributed to data generation. All authors contributed to the revising of the manuscript and read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
de Genaro, I.S., de Almeida, F.M., Hizume-Kunzler, D.C. et al. Low dose of chlorine exposure exacerbates nasal and pulmonary allergic inflammation in mice. Sci Rep 8, 12636 (2018). https://doi.org/10.1038/s41598-018-30851-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-30851-6
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
